

Abstract
By using a homology-based bioinformatics approach, a structural model of the vaccinia virus (VV) I7L proteinase was developed. A unique chemical library of ∼51,000 compounds was computationally queried to identify potential active site inhibitors. The resulting biased subset of compounds was assayed for both toxicity and the ability to inhibit the growth of VV in tissue culture cells. A family of chemotypically related compounds was found which exhibits selective activity against orthopoxviruses, inhibiting VV with 50% inhibitory concentrations of 3 to 12 μM. These compounds exhibited no significant cytotoxicity in the four cell lines tested and did not inhibit the growth of other organisms such as Saccharomyces cerevisiae, Pseudomonas aeruginosa, adenovirus, or encephalomyocarditis virus. Phenotypic analyses of virus-infected cells were conducted in the presence of active compounds to verify that the correct biochemical step (I7L-mediated core protein processing) was being inhibited. Electron microscopy of compound-treated VV-infected cells indicated a block in morphogenesis. Compound-resistant viruses were generated and resistance was mapped to the I7L open reading frame. Transient expression with the mutant I7L gene rescued the ability of wild-type virus to replicate in the presence of compound, indicating that this is the only gene necessary for resistance. This novel class of inhibitors has potential for development as an efficient antiviral drug against pathogenic orthopoxviruses, including smallpox.
The recent deliberate dispersal of anthrax spores into our environment has raised awareness and concern over our ability to effectively prevent or treat infections caused by infectious agents. Of particular concern is the Centers for Disease Control and Prevention list of category A agents, which includes variola virus, the causative agent of smallpox. A smallpox vaccine is available, but due to the small but significant risk of serious complications that can include encephalitis (19), myopericarditis, hemolytic anemia, and even death (10), the vaccine has only been administered to a select population (mainly laboratory personnel working with orthopoxviruses, military personnel, and some first-responders). While smallpox was eradicated from the natural biosphere in the 1970s, the subsequent 30 years have produced a population that is immunologically naïve and susceptible to infection. In the advent of a release of smallpox into the environment, the vaccine would need to be administered within 3 days of exposure to be effective, but there is a period of 10 to 14 days when a person is infected before obvious clinical symptoms become evident. In addition to smallpox concerns, there is the recent spate of monkeypox virus infections in North America. Likewise, there are health concerns due to complications to vaccination against smallpox in immunocompromised individuals, such as progressive vaccinia. Thus, there is a pressing need for effective antiorthopoxvirus therapeutics to address these problems and concerns.
No antiviral drug has been proven to be effective in the treatment of human smallpox. The only antiviral agent currently approved for use against orthopoxviruses is cidofovir, which has been shown to be useful against the molluscum contagiosum and orf viruses in humans (9). However, cidofovir has low oral bioavailability and must be administered intravenously (7), which limits its usefulness. Cidofovir has also been shown to cause nephrotoxicity affecting renal cells and must be coadministered with oral probenecid to reduce the toxic effects (16).
Development of an effective antiviral drug requires the identification of a specific interaction or activity whose disruption will be lethal to the virus and relatively benign to the host. Since viruses, such as orthopoxviruses, are obligate intracellular parasites which utilize many of the host cell's enzymes and metabolic pathways during their replication, this task is often quite difficult, and this fact is chiefly responsible for the relative paucity of successful antiviral drugs. The drugs that have proven effective, such as acyclovir, are typically directed against nucleotide-metabolizing or biosynthetic enzymes. Since many of the orthopoxvirus-encoded enzymes involved in nucleic acid biosynthesis are highly similar to their mammalian counterparts (for example, vaccinia virus [VV] thymidine kinase shares more than 90% identity with the human enzyme), it may prove difficult, if not impossible, to identify compounds that specifically block these viral enzymes.
Fortunately, it has recently been discovered that proteolysis catalyzed by viral-encoded proteinases is a necessary step in the developmental cycle of most viruses. This realization has created a new class of targets for antiviral drug development. Proteinase inhibitors have proven to be effective antiviral drugs that target human immunodeficiency virus, influenza, hepatitis C, and rhinovirus enzymes and that prevent disease in the human host. Small molecule inhibitors of the NS3 protease in hepatitis C virus have recently been shown to be effective both in animal and human trials (17). Thus, it is of particular interest to note that proteolytic maturation of orthopoxvirus core proteins appears to be required for infectious progeny to be produced (13). Studies in our laboratory over the past 10 years have identified the unique cis signals required to direct endoproteolytic cleavage of core protein precursors and established the contextual requirements of core protein maturation; in recent work we have identified the poxvirus gene (I7L) that encodes the viral core protein proteinase.
VV is a large double-stranded DNA virus that is a prototypic member of the orthopoxvirus family. It shares around 90% similarity with variola virus, the causative agent of smallpox, and it is believed that a compound that inhibits VV will also inhibit variola virus. Poxviruses are unique in that they replicate entirely in the cytoplasm of infected cells, encoding most of the genes necessary for their own replication. Briefly, the virus attaches to the outside of the cells, enters, uncoats, undergoes early gene expression, intermediate gene expression, DNA synthesis, late gene expression, virion formation, and virus maturation before egress from the cell. Morphogenic proteolysis occurs in the stage between the formation of the infectious intracellular mature virus from the noninfectious intracellular virus. The gene product of the I7L open reading frame (ORF) has been shown to be the cysteine proteinase responsible for the cleavage of the core protein precursors that occurs in this stage of the viral life cycle (4, 5).
The long-range goals of the experiments reported here are to discover and develop compounds capable of inhibiting the growth of pathogenic orthopoxviruses (such as smallpox) and disease-associated pathology in the human host. In this study, we describe the identification of a novel class of small molecule inhibitors that were modeled to fit into the predicted active site pocket of I7L. The inhibitory phenotype and genetic analysis are consistent with a defect in morphogenesis. Since these compounds are specific for orthopoxviruses and appear to have minimal toxicity to cells, they would appear to warrant further development as smallpox antiviral drug candidates.
MATERIALS AND METHODS
Chemical compounds.
Compound stocks were prepared at a concentration of 10 mM in 100% dimethyl sulfoxide. TTP-6171 will be made available to scientific colleagues under a Material Transfer Agreement for research purposes.
Computational modeling.
TransTech Pharma's Translational Technology was designed and developed for rapid lead generation and optimization into preclinical drug candidates. It consists of two subtechnologies: TTProbes and TTPredict. TTProbes is a set of >51,000 pharmacophorically diverse molecules with high information density. TTPredict, on the other hand, is a state-of-the-art computer-based technology that automates high-throughput three-dimensional target model building, binding site identification, and conformational analysis. It is used to dock, score, and rank members of TTProbes set into targets' binding sites.
Cells and viruses.
Table 1 provides a list of each cell line and virus strain used. BSC40 cells (21), HeLa cells, 293 cells, and L929 cells were grown in Eagle's minimal essential medium (MEM-E; Gibco-BRL, Rockville, Md.) containing 10% fetal calf serum (FCS) (Gibco-BRL), 2 mM glutamine, and 15 μg of gentamicin sulfate per ml in a 37°C incubator with 5% CO2. VV, cowpox virus, adenovirus, and encephalomyocarditis virus infections were carried out in MEM containing 5% FCS, 2 mM glutamine, and 15 μg of gentamicin sulfate per ml in a 37°C incubator with 5% CO2. Purified VV was prepared as previously described (13). Escherichia coli strains were grown in Luria-Bertani broth or on Luria-Bertani medium containing 1.5% agar and ampicillin (50 μg/ml). Recombinant VV expressing the green fluorescent protein (vvGFP) was constructed by inserting the GFP gene into the thymidine kinase locus in the Western Reserve strain of VV, driven by the VV 7,500-molecular-weight early-late promoter. Saccharomyces cerevisiae was grown in 1245 yeast extract-peptone-dextrose medium (American Type Culture Collection [ATCC], Manassas, Va.), and Pseudomonas aeruginosa was grown in nutrient broth (Difco, Detroit, Mich.).
TABLE 1.
Plasmids, oligonucleotides, cells, and strains used in this studya
| Name | Description | Source or reference |
|---|---|---|
| Plasmid | ||
| pCR2.1 | Kmr, Ampr | Invitrogen |
| pCB26-23 | pCR2.1 with I7L behind its native promoter | This work |
| pCB26-23-17 | pCB26-23 with aa 104 mutated Y→C and aa 324 mutated L→M | This work |
| pRB21 | puc derived with F13L flanks, MCS downstream of synthetic early-late promoter | 3 |
| pI7L | pRB21 plasmid with full-length I7L | 4 |
| pI7L-17 | pI7L with aa 104 mutated Y→C and aa 324 mutated L→M | This work |
| p78-104 | pI7L with aa 78 mutated to create an XbaI site; aa 104 mutated Y→C | This work |
| p78-324 | pI7L with aa 78 mutated to create an XbaI site and aa 324 mutated L→M | This work |
| p78-104-324 | pI7L with aa 78 mutated to create an XbaI site; aa 104 Y→C; aa 324 L→M | This work |
| Oligonucleotides | ||
| CB26 | 5′-GAG CTC GTT TTC CTA GTG ATG GAG GAG-3′ | This work |
| CB23 | 5′-AAG CTT TTA TTC ATC GTC GTC TAC-3′ | This work |
| CB84 | 5′-GAG TCG GGG CAC CTG TCT AGA CCC AAT AGT AGC G-3′ | This work |
| CB85 | 5′-CGC TAC TAT TGG GTC TAG ACA GGT GCC CCG ACT C-3′ | This work |
| CB86 | 5′-CGC GTT ACC ATT CTA TAT GTG ATG TTT TTG AGT TAC C-3′ | This work |
| CB87 | 5′-GGT AAC TCA AAA ACA TCA CAT ATA GAA TGG TAA CGC G-3′ | This work |
| CB88 | 5′-GTG GAA GTT AAT CAG CTG ATG GAA TCT GAA TGC GGG-3′ | This work |
| CB89 | 5′-CCC GCA TTC AGA TTC CAT CAG CTG ATT AAC TTC CAC-3′ | This work |
| Cell line | ||
| BSC40 | BSC1 African green monkey kidney cells adapted to grow at 40°C | 21 |
| 293 | Human embryonic kidney cells | Invitrogen |
| HeLa | Human cervical carcinoma cells | ATCC |
| L929 | Mouse fibroblast cells; ATCC CCL-1 | ATCC |
| Virus | ||
| VV WR | Western Reserve strain of vaccinia virus | ATCC |
| VV COP | Copenhagen strain of vaccinia virus | ATCC |
| VV IHDJ | IHDJ strain of vaccinia virus | ATCC |
| Cowpox | Brighton Red strain | Duke University |
| Adenovirus | Mastadenovirus, human adenovirus C | ATCC |
| MHV-A59 | Mouse hepatitis virus | ATCC |
| EMC | Encephalomyocarditis virus | ATCC |
| vvGFP | Western Reserve vaccinia virus with GFP in the TK locus | This work |
| v17-3 | vvGFP resistant to TTP-6171 with mutations in IL aa 104 and 324 | This work |
| vCB | Recombinant vvGFP with the I7L gene replaced with the mutant I7L gene | This work |
| ts16 | Temperature-sensitive VV with mutation mapped to I7L | 56 |
| Bacteria | ||
| E. coli INVαF′ | F′ endA1 recA1 hsdR17 (rK− mK+) supE44 thi-1 gyrA96rel A1O80 lacZAM15 A(lacZYA-arg-F)U16 | Invitrogen |
| P. aeruginosa | Broad spectrum of resistance to various commerical germicides | ATCC |
| S. cerevisiae | Budding yeast derived from X2180 | ATCC |
aa, amino acid; Tk, thymidine kinase.
Cytotoxicity assay.
Confluent monolayers of BSC40 cells were grown in 96-well black-sided view plates (Packard, Meriden, Conn.). Compounds were diluted to the desired concentration in MEM and applied to the cell monolayers in doubling dilutions. Controls included cells treated without compound and cells treated with hydroxyurea, rifampin, or cytosine arabinoside (AraC). The cells were incubated for 24 h at 37°C. Background fluorescence was measured by using a Wallac Victor2 V multilabel HTS counter (Perkin-Elmer, Turku, Finland) with an excitation wavelength of 485 nm, and plates were read at 535 nm. Cells were visualized under a light microscope for cytopathic effect (CPE) and treated with Alamar Blue (BioSource International, Inc., Camarillo, Calif.) to determine the toxicity of the compounds.
Fluorescence assay.
Confluent monolayers of BSC40 cells in 96-well black-sided view plates (Packard) were infected with vvGFP at a multiplicity of infection (MOI) of 0.1 for 30 min at 37°C before the addition of compounds. Controls included uninfected cells and vvGFP-infected cells treated individually with AraC, hydroxyurea, rifampin, or no compound. Compounds were applied to the infected cells in doubling dilutions. Monolayers were incubated at 37°C for 24 h. Plates were washed with 1× phosphate-buffered saline (PBS) and fluorescence was measured by using a Wallac Victor2 V multilabel HTC counter (Perkin-Elmer) with an excitation wavelength of 485 nm, and plates were read at 535 nm. Wells showing a reduction in fluorescence over the untreated vvGFP-infected control lane were visualized under a light microscope to verify loss of virus replication versus removal of cell monolayer from a productive and concomitant CPE.
Determination of TI.
The 50% inhibitory concentrations (IC50s) were determined by CPE inhibition as seen by fluorescence by using vvGFP and plaque reduction assays with crystal violet staining or neutral red uptake. The 50% cell toxicity concentrations (TC50s) were determined as the concentrations of compounds that caused 50% of the cells to round up and show signs of toxicity both visibly and through the indication of Alamar Blue dye. The therapeutic index (TI) was calculated as the value for the TC50 divided by the IC50.
Western blot analysis.
Confluent monolayers of BSC40 cells in six-well plates were infected with vvGFP at an MOI of 1 for 30 min prior to the addition of compound. One well of cells was pretreated with hydroxyurea for 1 h prior to infection. Each compounds was added to the infected cell monolayers at a concentration of 100 μM. Monolayers were incubated at 37°C for 24 h. Virus-infected cell extracts were harvested by scraping, and the total cell extract was collected in 1.5-ml microcentrifuge tubes. The total cell extract was centrifuged for 15 min at 4°C, and the pellet was resuspended in 100 μl of 1× PBS and subjected to three cycles of freeze-thaw to release the virus from the cell debris. A total of 15 μl of the whole cell extract was run on sodium dodecyl sulfate-polyacrylamide gels and transferred to polyvinylidene difluoride (Pall, Ann Arbor, Mich.) membranes. The membranes were incubated with a 1:1,000 dilution of anti-4b antisera (24) and then with a 1:2,000 dilution of goat anti-rabbit alkaline phosphatase antisera (Bio-Rad, Hercules, Calif.). The proteins were detected by using the alkaline phosphatase development system (Bio-Rad) according to the manufacturer's instructions.
Isolation of compound-resistant vvGFP isolates.
BSC40 cells in six-well plates were infected with vvGFP at an MOI of 0.05 for 30 min at 37°C prior to treatment with compound. The IC50 of the compound was added, and monolayers were incubated at 37°C for 24 h. Total cell extracts were harvested as above, and 10 μl of crude cell extract was used to infect fresh monolayers of BSC40 cells in the presence of compound. This process was repeated nine times with the titer of each passage determined. After the ninth passage, the titer had returned to a level similar to that of untreated virus. Compound-resistant virus was plaque purified to isolate individual resistant viruses and then amplified in six-well plates.
Genomic DNA preparation.
Confluent monolayerss of BSC40 cells in six-well plates were infected with compound-resistant virus, treated with the same compound at the IC50, and incubated at 37°C until about 90% CPE was observed. Total cell extracts were prepared by scraping and were placed into a 1.5-ml centrifuge tube and centrifuged for 10 min at 4°C; the pellet was resuspended in 1 ml of 1× PBS and subjected to three freeze-thaw cycles. Aliquots (100-μl) were removed for storage, and the remaining 900 μl was treated with 10 mM Tris-hydrochloride (pH 8), 1 mM EDTA, 5 mM β-mercaptoethanol, 150 μg of proteinase K per ml, 200 mM NaCl, 1% sodium dodecyl sulfate; the DNA was then extracted with Tris-EDTA buffer saturated with phenol-chloroform-isoamyl alcohol (25:24:1) and ethanol precipitated. The resulting DNA was used for PCR amplification of the VV I7L gene and for cloning.
Plasmids and site-directed mutagenesis.
Table 1 gives a description of all plasmids, oligonucleotides, and strains used. The VV I7L gene was amplified from Western Reserve strain genomic DNA and cloned into pRB21 (3) with PstI and HindIII flanking, yielding plasmid pI7L. Mutant I7L was amplified from the TTP-6171 compound-resistant virus DNA with primers CB26 and CB23 and cloned into pCR2.1-Topo Vector (Invitrogen, Carlsbad, Calif.) with PstI and HindIII flanking, yielding plasmid pCR2.1:I7L. Site-directed mutagenesis of the I7L gene was performed by using a QuikChange site-directed mutagenesis kit (Stratagene, Cedar Creek, Tex.) according to the manufacturer's instructions; primers CB84-85, CB86-87, and CB88-89 were used to mutate amino acids 78, 104, and 324, respectively, by using pI7L as a template, yielding pI7L-78/104, pI7L-78/324, and pI7L-78/104/324. Amino acid 78 of the I7L gene was mutated to create an XbaI site via a silent mutation to facilitate screening for recombinant viruses. Amino acid 104 of the I7L gene had a Y→C mutation, and amino acid 324 had an L→M mutation.
Transient expression.
Confluent monolayers of BSC40 cells in six-well plates were infected with vvGFP at an MOI of 0.01 PFU per cell and transfected with 2 μg of plasmid DNA by using DMRIE-C (1,2-dimyristyloxypropyl-3-dimethyl-hydroxy ethyl ammonium bromide and cholesterol) (Invitrogen) according to the manufacturer's instructions. Plasmids contained either no insert, the wild-type I7L ORF, or the mutant I7L ORF with each single mutation or the double mutation. Cells were untreated or treated with the IC50 of the compound and incubated at 37°C. Cells were harvested at 24 h postinfection by pipetting up and down to lift the cells from the surface. The crude extract was centrifuged at 15,000 rpm for 10 min, the supernatant was aspirated off, and the pellet was resuspended in 1 ml of 1× PBS, freeze-thawed three times, and then centrifuged at 600 × g for 3 min to sediment cellular debris. The supernatant was tittered to determine viral yield.
Electron microscopy.
Confluent monolayers of BSC40 cells in six-well plates were infected with vvGFP at an MOI of 3 PFU per cell for 30 min prior to treatment with compound. Compound was added at a concentration of 100 μM, and the cells were incubated at 37°C for 24 h. Cells were harvested as above and fixed with 2.5% glutaraldehyde and 1.2% paraformaldehyde in 0.1 M cacodylate buffer (pH 7.3), postfixed in osmium tetroxide, dehydrated, and embedded in Spurr's resin (23). Ultrathin sections were stained by using the double lead stain technique of Daddow (8), with lead citrate (25) and uranyl acetate. Sections were viewed with a Philips CM12 transmission electron microscope, operated at 60 KeV.
One step time course of viral replication.
Confluent monolayers of BSC40 cells in six-well plates were infected with either vvGFP or v17 at an MOI of 1. One hour after infection, the virus-infected cells were treated with TTP-6171 at a final concentration of either 0, 10, 25, or 60 μM. Cells were harvested at 2, 12, 24, and 48 h postinfection and subjected to three freeze-thaw cycles to release virus, and viral titers were determined in BSC40 cells.
Transfection and isolation of mutant I7L recombinant virus.
Confluent monolayers of BSC40 cells in six-well plates were infected with vvGFP at an MOI of 0.05 at 37°C. At 3 h postinfection, 1.5 μg of DNA was transfected into the cells by using DMRIE-C reagent (Invitrogen). Infected cells were harvested 24 h postinfection and the virus was released by three freeze-thaw cycles. After two rounds of plaque purification in the presence of TTP-6171 to select for recombinant virus (vCB), incorporation of the mutated I7L gene into the viral isolates was confirmed by PCR, digest with XbaI, and sequencing.
RESULTS
Identification of a new class of orthopoxvirus antiviral compounds.
TTPredict was used to construct threading and homology models for I7L (Fig. 1A). A sequence comparison to proteins with experimentally determined three-dimensional structures showed that the highest sequence identity with VV I7L was to the C-terminal domain of the ULP1 protease. The latter consists of 221 amino acids and has a 22% sequence identity with I7L. ULP1 protease served as a template for building the three-dimensional structure of I7L. TTPredict site search algorithms readily identified the catalytic site of I7L (Fig. 1B), based on the location of the active site residues, H241, D248, and C328, that are essential for activity (5). TTProbes were docked into this site. The fit of every docked TTProbe was computed by using several scoring functions. High-scoring probes were identified, and the highest ranking TTProbes were submitted for in vivo screening.
FIG. 1.
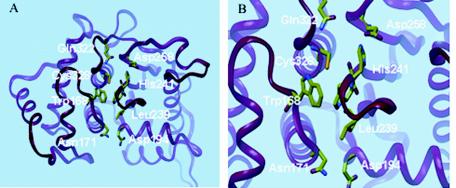
TTPredict three-dimensional model of the VV I7L cysteine proteinase modeled by using the structure of the C-terminal ULP1 protease. (A) Critical binding site residues are shown in colored tubes. (B) Close-up view of the I7L ligand binding and catalytic domain.
In order to screen these compounds for their ability to inhibit VV replication, an in vivo tissue culture-based screening assay was developed. Though more cumbersome than a biochemical assay, this approach had the advantage of providing information regarding compound toxicity and cellular uptake, along with the antiviral activity read-out. For this purpose, a fluorescence assay was developed that used vvGFP as the read-out in a 96-well-plate format. vvGFP-infected BSC40 cells in 96-well plates were treated with compound and analyzed 24 h later for relative fluorescence. Considerable effort was placed on determining the appropriate cell density, MOI, and incubation time needed to provide a validated assay capable of providing reproducible results (data not shown). Controls included uninfected cells, which have a low fluorescence background (Fig. 2), and vvGFP-infected cells treated with either rifampin, an inhibitor of viral assembly (11, 20), hydroxyurea, which blocks DNA replication (22), or AraC, an inhibitor of DNA synthesis in VV (12, 2). These control treatments display low levels of fluorescence regardless of whether they inhibit late or early in the replication cycle. As a negative control, cells were infected with vvGFP and not treated with any compound. As the data in Fig. 2 demonstrate, a robust signal of approximately 25-fold over background was observed in vvGFP-infected cells versus uninfected cells.
FIG. 2.
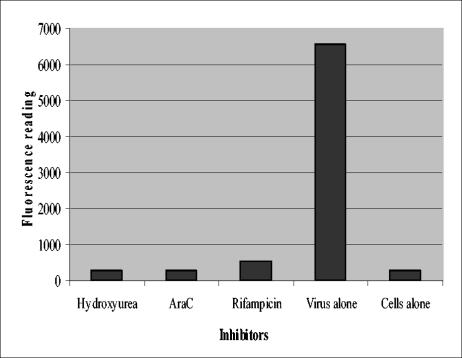
vvGFP assay. Graphical representation of the relative fluorescence units from 96-well plates of vvGFP-infected BSC40 cells treated with various compounds with an excitation wavelength of 485 nm and read-out at 535 nm. Each bar represents the average of 10 separate readings.
A total of 3,460 compounds were identified as potential I7L inhibitors by in silico screening. This biased library was evaluated by using the vvGFP fluorescence assay to identify compounds which were nontoxic to cells and which inhibited virus growth. Of the compounds that were initially found to be toxic to the cells at a concentration of 100 μM, serial dilutions were performed until a concentration was found that inhibited virus replication but was no longer toxic. Of the 3,460 compounds, 136 were found to inhibit viral replication without being toxic. These compounds were then assessed to determine which stage of the virus life cycle they inhibit by looking for morphogenesis inhibitors as well as by looking at the relative IC50, which narrowed the lead compound list down to 19. The TI was then taken into consideration, and the list was reduced to TTP-6171 and several other chemically related compounds (Table 2). During initial screening, TTP-6171 demonstrated an IC50 of 12 and a TC50 of 900, with a calculated TI of 75. For the rest of the experiments reported here, only TTP-6171 was tested as the prototype member of this compound family. Figure 3 shows the chemical structure of TTP-6171.
TABLE 2.
TI values of selected compounds
| Compound | TC50 (μM) | IC50 (μM) | TI |
|---|---|---|---|
| 6171 | 900 | 12 | 75 |
| 201018 | 600 | 200 | 3 |
| 200480 | 55 | 14 | 4 |
| 174878 | 400 | 50 | 8 |
| 130961 | 150 | 10 | 15 |
| 176510 | 400 | 56 | 7 |
| 123045 | 200 | 50 | 4 |
FIG. 3.
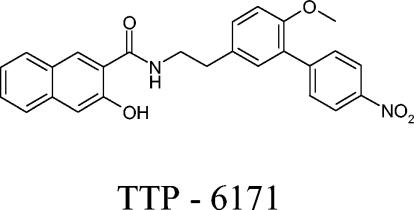
Chemical structure of TTP-6171.
Figure 4 illustrates the screening data obtained with TTP-6171. The top left panel shows light microscopy of vvGFP-infected BSC40 cells with characteristic rounding up and CPE. The top right panel is the same field viewed with fluorescence microscopy to demonstrate a vigorous viral infection as demonstrated by GFP expression. In contrast, the bottom panels show the results of similar analyses carried out in the presence of TTP-6171. After 24 h no CPE (or toxicity) is evident, nor is there any demonstrable vvGFP replication. At high concentrations and upon prolonged incubation (48 to 72 h), TTP-6171 has toxic effects on cells, and plaque assays cannot be carried out in the presence of inhibitory concentrations of the drug.
FIG. 4.
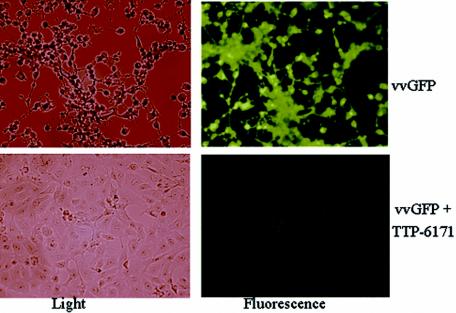
Light and fluorescent images of vvGFP-infected cells with and without compound TTP-6171.
TTP-6171 is specific for orthopoxviruses.
To determine the target specificity of TTP-6171, we tested the compound against a variety of organisms including those that contain a cysteine protease with partial similarity to I7L, such as adenovirus (1, 18). It was found that TTP-6171 inhibited the growth of various strains of VV including Western Reserve, Copenhagen, and IHDJ, as well as cowpox virus at a concentration of 25 μM. In contrast, no inhibition was observed in the growth of P. aeruginosa, S. cerevisiae, or mouse hepatitis virus, each of which expresses a cysteine protease. Likewise, TTP-6171 did not inhibit the replication of encephalomyocarditis virus or adenovirus at a concentration of 100 μM (data not shown). These results suggest that TTP-6171-mediated inhibition appears to be specific for members of the Orthopoxviridae.
TTP-6171 inhibits a late stage in the viral life cycle.
To investigate which stage of the viral life cycle TTP-6171 was inhibiting, immunoblot analyses of extracts from cells infected with vvGFP (with or without candidate drugs) were conducted by using a monospecific antiserum against the VV A3L protein. The gene product of the A3L ORF, P4b, is a 72-kDa late protein that is cleaved by I7L to form the mature core protein 4b during a late stage of morphogenesis (5). As A3L is a late gene, no P4b signal on the immunoblot would indicate that TTP-6171 inhibited an early step in viral replication (binding, penetration, early or delayed-early transcription, uncoating, or DNA replication). If both the P4b precursor and 4b product were observed, this would suggest that TTP-6171 acted at a very late stage of viral assembly or egress. In contrast, expression of P4b with no, or reduced, processing to the 4b product would be the phenotype expected if TTP-6171 were an I7L inhibitor. In control experiments the expected phenotypes were observed. Cells infected with vvGFP in the absence of compound express both P4b precursor (apparent molecular mass of 66 kDa) and the processed 4b product (apparent molecular mass of 62 kDa) (Fig. 5, lane 2). When vvGFP-infected cells are treated with rifampin, a morphogenesis inhibitor, P4b is made but not processed (Fig. 5, lane 3). When vvGFP-infected cells are treated with hydroxyurea, an inhibitor of viral DNA replication, no P4b is observed (Fig. 5, lane 4). Lanes 5 to 8 show representative results with a number of the drug candidates tested. In the presence of the drug tested in lane 8, P4b is still being processed to 4b, indicating that inhibition of virus replication is occurring at some stage after morphogenic proteolysis and, therefore, the drug is likely not targeting I7L. Likewise, the drug tested in lane 6 inhibits the production of P4b entirely and therefore blocks some early stage of replication prior to I7L expression. In contrast, the compounds tested in lanes 5 and 7 appear to be hitting the correct stage of the viral life cycle since P4b is made but not processed. The compound tested in lane 5 is TTP-6171.
FIG. 5.
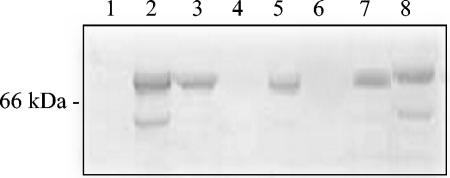
Processing of P4b core protein precursor. Western blot of virus-infected whole-cell extracts probed with P4b antisera. Molecular masses in kDa are indicated on the left side of the blot. In each lane cells are infected with vvGFP in the presence of 100 μM compound as follows: lane 1, mock-infected cells; lane 2, vvGFP alone; lane 3, rifampin (100 μg/ml); lane 4, hydroxyurea (5 mM); lane 5, TTP-6171; lane 6, TTP-130961; lane 7, TTP-176510; and lane 8, TTP-123045.
Phenotypic analysis via electron microscopy.
Ts16 is a temperature-sensitive mutant VV in which the responsible mutation has been mapped to the I7L gene (6, 14). At the permissive temperature, a variety of typical poxvirus assembly intermediates are observed including dense oval and brick-shaped virions that are consistent with infectious intracellular mature virions. At the nonpermissive temperature, assembly is aborted with the accumulation of immature particles containing enveloped viroplasm but with little or no subsequent nucleoid condensation. An electron microscopic examination of v17- or vvGFP-infected cells in the absence or presence of TTP-6171 is shown in Fig. 6, panels A to P. As seen in panels A through H of Fig. 6, the phenotypes of v17 observed in the presence or absence of TTP-6171 are identical to wild-type virus, with both immature virus and intracellular mature virus particles present. The phenotype of the parental virus vvGFP treated with TTP-6171 shows that no intracellular mature virus is observed in any of the cells. There was an accumulation of immature particles with numerous crescent-shaped particles present. This result supports the hypothesis that TTP-6171 is an inhibitor of I7L activity.
FIG. 6.
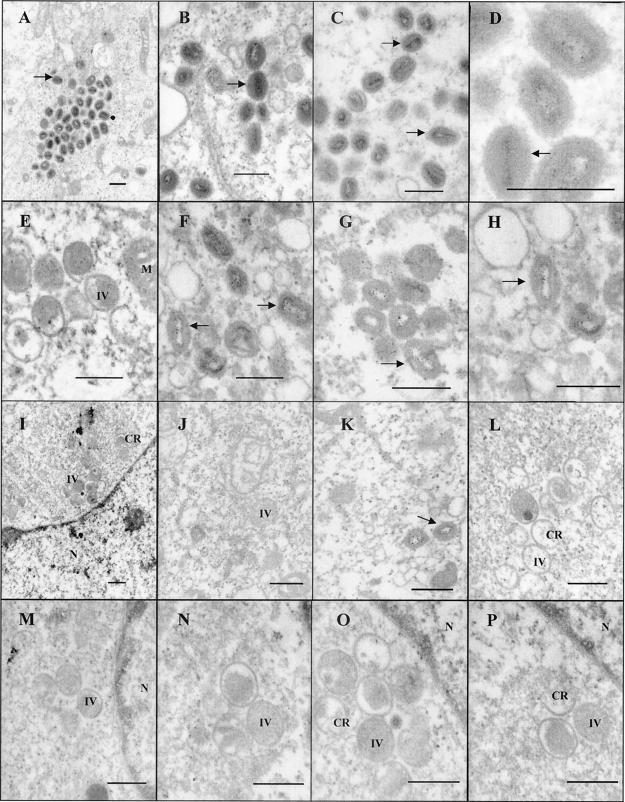
Electron micrographs of BSC40 cells infected with virus at an MOI of 3. Cells were harvested 24 h after infection and fixed, and ultrathin sections were prepared for transmission electron microscopy. Bar represents 400 nm. Arrows indicate intracellular mature virus. IV, intracellular virus; N, nucleus; CR, crescent-shaped viral particles. Panels A to D show v17 in the absence of compound, panels E to H show v17 treated with TTP-6171 (100 μM), and panels I to P show vvGFP treated with TTP-6171 (100 μM).
Isolation of drug-resistant virus.
In order to directly demonstrate that the target of TTP-6171-mediated inhibition was the I7L gene product, vvGFP was subjected to numerous passages in the presence of TTP-6171 to generate drug-resistant viral mutants. Cells were infected with vvGFP at an MOI of 0.1 in the presence of the IC50 concentration of compound for 24 h prior to being harvested. The titer of the virus-infected cellular cytoplasmic extract was determined, and a portion of this extract was used to infect fresh BSC40 cells. As the data in Fig. 7A show, the titer of infectious progeny dropped 7 logs from passage 0 to 4. Starting with passage 5 the progeny titer began to rise in the presence of the drug until a 4 log increase was observed by passage seven, presumably due to the emergence of a drug-resistant mutant population. After passage 9, individual resistant viral plaques were subjected to several rounds of plaque purification, the genomic DNA was extracted, and the I7L gene was amplified by PCR and sequenced. All the resistant viruses, such as v17, were found to have mutations in positions 104 and 324 within the I7L open reading frame with a Y to C mutation at 104 and an L to M mutation at position 324 (Fig. 7B). Because of the manner in which they were isolated, it is likely that the drug-resistant mutant viruses are siblings of each other and that these common mutations do not necessarily represent a common point for mutation. Interestingly, the latter mutation is in close proximity to the C328 residue that is part of the catalytic triad, so that it might be expected to influence active site binding specificity. Once purified, drug-resistant v17 grows with the same kinetics and produces the same yield as wild-type virus in the absence of drug (data not shown), indicating that the mutations did not have deleterious effects on viral replication per se. To compare the growth of the parental virus vvGFP with the drug-resistant virus v17, a one-step growth experiment was performed where cells were infected with either vvGFP or v17 at an MOI of 1, treated with various concentrations of TTP-6171 1 h postinfection, and harvested at 2, 12, 24, and 48 h postinfection. Figure 7C shows that there is no difference in viral replication between the two viruses at any drug concentration at 2 h postinfection but that at 12, 24, and 48 h postinfection, v17 replicates to a higher titer at all drug concentrations. An immunoblot analysis of extracts from cells infected with v17 (with or without TTP-6171) and using antiserum against the VV A3L protein indicates that P4b to 4b processing is restored in the resistant mutant, although not to the same level as in the absence of compound (data not shown). Pulse-label and pulse-chase analyses were carried out and support the results of the immunoblotting (data not shown). However, at late times TTP-6171 has general inhibitory effects on late protein synthesis, which, in conjunction with toxicity at high concentrations, makes interpretation of processing difficult. It is possible that this compound inhibits another aspect of viral replication in addition to proteolytic processing.
FIG. 7.
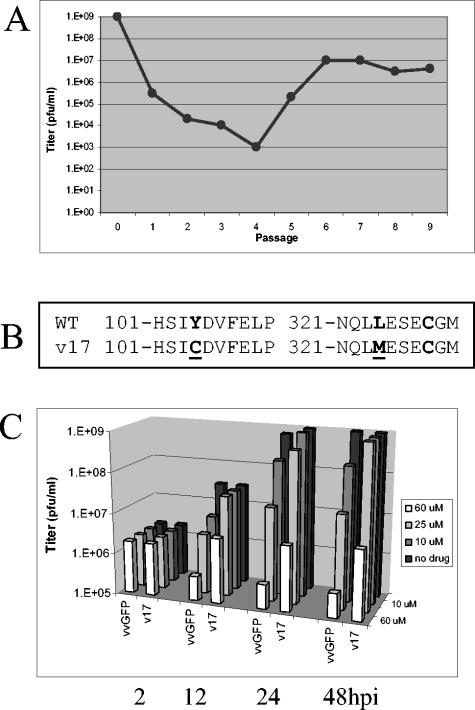
Passaging for drug resistance. (A) Viral titer of vvGFP passaged in the presence of TTP-6171. (B) Locations of mutations within the I7L ORF found in the TTP-6171-resistant virus isolate. Mutated residues are underlined. WT is wild-type virus; v17 is a TTP-6171-resistant isolate. (C) One-step growth curve of vvGFP and v17. Cells were infected at an MOI of 1 and treated with either 0, 10, 25, or 60 μM TTP-6171; cells were harvested at 2, 12, 24, or 48 h postinfection, and the viral titers were determined.
Transient expression and recombinant virus.
To confirm the hypothesis that the TTP-6171 resistance phenotype of v17 was due to the observed mutations in I7L and not another second site mutation and to determine whether one or both of the observed mutations contributed to resistance, transient expression and marker rescue procedures were employed. The mutant I7L gene from v17 was cloned into a plasmid vector, pRB21 (3), resulting in plasmid pI7L-17, such that its expression would be driven by a strong synthetic early-late VV promoter. By using this plasmid (pI7L-17) as a substrate, site-directed mutagenesis was used to create I7L genes containing only one of the observed mutations, both of them, or neither (wild type). First, these plasmids were tested for their ability to rescue the replication of vvGFP in the presence of TTP-6171. The data in Fig. 8 show that, as expected, overexpression of wild-type I7L provides some rescue, suggesting that TTP-6171 is a competitive inhibitor of I7L. Expression of the v17 I7L gene product provided a 10-fold higher level of rescue, consistent with the hypothesis that it is responsible for the TTP-6171 resistance of v17. Neither of the single mutations in v17 provided enhanced rescue when compared to wild-type I7L or the double mutant (v17 I7L). This result suggests that both mutations contribute to the full resistance phenotype of v17. As a final biological proof, the mutant alleles were recombined into the vvGFP genome, replacing the genomic I7L locus. In agreement with the transient expression results, both single mutants provide some degree of resistance to TTP-6171 inhibition, whereas the double mutant has the full resistance phenotype of v17 (data not shown). Furthermore, since the mutations were selected for by passage in the presence of TTP-6171, this strongly suggests that the I7L cysteine proteinase is the target of this drug.
FIG. 8.
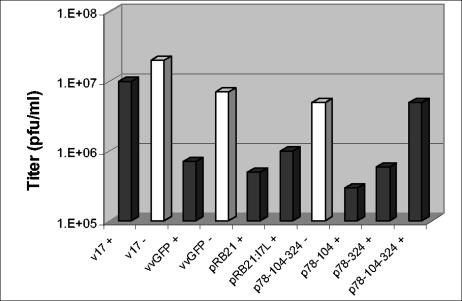
Transient expression. Wild-type or mutant viruses were grown in BSC40 cells and transfected with either plasmids containing no insert, wild-type I7L, or mutant I7L. The graph shows viral titers obtained in the presence or absence of TTP-6171. Transient expression with each plasmid is with wild-type virus. Light bars indicate no drug treatment (−), while dark bars indicate treatment with 25 μM TTP-6171 (+).
DISCUSSION
The classic approach to antiviral drug discovery involves high-throughput screening of large libraries of chemical compounds to look for inhibitors of viral replication. While effective, this approach has proven to be slow and tedious. An alternative approach, which we have utilized here, involves modeling of the target structure and computational docking of potential inhibitor molecules, followed by in vitro screening to discover candidate drugs. Drugs that do not hit the target will also be discovered by this approach, and further investigation into their mode of action will be necessary to determine their utility.
By using homology-based computational modeling, a structural model of the VV I7L cysteine proteinase, which is responsible for essential morphogenic cleavage reactions during viral maturation, was developed (Fig. 1). This model was used in concert with in silico drug docking procedures to query a combinatorial chemical library. A biased subset of compounds that was predicted to bind to the I7L catalytic site was directly tested for their ability to inhibit VV replication in vivo (Fig. 2 and 3). A number of chemically related effective inhibitors were identified, with the prototype being compound TTP-6171 (Table 2). Investigation of the mechanism of TTP-6171-mediated inhibition of VV replication indicated that although early stages of VV replication were unaffected, cleavage of the major core protein precursors and subsequent maturation of the immature viral intermediates into infectious intracellular mature virions were blocked (Fig. 5 and 6). These data were consistent with the idea that I7L catalytic activity is the target of TTP-6171 inhibition. To confirm this hypothesis, TTP-6171-resistant mutants were selected, and mutations within the I7L gene were shown to be responsible for the resistance phenotypes (Fig. 7 and 8).
TTP-6171 represents a very promising poxvirus antiviral drug candidate. When compared to cidofovir, which is the best-known poxvirus antiviral drug, TTP-6171 appears to have superior characteristics in tissue culture. Whereas cidofovir has a TC50 of 280 μM, IC50 of 33 μM, and a TI of 8 against VV (15), under similar conditions TTP-6171 has an IC50 of 12 μM and almost undetectable toxicity against a variety of tissue culture cells from a number of species, including monkey (BSC40), human (HeLa and 293), and mouse (L929) when examined 24 h after exposure. TTP-6171 does show some toxicity at high concentrations upon prolonged exposure, and hit-to-lead chemical optimization is under way to improve the pharmacological profile of this compound. TTP-6171 appears to exhibit an excellent specificity in that it does not inhibit growth of any of the other nonorthopox organisms tested, which include RNA viruses (ecotropic murine virus and mouse hepatitis virus), yeast, bacteria, or adenovirus. The latter result is particularly noteworthy because adenovirus has been shown to have a 90-amino-acid region with homology to the cysteine proteinase of I7L (1, 18). Although in this study TTP-6171 was only tested against several strains of VV and cowpox virus, it is highly likely that TTP-6171 will be broadly effective against other orthopoxviruses. As shown in Table 3, it appears that virtually all poxviruses require I7L-mediated essential morphogenic maturation of their core proteins as all contain an AGX motif at the same location within the P4b precursor. Within the Orthopoxviridae, sequence identity of the I7L gene is between 95 to 99% (including variola and monkeypox virus), and the residues flanking the catalytic site are completely conserved. Furthermore, the sequence conservation of the I7L gene remains considerable in other poxvirus genera, especially around the catalytic site, suggesting that TTP-6171 may have promise as an antiviral drug to treat more exotic poxvirus diseases as well as those of present concern, such as smallpox and monkeypox.
TABLE 3.
Sequence identity of catalytic region of I7L among various poxviruses
| Virus | Family | Catalytic triad sequence | % Identity with vvI7L ORF | P4b (AGA) |
|---|---|---|---|---|
| VV I7L | Orthopoxvirus | 241- HWKCVIYDKK QCLVSFYDSG 321-NQLLESECGM | 100 | Yes |
| Cowpox | Orthopoxvirus | 241- HWKCVIYDKK QCLVSFYDSG 321- NQLLESECGM | 96 | Yes |
| Camelpox | Orthopoxvirus | 241- HWKCVIYDKK QCLVSFYDSG 321- NQLLESECGM | 99 | Yes |
| Variola major | Orthopoxvirus | 241- HWKCVIYDKK QCLVSFYDSG 321- NQLLESECGM | 99 | Yes |
| Variola minor | Orthopoxvirus | 241- HWKCVIYDKK QCLVSFYDSG 321- NQLLESECGM | 99 | Yes |
| Monkeypox | Orthopoxvirus | 241- HWKCVIYDKK QCLVSFYDSG 321- NQLLESECGM | 99 | Yes |
| Ectromelia | Orthopoxvirus | 241- HWKCVIYDKK QCLVSFYDSG 321- NQLLESECGM | 95 | Yes |
| Sheeppox | Capripoxvirus | 251- HWKCVIFDKE KLVVCFYDSG 332- NQLLESECGM | 63 | Yes |
| Lumpy skin | Capripoxvirus | 251- HWKCVIFDKE KLVVCFYDSG 332- NQLLESECGM | 66 | Yes |
| Yaba-like | Yatapoxvirus | 246- HWKCVIINKE KLFVAFYDSG 327- NQLLESECGM | 69 | Yes |
| Swinepox | Suipoxvirus | 249- HWKCVIFDKE HHIVCFYDSG 330- NQLLESECGM | 68 | Yes |
| Rabbit fibroma | Leporipoxvirus | 245- HWKCVIFDKE KQIACFYDSG 321- NQLLESECGM | 66 | AGV |
| Myxoma virus | Leporipoxvirus | 245- HWKCVIFDKE KQIVCFYDSG 326- NQLLESECGM | 66 | AGV |
| Molluscum contagiosum | Molluscipox | 242- HWKSLVFDRR QRLVAFYDSG 322- NQLLESECGM | 62 | Yes |
| Fowlpox virus | Avipoxvirus | 242- HWKCAIYDKN RDFICFYDSG 322- NQLLESECGM | 58 | Yes |
| Canarypox virus | Avipoxvirus | 242- HWKCLIYDRE NDFVCFYDSG 323- NQLMESECGM | 55 | NPa |
| Amsacta moorei | Entomopoxvirus | 254- HFTSAVIDKK RKICYLFNSS 336- IQYDSPDCGM | 25 | NP |
NP, not published.
Based on the results reported here, the chemical compound family represented by TTP-6171 represents a promising avenue toward developing an effective antiviral drug that can be used to prevent or treat diseases caused by orthopoxviruses, such as smallpox. Although the development of effective antiviral drugs has proved challenging for some viruses, the 10- to 14-day incubation period between exposure to infectious smallpox and the development of obvious serious disease symptoms may provide a therapeutic window. It is envisioned that an effective smallpox antiviral drug will have several utilities including prophylaxis of individuals at risk to exposure, treatment of individuals already exposed, and as an adjunct to vaccination in immunocompromised patients. In any case, TTP-6171 represents an appropriate launch point for initiating hit-to-lead chemical optimization to improve drug activity as a prequel to initiation of animal efficacy studies in appropriate surrogate poxvirus animal challenge models.
Acknowledgments
This work was funded by NIH grant AI-48486 and Department of the Army contract DAMD-17-080C-0040. Partial funding was also provided by SIGA Technologies, Inc., and TransTech Pharma, Inc.
We thank Brita Hanson, Shirley Kickner, Stephen Ireland, and Guoxiang Huang for technical assistance. We thank Mike Nisson for assistance with the electron microscopy. We also thank R. C. Condit for ts16 and D. Pickup for cowpox virus.
REFERENCES
- 1.Andres, G., A. Alejo, C. Simon-Mateo, and M. L. Salas. 2001. African swine fever virus protease, a new viral member of the SUMO-1 specific protease family. J. Biol. Chem. 276:780-787. [DOI] [PubMed] [Google Scholar]
- 2.Berger, N. A., G. Weber, A. S. Kaichi, and S. J. Petzold. 1978. Relation of poly(adenosine dipohosporibose) synthesis to DNA synthesis and cell growth. Biochim. Biophys. Acta 519:105-117. [DOI] [PubMed] [Google Scholar]
- 3.Blasco, R., and B. Moss. 1995. Selection of recombinant vaccinia viruses on the basis of plaque formation. Gene 158:157-162. [DOI] [PubMed] [Google Scholar]
- 4.Byrd, C. M., T. C. Bolken, and D. E. Hruby. 2002. The vaccinia virus I7L gene product is the core protein proteinase. J. Virol. 76:8973-8976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Byrd, C. M., T. C. Bolken, and D. E. Hruby. 2003. Molecular dissection of the vaccinia virus I7L core protein proteinase. J. Virol. 77:11279-11283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Condit, R. C., A. Motyczka, and G. Spizz. 1983. Isolation, characterization, and physical mapping of temperature-sensitive mutants of vaccinia virus. Virology 128:429-443. [DOI] [PubMed] [Google Scholar]
- 7.Cundy, K. C. 1999. Clinical pharmacokinetics of the antiviral nucleotide analogues cidofovir and adefovir. Clin. Pharmacokinet. 36:127-143. [DOI] [PubMed] [Google Scholar]
- 8.Daddow, L. Y. 1986. An abbreviated method of the double lead stain technique. J. Submicrosc. Cytol. 18:221-224. [PubMed] [Google Scholar]
- 9.De Clercq, E. 2002. Cidofovir in the treatment of poxvirus infections. Antivir. Res. 55:1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fulginiti, V. A., A. Papier, J. M. Lane, J. M. Neff, and D. A. Henderson. 2003. Smallpox vaccination: a review, part II. Adverse events. Clin. Infect. Dis. 37:251-271. [DOI] [PubMed] [Google Scholar]
- 11.Heller, E., M. Argaman, H. Levy, and N. Goldblum. 1969. Selective inhibition of vaccinia virus by the antibiotic rifampicin. Nature 222:273-274. [DOI] [PubMed] [Google Scholar]
- 12.Herrmann E. C., Jr. 1968. Sensitivity of herpes simplex virus, vaccinia virus, and adenoviruses to deoxyribonucleic acid inhibitors and thiosemicarbazones in a plaque suppression test. Appl. Microbiol. 16:1151-1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hruby, D. E., L. A. Guarino, and J. R. Kates. 1979. Vaccinia virus replication. I. Requirement for the host-cell nucleus. J. Virol. 29:705-715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kane, E. M., and S. Shuman. 1993. Vaccinia virus morphogenesis is blocked by a temperature-sensitive mutation in the I7 gene that encodes a virion component. J. Virol. 67:2689-2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kern, E. R. 2003. In vitro activity of potential anti-poxvirus agents. Antivir. Res. 57:35-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lalezari, J. P., and B. D. Kuppermann. 1997. Clinical experience with cidofovir in the treatment of cytomegalovirus retinitis. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 14(Suppl. 1):S27-S31. [DOI] [PubMed] [Google Scholar]
- 17.Lamarre, D., P. C. Anderson, M. Bailey, P. Beaulieu, G. Bolger, P. Bonneau, M. Bos, D. R. Cameron, M. Cartier, M. G. Cordingley, A.-M. Faucher, N. Goudreau, S. H. Kawai, G. Kukolj, L. Lagace, S. R. LaPlante, H. Narjes, M.-A. Poupart, J. Rancourt, R. E. Sentjens, R. St George, B. Simoneau, G. Steinmann, D. Thibeault, Y. S. Tsantrizos, S. M. Weldon, C.-L. Yong, and M. Llinas-Brunet. 2003. An NS3 protease inhibitor with antiviral effects in humans infected with hepatitis C virus. Nature 426:186-189. [DOI] [PubMed] [Google Scholar]
- 18.Li, S. J., and M. Hochstrasser. 1999. A new protease required for cell-cycle progression in yeast. Nature 398:246-251. [DOI] [PubMed] [Google Scholar]
- 19.Miravalle, A., and K. L. Roos. 2003. Encephalitis complicating smallpox vaccination. Arch. Neurol. 60:925-928. [DOI] [PubMed] [Google Scholar]
- 20.Moss, B., E. N. Rosenblum, E. Katz, and P. M. Grimley. 1969. Rifampicin: a specific inhibitor of vaccinia virus assembly. Nature 224:1280-1284. [DOI] [PubMed] [Google Scholar]
- 21.Raczynski, P., and R. C. Condit. 1983. Specific inhibition of vaccinia virus growth by 2′-O-methyladenosine: isolation of a drug-resistant virus mutant. Virology 128:458-462. [DOI] [PubMed] [Google Scholar]
- 22.Rosenkranz, H. S., H. M. Rose, C. Morgan, and K. C. Hsu. 1966. The effect of hydroxyurea on virus development II. Vaccinia virus. Virology 28:510-519. [DOI] [PubMed] [Google Scholar]
- 23.Spurr, A. R. 1969. A low-viscosity epoxy resin embedding medium for electron microscopy. J. Ultrastruct. Res. 26:31-43. [DOI] [PubMed] [Google Scholar]
- 24.VanSlyke, J. K., and D. E. Hruby. 1994. Immunolocalization of vaccinia virus structural proteins during virion formation. Virology 198:624-635. [DOI] [PubMed] [Google Scholar]
- 25.Venable, J. H., and R. Coggeshall. 1965. A simplified lead citrate stain for use in electron microscopy. J. Cell Biol. 25:407-408. [DOI] [PMC free article] [PubMed] [Google Scholar]