

Abstract
The zinc finger antiviral protein (ZAP) is a recently isolated host antiviral factor. It specifically inhibits the replication of Moloney murine leukemia virus (MLV) and Sindbis virus (SIN) by preventing the accumulation of viral RNA in the cytoplasm. For this report, we mapped the viral sequences that are sensitive to ZAP inhibition. The viral sequences were cloned into a luciferase reporter and analyzed for the ability to mediate ZAP-dependent destabilization of the reporter. The sensitive sequence in MLV was mapped to the 3′ long terminal repeat; the sensitive sequences in SIN were mapped to multiple fragments. The fragment of SIN that displayed the highest destabilizing activity was further analyzed by deletion mutagenesis for the minimal sequence that retained the activity. This led to the identification of a fragment of 653 nucleotides. Any further deletion of this fragment resulted in significantly lower activity. We provide evidence that ZAP directly binds to the active but not the inactive fragments. The CCCH zinc finger motifs of ZAP play important roles in RNA binding and antiviral activity. Disruption of the second and fourth zinc fingers abolished ZAP's activity, whereas disruption of the first and third fingers just slightly lowered its activity.
The zinc finger antiviral protein (ZAP) was originally recovered from a screen for genes conferring resistance to the infection of cells by Moloney murine leukemia virus (MLV) (11). The overexpression of ZAP rendered cells 30-fold more resistant to viral infection. An analysis to determine the step at which ZAP blocked virus infection revealed that in ZAP-expressing cells, reverse transcription and nuclear entry of the viral DNA were normal but the production of viral RNA in the cytoplasm was inhibited (11). In addition to its inhibition of MLV, ZAP potently inhibits the replication of multiple members of the Alphavirus genus of the Togaviridae family, including Sindbis virus (SIN), Semliki Forest virus, Ross River virus, and Venezuelan equine encephalitis virus (3). The expression of ZAP does not induce a broad-spectrum antiviral state, as some viruses, including herpes simplex virus type 1 and yellow fever virus, grow normally in ZAP-expressing cells (3). ZAP targets SIN at a stage after binding and penetration, and it prevents translation of the incoming viral RNA (3). Given that alphaviruses are replicated entirely in an RNA state in the cytoplasm (19) and that the production of MLV viral RNA was inhibited only in the cytoplasm, it is tempting to propose that a common mechanism occurring in the cytoplasm underlies the ZAP-mediated elimination of MLV and SIN viral RNAs. However, since there is no obvious sequence homology between MLV and SIN, the common feature(s) shared by these two divergent viruses to account for their sensitivity to ZAP remained elusive.
Sequence analysis revealed that in the N terminus of ZAP there are four CCCH-type zinc finger motifs. A fragment of 254 amino acids of the N terminus (NZAP) containing the four zinc finger motifs displayed the same antiviral activity as full-length ZAP when fused to the Zeocin resistance gene (NZAP-Zeo) (11), further highlighting the importance of these motifs. Similar CCCH-type zinc finger motifs are also found in tristetraprolin (TTP) (9), which specifically binds to the AU-rich element (ARE) (12) and recruits the exosome to degrade ARE-containing RNAs (6). Disruption of the zinc fingers results in the loss of the binding capability of TTP to the target RNA and thereby the destabilizing activity (5, 12, 14). The similar CCCH-type zinc finger motifs shared by ZAP and TTP, along with the observation that ZAP specifically prevents the accumulation of viral RNA in the cytoplasm, suggested that ZAP and TTP may share a similar RNA destabilizing mechanism.
In the present study, we describe the evidence that ZAP, unlike TTP, does not destabilize ARE-containing RNAs. The target sequences of ZAP in MLV and SIN were mapped to the 3′ long terminal repeat (3′-LTR) of MLV and to multiple fragments in the SIN genome. ZAP binds directly to these RNAs, and the CCCH finger motifs are required for binding and for ZAP's antiviral activity.
MATERIALS AND METHODS
Plasmid construction.
The plasmid pBabe-NZAP-Zeo has been described previously (11). pBabe-H86K, pBabe-C88R, pBabe-C168R, and pBabe-H191R express each of the four zinc finger mutants of NZAP-Zeo. The H86K mutant, in which the histidine of the first CCCH zinc finger motif was replaced with lysine, was generated by replacing the EcoRI-NotI fragment of NZAP with EcoRI-AflII and AflII-NotI PCR-derived fragments. The EcoRI-AflII fragment was generated by using the forward primer SP (5′-GCTTATCCATATGATGTTCCA-3′) upstream of the EcoRI site and the reverse primer H86K-AP (5′-ATATAGCTTAAGGCTGTCGCAGGGTCTCTG-3′), bearing silent mutations to create an AflII site (underlined) and the mutant Lys codon (in bold); the AflII-NotI fragment was generated by using the forward primer H86K-FP (5′-ATATAGCTTAAGCTCTGCAAGCTTAATCTGC-3′), bearing silent mutations to create an AflII site (underlined), and the reverse primer AP (5′-TCCAGAACTCGACCGCT-3′) downstream of the NotI site. The same strategy was employed to generate the other zinc finger mutants. In the following description of plasmid construction, the restriction sites built into the primers are underlined and the mutation codons are shown in bold. The primers used to generate the mutations were as follows: C88R-AP (5′-ATATAGTTCGAAGGTGCAGGCTGTCGCAG-3′) and C88R-SP (5′-ATATAGTTCGAAAGCTTAATCTGCTCGGC-3′), bearing a BstBI site to generate the C88R mutant, in which the first cysteine residue of the second CCCH zinc finger motif was replaced with arginine; C168R-AP (5′-ATATAGCCTAGGCTGTGGCTGCCCACAG-3′) and C168R-SP (5′-ATATAGCCTAGGGAGAGACTCCACATCTGT-3′), bearing an AvrII site to generate the C168R mutant, in which the third cysteine of the third CCCH zinc finger motif was replaced with arginine; and H191R-AP (5′-ATATAGTCTAGACCTGAGACAGTTGAGG-3′) and H191R-SP (5′-ATATAGTCTAGAAACCTGATGGACAGAAAGG-3′), bearing an XbaI site to generate the H191R mutant, in which the histidine of the fourth CCCH zinc finger motif was replaced with arginine.
pcDNA4/TO/myc-ZAP was previously described as pZAP-myc (11). pcDNA4/TO/myc-ZAP-C88R expresses Myc-tagged full-length ZAP containing the C88R mutation. To create pcDNA4/TO/myc-ZAP-C88R, we replaced the EcoRI-NheI fragment of pcDNA4/TO/myc-ZAP with the EcoRI-NheI fragment of pBabe-C88R.
pcDNA4/TO/myc-NZAP was previously described as pNZAP-myc (11). pcDNA4/TO/myc-NZAP-Zeo expresses NZAP-Zeo with a Myc tag fused to the C terminus. To generate NZAP-Zeo-myc, we PCR amplified the Zeo coding sequence from pBabe-NZAP-Zeo, using the forward primer 5′-GACAGAAGCAAAAGCAGAGAC-3′ (right before the NotI site) and the reverse primer 5′-ATATAGTCTAGAGGGTCCTGCTCCTCGGCCACGAA-3′ to introduce an XbaI site. The PCR product was inserted into pcDNA4/TO/myc-NZAP by use of the NotI and XbaI sites to generate pcDNA4/TO/myc-NZAP-Zeo.
pcDNA4/TO/myc-TTP expresses human TTP with a Myc tag fused to the C terminus. The TTP coding sequence was PCR amplified from an EST clone by use of the forward primer TTP-SP (5′-ATATAGCTTAAGCCACCATGGATCTGACTGCC-3′), bearing an AlfII site, and the reverse primer TTP-AP (5′-ATATAGGCGGCCGCCCTCAGAAACAGAGATGCG-3′), by which a NotI site was built in frame with the Myc tag coding sequence in the expression vector. The PCR product was cloned into the pcDNA4/TO/myc-HisB (Invitrogen) vector to generate pcDNA4/TO/myc-TTP.
pGL3-Luc and MLV-Luc have been described previously (11). pGL3-5′LTR was generated by replacing the SacI-HindIII fragment with the EcoRI-HindIII 5′-LTR fragment of MLV-Luc. For the convenience of constructing pGL3-3′LTR, pGL3-Luc was first modified to destroy the BamHI site in the vector. A linker bearing multiple cloning sites, including a new BamHI site, was inserted into the XbaI site located downstream of the luciferase coding sequence to create pGL3-Luc-linker. The SphI-BamHI fragment of pGL3-Luc-linker was replaced with a 2.3-kb SphI-HindIII fragment containing part of the luciferase coding sequence and the 3′-LTR of MLV-Luc to generate pGL3-3′LTR.
To map the sequences that were responsive to ZAP, we digested pToto1101, an infectious clone of SIN (18), with BstYI to generate seven fragments and inserted each fragment into the BamHI site in the pGL3-Luc-linker in either a sense or antisense orientation. The deletion mutants of the SIN fragments were generated by PCRs with primers bearing a BamHI site upstream of the matching sequences. The resulting fragments were inserted into the BamHI site in pGL3-Luc-linker.
To test whether ZAP targets ARE sequences, we cloned three different types of AREs into pGL3-Luc-linker downstream of the luciferase coding sequence. A stretch of sequence containing the ARE of tumor necrosis factor alpha was generated by annealing two oligonucleotides, 5′-GATCTTATTTATTATTTATTTATTATTTATTTATT-3′ and 3′-AATAAATAATAAATAAATAATAAATAAATAACTAG-5′, and was cloned into the BamHI site to create pGL3-ARE(II)-Luc. The c-fos ARE was amplified from the total RNA of HeLa cells by reverse transcription-PCR using the forward primer 5′-CGGAATTCGGCCTGGGTCTGTGTCTCTTTTC-3′ and the reverse primer 5′-ATATAGGGATCCGACAATGTCTTGGAACAATAAGC-3′. The c-jun ARE was amplified from the total RNA of NIH 3T3 cells by reverse transcription-PCR using the forward primer 5′-CGGAATTCCATTGACCAAGAACTGCATGG-3′ and the reverse primer 5′-ATATAGGGATCCGGTATTTGAATACATTTATTGTG-3′. The PCR products amplified from c-fos and c-jun were cloned into pGL3-Luc-linker to generate pGL3-ARE(I)-Luc and pGL3-ARE(III)-Luc, respectively.
Cell culture.
All cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum. Rat2-HA-Zeo and Rat2-NZAP-Zeo cells were described previously (11). The pBabe-NZAP-Zeo-based plasmid constructs containing mutations in the zinc finger motifs were packaged into MLV pseudoviruses to transduce Rat2 cells. The cells were selected with 100 μg of Zeocin/ml, and Zeocin-resistant cells were pooled to generate Rat2 cells expressing the NZAP-Zeo mutants. The ecotropic MLV-Luc pseudovirus, Eco-Luc, has been previously reported (10). Retroviral infection was conducted for 3 h followed by replacement of the infection medium with fresh medium. SIN infection and titration have been previously described (19). Briefly, cells were seeded in six-well dishes the day prior to infection. The next day, the cells were infected with the Toto1101 virus for 1 h at a multiplicity of infection of 0.01 PFU per cell. The titer of the stock was determined on Rat2-HA-Zeo cells. After infection, the cells were washed twice with medium, and 3 ml of fresh medium was added. At 24 h postinfection, the supernatants were collected and titrated in duplicate wells of permissive BHK-J cells. Transfection was performed by the use of Fugene 6 (Roche Diagnostics) according to the manufacturer's instructions. For all transfection experiments, pRL-TK (Promega), a Renilla luciferase reporter that is not sensitive to ZAP at all, was included to normalize the transfection efficiencies. The luciferase activity was measured 48 h after transfection or infection by use of a luciferase assay system (Promega).
293TRex cells (Invitrogen), which stably express the repressor of the Tet operon, were maintained according to the provider's instructions. To generate the 293TRex-ZAP cell line, we stably transfected 293TRex cells with pcDNA4/TO/myc-ZAP and selected for Zeocin resistance. Individual clones were picked, expanded, and tested for tetracycline-inducible protein expression by Western blotting. To generate Rat2-NZAP-Zeo-myc cells, we stably transfected Rat2 cells with pcDNA4/TO/myc-NZAP-Zeo and used Zeocin for selection. The Zeocin-resistant cells were pooled.
Northern blotting.
Rat2 cells were infected with the Eco-Luc virus or transfected with the pGL3-SIN-D(−)-Luc reporter. Forty-eight hours later, total RNAs were isolated from the cells by use of an RNeasy kit (Qiagen) according to the manufacturer's instructions. The RNA samples were separated by electrophoresis, transferred to a nylon membrane, and hybridized for 15 to 20 h with 32P-labeled probes prepared by use of a random primer labeling kit (Stratagene). The nylon membrane was washed three times with 0.1× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate) and 0.1% sodium dodecyl sulfate at 42°C and was then exposed to X-ray film.
In vitro RNA binding assay.
Rat2-NZAP-Zeo-myc cells or 293TRex-ZAP cells treated with tetracycline to induce ZAP expression were lysed in lysis buffer A (25 mM Tris-phosphate [pH 7.8], 2 mM dithiothreitol, 2 mM 1,2-diaminocyclohexane-N,N,N′,N′-tetraacetic acid, 10% glycerol, 1% Triton X-100). The lysates were clarified by centrifugation at 13,000 rpm (Sorvall Biofuge, Fresco) for 10 min. The Myc-tagged proteins were immobilized to protein G-agarose resin (Santa Cruz) by use of the 9E10 antibody (Santa Cruz). The pellets were washed three times in RNase-free binding buffer (10 mM Tris-HCl [pH 7.5], 50 mM NaCl, 1 mM EDTA, and 10 μM ZnCl2) and resuspended in 30 μl of binding buffer supplemented with 40 U of RNasin (Promega), 1 μg of heparin/μl, and 200 ng of yeast tRNA/μl. Special care was taken during washing not to lose any of the resin.
For the preparation of RNA probes, the cDNAs were cloned into pBluescript. The plasmids were linearized by restriction digestion and used as templates to transcribe the RNA probes in the presence of [α-32P]UTP (Amersham) by use of the Riboprobe system (Promega) according to the manufacturer's instructions. The labeled RNA probes were purified with Sephadex G-25 spin columns (Roche Diagnostics).
The 32P-labeled RNA probe (2 × 105 cpm in a total volume of 1 μl) was incubated with 30 μl of the ZAP protein immobilized on the agarose resin in binding buffer supplemented with 40 U of RNasin (Promega), 1 μg of heparin/μl, and 200 ng of yeast tRNA/μl at room temperature for 30 min. The resins were washed three times with the binding buffer, resuspended, and divided into two equal aliquots, one for detection of the bound RNA by urea-polyacrylamide gel electrophoresis and the other for detection of the ZAP protein by Western blotting.
RESULTS
Mapping the target RNA sequence in the MLV vector.
ZAP can specifically prevent the accumulation of MLV and SIN viral RNAs (3, 11), suggesting that ZAP may target some specific viral RNA sequences. To map the target sequence of ZAP in the MLV vector, we cloned the 5′-LTR and 3′-LTR of the MLV-Luc retroviral vector into the pGL3-Luc reporter, which is nearly insensitive to ZAP (Fig. 1), and assayed the clones for their ability to mediate the response to ZAP. The constructs were assayed in both Rat2 cells expressing NZAP-Zeo (Fig. 1) and 293 cells expressing full-length ZAP (data not shown), and similar results were obtained. A Renilla luciferase reporter, pRL-TK, which is not inhibited by ZAP (Fig. 1), served to normalize the transfection efficiencies. The inhibition (n-fold) was calculated as the normalized luciferase activity expressed in Rat2-HA-Zeo cells divided by the normalized luciferase activity expressed in Rat2-NZAP-Zeo cells. As shown in Fig. 1, replacement of the promoter sequence of pGL3-Luc with the 5′-LTR did not change the response to ZAP at all. However, cloning of the 3′-LTR downstream of the luciferase coding sequence rendered the reporter responsive to ZAP to a similar extent as MLV-Luc. These results indicated that the target sequence of ZAP in MLV-Luc is located in the 3′-LTR. Furthermore, these results also indicated that the target sequence of ZAP functions when it is cloned downstream of the luciferase coding sequence.
FIG. 1.
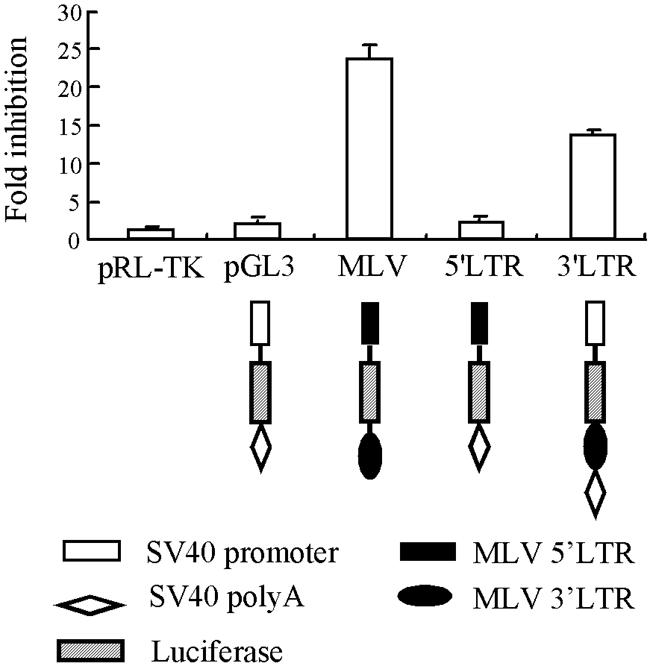
Mapping the target sequence of ZAP in the MLV-Luc vector. The 5′-LTR or 3′-LTR of MLV-Luc was cloned into the control reporter pGL3-Luc as indicated. The resultant constructs were transfected into Rat2-HA-Zeo or Rat2-NZAP-Zeo cells. At 48 h posttransfection, the cells were lysed and the luciferase activities were measured. The expression level of pRL-TK, a plasmid expressing Renilla luciferase, was used to normalize the transfection efficiencies. The inhibition (n-fold) was calculated as the normalized luciferase activity expressed in Rat2-HA-Zeo cells divided by the normalized luciferase activity expressed in Rat2-NZAP-Zeo cells. The data are means plus standard deviations SD of at least three independent experiments.
Mapping the RNA target sequence in the SIN genome.
To further characterize the features of the target sequence of ZAP, we digested the infectious clone of SIN (pToto1101) (18) into seven fragments by use of the restriction endonuclease BstYI (Fig. 2A) and individually cloned the fragments into the pGL3-Luc reporter between the luciferase coding sequence and the poly(A) signal. ZAP is able to prevent translation of the incoming SIN viral RNA (3) and thus likely affects the SIN plus-strand RNA. However, SIN replication involves the production of a minus-strand RNA which is utilized to generate an additional plus-strand genomic RNA (19), and this minus-strand RNA might also be targeted by ZAP. The fragments were therefore cloned into the reporter in both sense [(+)] and antisense [(−)] orientations and analyzed for sensitivity to ZAP in the luciferase reporter assay.
FIG. 2.

Mapping the target sequences of ZAP in the SIN genome. The test sequences of SIN were cloned into pGL3-Luc-linker between the luciferase coding sequence and the poly(A) signal in either a (+) or (−) orientation. The numbers indicate the positions of the ends of the fragments in the SIN genomic clone. The constructs were transfected into Rat2-HA-Zeo or Rat2-NZAP-Zeo cells. At 48 h posttransfection, the cells were lysed and analyzed for inhibition as described in the legend to Fig. 1. The data are means plus SD of at least three independent experiments. (A) The infectious clone of SIN was divided by restriction digestion into fragments, designated as indicated, and each fragment was tested for inhibition. V, control vector pGL-3-Luc-linker. (B) The D fragment from panel A was truncated from the 5′ or 3′ end, as indicated, and analyzed for inhibition. M, a fragment of D retaining most of the sensitivity. (C) The M fragment from panel B was truncated from the 5′ or 3′ end, as indicated. The fragments were cloned into pGL3-Luc-linker in the antisense orientation. N, a fragment retaining most of the sensitivity of M. (D) The N fragment from panel C was further analyzed by deletion mutagenesis, as indicated. The fragments were cloned into pGL3-Luc-linker in the antisense orientation.
Most of the fragments destabilized the reporter in the ZAP-expressing cells to various extents in either orientation, suggesting that the target sequences of ZAP are located at multiple sites in the SIN genome (Fig. 2A). Fragment D displayed the highest level of inhibition, about 50-fold, in either orientation (Fig. 2A). This fragment, which spans nucleotides (nt) 4633 to 7334 of the SIN genome, was further analyzed to identify the minimal functional domain. A series of deletion mutants from both the 5′ and 3′ ends of fragment D were cloned into the pGL3-Luc reporter in either orientation and then analyzed for sensitivity to ZAP (Fig. 2B). Removal of the sequence up to nt 7018 from the 3′ end (fragment Da) had no effect in either orientation, but further deletion resulted in a somewhat lower sensitivity in the (+) orientation. Deletion of the 5′ end sequence affected the (+) orientation more than the − orientation (Fig. 2B). In the (−) orientation, deletion of the sequence up to nt 5628 from the 5′ end (fragment Dg) had little effect, but further deletion (fragment Dh) significantly lowered the sensitivity. These results indicated that the sequence between nt 5646 and 7018 contains the element that is responsive to ZAP and is more effective when cloned in the (−) orientation than in the (+) orientation. Indeed, this fragment (M) conferred almost the same sensitivity as fragment D (Fig. 2B) to the reporter.
Fragment M was further mapped by finer serial deletions (Fig. 2C). Since most fragments displayed a higher or the same destabilizing activity in the (−) orientation than in the (+) orientation, for the following mapping experiments the test fragments were cloned only in the (−) orientation. Deletion from the 5′ end up to nt 6062 (fragment Md) did not significantly affect the sensitivity to ZAP, but further deletion (fragment Me) resulted in a dramatic drop in the sensitivity. The sequence at the 3′ end of fragment M that contributed to the sensitivity was mapped with another series of deletions made from a fragment spanning nt 5943 to 6915 (fragment Mi), which retained most of the activity of fragment M (Fig. 2C). Deletions up to nt 6715 caused little change in the inhibition. This analysis highlighted the sequence between nt 6062 and 6715 as the core of the target sequence of ZAP. Indeed, fragment N (spanning nt 6062 to 6715) displayed a 33-fold level of inhibition (Fig. 2C), in comparison with the 45-fold level of inhibition displayed by fragment M. Any further truncation of fragment N resulted in a dramatic drop in sensitivity (Fig. 2D).
ZAP does not destabilize ARE-containing RNAs.
A sequence analysis of the MLV 3′-LTR and the ZAP-sensitive SIN fragments did not identify any obvious common motifs or the presence of potential AREs. Nevertheless, based on the wide distribution of AREs in mRNAs and their engagement in mediating RNA destabilization (1, 2, 8, 15, 21), we were tempted to analyze whether ZAP destabilizes ARE-containing RNAs. AREs are classified into three types (8, 21). Type I AREs contain one to three AUUUA pentamers and a nearby U-rich region, type II AREs contain at least two overlapping copies of a UUAUUU(U/A)(U/A)U nonamer in a U-rich environment, and type III AREs contain U-rich stretches but not any AUUUA pentamers. All three types of AREs were tested. The AREs of the c-fos, tumor necrosis factor alpha, and c-jun mRNAs were chosen as representatives of type I, II, and III AREs (7, 8, 12, 20), respectively, and were cloned into the pGL3-Luc reporter downstream of the luciferase coding sequence. The ARE-containing reporters were cotransfected with an empty vector or a TTP- or ZAP-expressing vector into 293A cells and analyzed for luciferase activity. When the ARE-containing reporters were cotransfected with the empty vector, their expression levels were obviously lower than that of the pGL3-Luc reporter (Fig. 3, open bars), presumably due to the endogenous ARE targeting mechanisms. The overexpression of TTP further reduced the expression of type I and type II ARE reporters but had little effect on the type III ARE reporter (Fig. 3, hatched bars). In contrast, the overexpression of ZAP significantly increased the expression of the ARE reporters, especially the type I and type II ARE reporters (Fig. 3, solid bars). Based on these results, we concluded that ZAP does not target any of the three types of AREs. The increased reporter expression may have been due to the sequestration of the endogenous ARE decay machinery by ZAP overexpression.
FIG. 3.
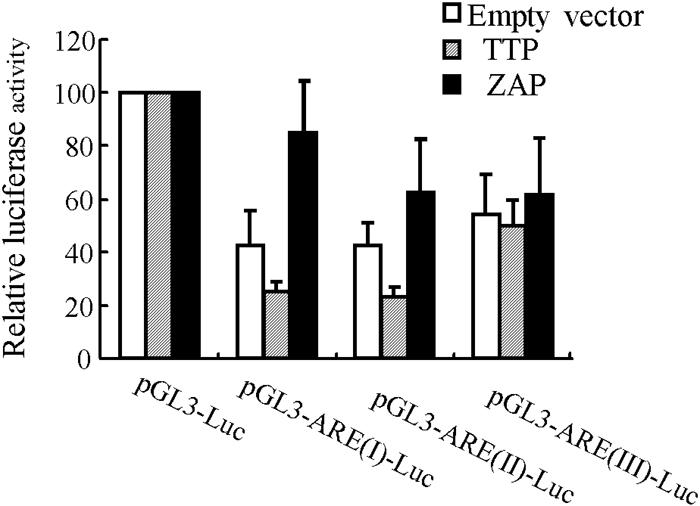
ZAP does not target AREs. pGL3-Luc reporters containing different types of AREs were cotransfected into 293A cells with an empty vector or a vector expressing TTP or ZAP. At 48 h posttransfection, the cells were lysed and the luciferase activities were measured. The luciferase activity in cells transfected with the pGL3-Luc reporters was arbitrarily defined as 100. The data are means plus SD of three independent experiments.
The integrity of the second and fourth zinc finger motifs is required for the activity of ZAP.
In the N terminus of ZAP there are four CCCH-type zinc finger motifs. Similar motifs have been shown to be critical for the sequence-specific RNA-destabilizing activity of TTP (5, 12, 14). To evaluate the importance of the zinc fingers for ZAP's function, we individually disrupted each finger by site-directed mutagenesis in the context of NZAP-Zeo (Fig. 4) and full-length ZAP (data not shown). The resulting mutants, designated H86K, C88R, C168R, and H191R to indicate the substitutions, were analyzed for antiviral activity. Disruption of the second (C88R) or fourth (H191R) finger almost completely abolished the capability of ZAP to inhibit SIN replication (Fig. 4A). In contrast, disruption of the first (H86K) or third (C168R) finger had just a modest effect (Fig. 4A). When these mutants were analyzed for their activities on MLV-Luc or the SIN fragment D-containing reporter, similar patterns were observed (Fig. 4B). Northern blotting analysis of the mRNA levels of luciferase reporters in the mutant-expressing cells revealed that there was a perfect correlation between the fold inhibition and the mRNA levels for both MLV-Luc (Fig. 4C, MLV-Luc) and the SIN fragment D-containing reporter [Fig. 4C, SIN-D(−)-Luc], further suggesting that ZAP inhibits the replication of MLV and SIN by the same mechanism of destabilizing RNA.
FIG. 4.
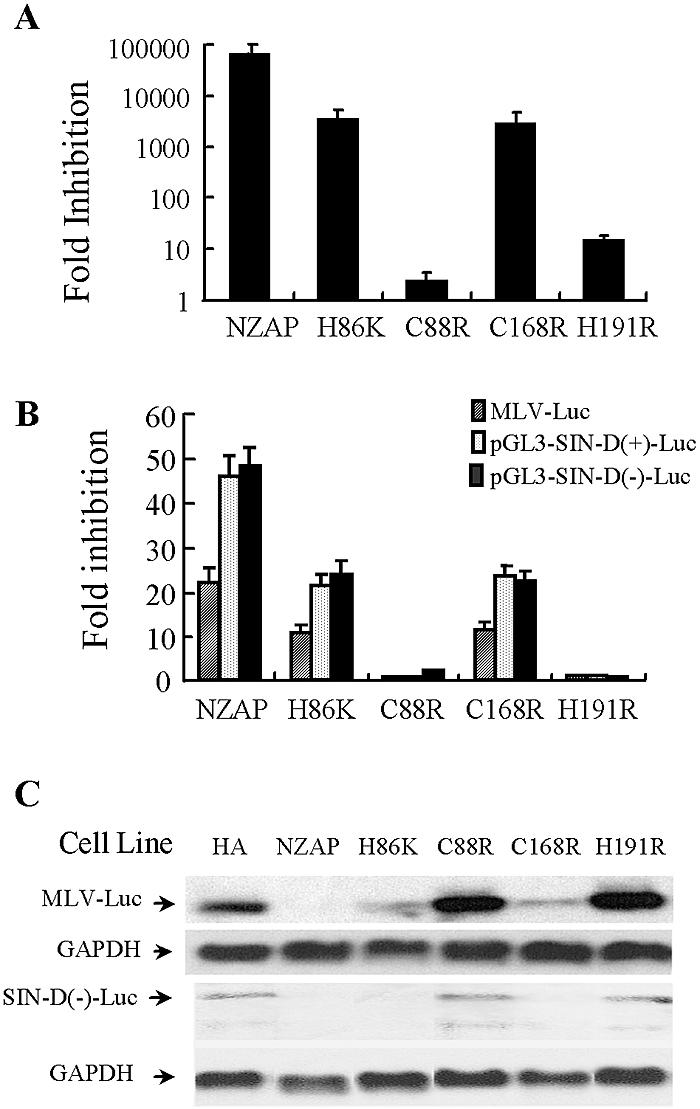
Analysis of the effect of each zinc finger mutation on the function of ZAP. Each zinc finger of NZAP-Zeo was disrupted by the mutation of a critical residue in the CCCH motif. The mutants were packaged into MLV pseudoviruses to transduce Rat2 cells and analyzed for the inhibition of viruses or luciferase reporters. NZAP, NZAP-Zeo; H86K, NZAP-Zeo-H86K; C88R, NZAP-Zeo-C88R; C168R, NZAP-Zeo-C168R; C191R, NZAP-Zeo-C191R; HA, HA-Zeo, empty vector control. (A) The cells were infected in duplicate wells with SIN for 1 h at a multiplicity of infection of 0.01 PFU per cell. At 24 h postinfection, the supernatants were collected. For each well, the mean virus titer was obtained by titration in duplicate on BHK-J cells. For each set of infections, the inhibition (n-fold) was calculated as the mean titer of the virus produced by infection of Rat2-HA-Zeo cells divided by the mean titer of the virus produced by infection of Rat2 cells expressing NZAP-Zeo or an NZAP-Zeo mutant. The data are mean fold inhibitions plus SD. (B) Cells were infected with the Eco-Luc virus (hatched bars) or transiently transfected with the pGL3-SIN-D(+)-Luc (gray bars) or pGL3-SIN-D(−)-Luc (solid bars) reporter and then analyzed for inhibition as described in the legend to Fig. 1. (C) Cells were infected with the Eco-Luc virus or transiently transfected with pGL3-SIN-D(−)-Luc. At 48 h postinfection or posttransfection, the total RNA was extracted from the cells and the reporter RNA level was analyzed by Northern blotting using a 32P-labeled luciferase probe. The expression level of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA served as a loading control. The arrows indicate the positions of the RNAs of interest.
ZAP directly binds to target RNA.
CCCH-type zinc fingers have been reported to be capable of binding to RNA (5, 12, 13). The foregoing results showing that ZAP specifically eliminates cytoplasmic mRNA and that the zinc finger motifs are essential for its function suggested that ZAP may also bind to its target RNA through the zinc finger motifs. To test this hypothesis, we performed an in vitro RNA binding assay. Four SIN fragments from the above luciferase reporter assay were chosen, with two being insensitive (fragments F and Di) and two being sensitive (fragments Md and Na), and were assayed for binding to NZAP-Zeo or ZAP. Since neither ZAP nor NZAP could be expressed in Escherichia coli in a soluble form, even when fused with maltose binding protein or glutathione S-transferase (data not shown), we chose to express the Myc-tagged proteins in mammalian cells. NZAP-Zeo-myc was expressed in Rat2 cells (see Materials and Methods). For the expression of ZAP-myc, 293TRex cells were stably transfected with pcDNA4/TO-myc-ZAP and selected with Zeocin. Individual Zeocin-resistant clones were expanded and tested for the inducible expression of ZAP by tetracycline. One clone displayed significant inducible ZAP expression, although without induction the protein was also expressed at a low level. Because of the leaky ZAP expression in the 293TRex-ZAP cells, in the binding assay the parental 293TRex cells were used as a negative control instead of the 293TRex-ZAP cells without tetracycline treatment. The ZAP proteins were immobilized on agarose resin by use of the Myc tag and an anti-Myc antibody and were assayed for binding to RNA probes. As predicted, the sensitive RNA fragments, Md and Na, bound to both NZAP-Zeo (Fig. 5A, top panel) and ZAP (Fig. 5B, top panel). In contrast, the insensitive fragments, F and Di, failed to bind to either protein (Fig. 5A and B, top panels), even though equivalent amounts of the ZAP proteins remained on the resins, as measured by Western blotting (Fig. 5A and B, bottom panels). When the second zinc finger was disrupted, ZAP lost its binding capability (Fig. 5C), consistent with the above observation that mutation of the second zinc finger abolished the function of ZAP.
FIG. 5.

In vitro binding of ZAP to target RNA. The lysates of ZAP-expressing or control cells were mixed with 9E10 anti-Myc antibody and protein G-agarose resin for 2 h to immobilize ZAP to the resin. The resins were washed and incubated with the indicated 32P-labeled RNA probes for 30 min in binding buffer. Bound RNAs were eluted by boiling in RNA sample buffer, subjected to urea-polyacrylamide gel electrophoresis, and detected by autoradiography; bound ZAP proteins were eluted by boiling in protein sample buffer and detected by Western blotting with the 9E10 antibody. (A) Binding of RNAs to NZAP-Zeo-myc. The NZAP-Zeo-myc protein was expressed in Rat2 cells and analyzed for binding to the indicated RNA probes. Rat2-vector, lysate of Rat2 cells transfected with the empty vector pcDNA4/TO-myc-His; Rat2-NZAP-Zeo-myc, lysate of Rat2 cells transfected with pcDNA4/TO/myc-NZAP-Zeo. The arrow in the lower panel indicates the position of NZAP-Zeo-myc. (B) Binding of RNAs to full-length ZAP. 293TRex, lysate of 293TRex control cells treated with tetracycline; 293TRex-ZAP, lysate of 293TRex-ZAP cells treated with tetracycline. The arrow in the lower panel indicates the position of ZAP-myc. (C) A mutation in the second zinc finger of ZAP abolished protein binding to the target RNA. 293TRex-ZAP-C88R, lysate of 293TRex cells expressing the second zinc finger mutant ZAP-C88R. The arrow in the lower panel indicates the position of ZAP-C88R-myc.
DISCUSSION
ZAP has been shown to selectively inhibit the replication of MLV and SIN. For MLV, ZAP markedly reduces the level of cytoplasmic viral mRNA (11), while for SIN, translation of the viral RNA is prevented by unknown mechanisms (3). For this report, using a luciferase reporter assay, we mapped the 3′-LTR of MLV and multiple fragments of SIN as the target sequences of ZAP. In addition, we showed that the presence of both MLV sequences and a SIN inhibitory fragment results in lower levels of reporter RNA. This suggests that ZAP may affect SIN RNA levels, resulting in decreased translation, rather than affecting translation per se. The multiple ZAP-sensitive sequences in the SIN genome may explain why ZAP has a more profound inhibitory effect on SIN than on MLV. Furthermore, since the SIN genomic clone was fragmented by single restriction enzyme digestion, other possible ZAP-sensitive fragments may have been disrupted by the digestion. Therefore, unidentified ZAP-sensitive fragments in the SIN genome may also exist. A common feature of the ZAP-sensitive fragments is that an extended length is required for conferring destabilizing activity on the reporter. A sequence analysis of these fragments did not identify any obvious homology or common motifs. Computer predictions of the secondary structures of these relatively long fragments turned out to be very complicated and fruitless; different structures were obtained when different types of software were used, and no obvious common characteristic structures such as stem-loops, hairpins, or knuckles were shared by these fragments. Based on these results, we speculate that ZAP may recognize a specific tertiary RNA structure. Alternatively, ZAP may recognize as yet unidentified short motifs or secondary structures, with the affinity for any such single motif being too low to support sustained binding; the clustering of these sequences would synergistically increase the affinity. The latter speculation is supported by the facts that there are four CCCH-type zinc finger motifs, each one of which is presumably an RNA binding unit (16, 17), and that disruption of any of the zinc fingers reduced ZAP's activity to some extent (Fig. 4). Further investigations using other methods, such as systematic evolution of ligands by exponential enrichment, may help to identify ZAP binding motifs.
CCCH-type zinc finger motifs are also found in TTP and its family members, which specifically destabilize ARE-containing mRNAs (4). Nonetheless, ZAP does not recognize any of the three types of AREs (Fig. 3). A detailed sequence comparison revealed that the CCCH-type zinc finger motifs of the TTP family are very conserved and that they all have a CX8CX5CX3H consensus sequence (13). However, no such consensus motifs were found in ZAP. Except for CX3H, the spacing between the cysteines was not conserved among the four zinc finger motifs of ZAP. The heterogeneity of the zinc finger motifs and the varied inhibitory capabilities of ZAP proteins containing mutations in the CCCH motifs suggest that the individual motifs may play different roles in ZAP's biological functions.
An important issue that remains to be addressed is the mechanism by which ZAP downregulates mRNAs. In this study, we described the evidence that ZAP, like TTP, directly binds to target RNAs through the CCCH-type zinc finger motifs. These results suggest that ZAP and TTP may share a common mechanism to destabilize target RNAs. Indeed, our preliminary data indicate that ZAP directly interacts with the exosome, as TTP does (6), and that downregulation of the exosome components significantly reduces ZAP's activity (X. Guo, J. Ma, and G. Gao, unpublished data). We suggest that ZAP may represent a novel type of trans-acting factors that modulate the stability of non-ARE-containing mRNAs.
Acknowledgments
This work was supported by grants to Guangxia Gao from the National Science Foundation (30225002), CAS Knowledge Innovation Projects (KSCX2-SW-216), and the Ministry of Science and Technology of China (2003AA219142).
We thank Matthew J. Bick for technical assistance and Hong Tang and Quan Chen for helpful discussions during the course of this work.
REFERENCES
- 1.Bakheet, T., M. Frevel, B. R. Williams, W. Greer, and K. S. Khabar. 2001. ARED: human AU-rich element-containing mRNA database reveals an unexpectedly diverse functional repertoire of encoded proteins. Nucleic Acids Res. 29:246-254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bakheet, T., B. R. Williams, and K. S. Khabar. 2003. ARED 2.0: an update of AU-rich element mRNA database. Nucleic Acids Res. 31:421-423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bick, M. J., J. W. Carroll, G. Gao, S. P. Goff, C. M. Rice, and M. R. MacDonald. 2003. Expression of the zinc-finger antiviral protein inhibits alphavirus replication. J. Virol. 77:11555-11562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blackshear, P. J. 2002. Tristetraprolin and other CCCH tandem zinc-finger proteins in the regulation of mRNA turnover. Biochem. Soc. Trans. 30:945-952. [DOI] [PubMed] [Google Scholar]
- 5.Carballo, E., W. S. Lai, and P. J. Blackshear. 1998. Feedback inhibition of macrophage tumor necrosis factor-alpha production by tristetraprolin. Science 281:1001-1005. [DOI] [PubMed] [Google Scholar]
- 6.Chen, C. Y., R. Gherzi, S. E. Ong, E. L. Chan, R. Raijmakers, G. J. Pruijn, G. Stoecklin, C. Moroni, M. Mann, and M. Karin. 2001. AU binding proteins recruit the exosome to degrade ARE-containing mRNAs. Cell 107:451-464. [DOI] [PubMed] [Google Scholar]
- 7.Chen, C. Y., and A. B. Shyu. 1994. Selective degradation of early-response-gene mRNAs: functional analyses of sequence features of the AU-rich elements. Mol. Cell. Biol. 14:8471-8482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen, C. Y., and A. B. Shyu. 1995. AU-rich elements: characterization and importance in mRNA degradation. Trends Biochem. Sci. 20:465-470. [DOI] [PubMed] [Google Scholar]
- 9.DuBois, R. N., M. W. McLane, K. Ryder, L. F. Lau, and D. Nathans. 1990. A growth factor-inducible nuclear protein with a novel cysteine/histidine repetitive sequence. J. Biol. Chem. 265:19185-19191. [PubMed] [Google Scholar]
- 10.Gao, G., and S. P. Goff. 1999. Somatic cell mutants resistant to retrovirus replication: intracellular blocks during the early stages of infection. Mol. Biol. Cell 10:1705-1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao, G., X. Guo, and S. P. Goff. 2002. Inhibition of retroviral RNA production by ZAP, a CCCH-type zinc finger protein. Science 297:1703-1706. [DOI] [PubMed] [Google Scholar]
- 12.Lai, W. S., E. Carballo, J. R. Strum, E. A. Kennington, R. S. Phillips, and P. J. Blackshear. 1999. Evidence that tristetraprolin binds to AU-rich elements and promotes the deadenylation and destabilization of tumor necrosis factor alpha mRNA. Mol. Cell. Biol. 19:4311-4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lai, W. S., E. Carballo, J. M. Thorn, E. A. Kennington, and P. J. Blackshear. 2000. Interactions of CCCH zinc finger proteins with mRNA. Binding of tristetraprolin-related zinc finger proteins to AU-rich elements and destabilization of mRNA. J. Biol. Chem. 275:17827-17837. [DOI] [PubMed] [Google Scholar]
- 14.Lai, W. S., E. A. Kennington, and P. J. Blackshear. 2002. Interactions of CCCH zinc finger proteins with mRNA: non-binding tristetraprolin mutants exert an inhibitory effect on degradation of AU-rich element-containing mRNAs. J. Biol. Chem. 277:9606-9613. [DOI] [PubMed] [Google Scholar]
- 15.Laroia, G., B. Sarkar, and R. J. Schneider. 2002. Ubiquitin-dependent mechanism regulates rapid turnover of AU-rich cytokine mRNAs. Proc. Natl. Acad. Sci. USA 99:1842-1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McColl, D. J., C. D. Honchell, and A. D. Frankel. 1999. Structure-based design of an RNA-binding zinc finger. Proc. Natl. Acad. Sci. USA 96:9521-9526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Michel, S. L., A. L. Guerrerio, and J. M. Berg. 2003. Selective RNA binding by a single CCCH zinc-binding domain from Nup475 (tristetraprolin). Biochemistry 42:4626-4630. [DOI] [PubMed] [Google Scholar]
- 18.Rice, C. M., R. Levis, J. H. Strauss, and H. V. Huang. 1987. Production of infectious RNA transcripts from Sindbis virus cDNA clones: mapping of lethal mutations, rescue of a temperature-sensitive marker, and in vitro mutagenesis to generate defined mutants. J. Virol. 61:3809-3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Strauss, J. H., and E. G. Strauss. 1994. The alphaviruses: gene expression, replication, and evolution. Microbiol. Rev. 58:491-562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Treisman, R. 1985. Transient accumulation of c-fos RNA following serum stimulation requires a conserved 5′ element and c-fos 3′ sequences. Cell 42:889-902. [DOI] [PubMed] [Google Scholar]
- 21.van Hoof, A., and R. Parker. 2002. Messenger RNA degradation: beginning at the end. Curr. Biol. 12:R285-R287. [DOI] [PubMed] [Google Scholar]