

Abstract
A polyomavirus mutant isolated by the tumor host range selection procedure (19) has a three-amino-acid deletion (Δ2-4) in the common N terminus of the T antigens. To search for a cellular protein bound by wild-type but not the mutant T antigen(s), a yeast two-hybrid screen of a mouse embryo cDNA library was carried out with a bait of wild-type small T antigen (sT) fused N terminally to the DNA-binding domain of Gal4. TAZ, a transcriptional coactivator with a WW domain and PDZ-binding motif (17), was identified as a binding partner. TAZ bound in vivo to all three T antigens with different apparent affinities estimated as 1:7:100 (large T antigen [lT]:middle T antigen [mT]:sT). The Δ2-4 mutant T antigens showed no detectable binding. The sT and mT of the host range transformation-defective (hr-t) mutant NG59 with an alteration in the common sT/mT region (179 D→NI) and a normal N terminus also failed to bind TAZ, while the unaltered lT bound but with reduced affinity compared to that seen in a wild-type virus infection. The WW domain but not the PDZ-binding motif of TAZ was essential for T antigen binding. The Δ2-4 mutant was defective in viral DNA replication. Forced overexpression of TAZ blocked wild-type DNA replication in a manner dependent on the binding site for the polyomavirus enhancer-binding protein 2α. Wild-type polyomavirus T antigens effectively block transactivation by TAZ. The functional significance of TAZ interactions with polyomavirus T antigens is discussed.
Oncogenes of DNA tumor viruses exert essential functions in the virus life cycle by binding and altering the activities of specific cellular factors. The latter may be regulators of the cell cycle or apoptosis or have other functions that must be activated or inhibited by the virus in order to grow. What is known to date about the highly oncogenic mouse polyomavirus and its interactions with host factors gives an incomplete account of its replicative and oncogenic functions (2). A tumor host range (thr) selection procedure was previously devised to expand the search for cellular factors targeted by polyomavirus. The rationale behind this procedure is based on the supposition that cells from spontaneous or carcinogen-induced tumors may have undergone the loss or alteration of a cellular factor(s) that the virus normally targets to grow efficiently. thr mutants are selected to grow in certain tumor or transformed cells but not in normal cells and are presumed to have lost the targeting function(s). The wild-type T antigen corresponding to that altered in such mutants may be used to identify a putative cellular target(s). The homeotic transcription factor Sal2 was discovered as a target of the polyomavirus large T antigen (lT), and the binding was shown to be essential for virus replication, using this procedure (19).
Here, we have used a chemically transformed mouse liver tumor cell line (25) as a permissive host and primary baby mouse kidney (BMK) cells as a nonpermissive host to isolate a new polyomavirus thr mutant. The alteration giving rise to this host range is in the N-terminal sequence common to the three T antigens. A search for a cellular target has identified the TAZ protein as interacting with all three T antigens, depending in part on their N terminus. TAZ, first described as a 14-3-3-binding protein, has a PDZ-binding motif and a WW domain critical to its function as a transcriptional coactivator (17). The RUNX (runt domain-containing) family of transcription factors is among those activated by TAZ. RUNX gene products include the polyomavirus enhancer-binding protein 2α (PEBP-2α) (RUNX1/AML1/Cbfa2) (4). RUNX1/AML1 and other members of the RUNX family are frequently altered in human cancers (15, 20, 22, 33).
MATERIALS AND METHODS
Cells and viruses.
BNL 1ME (BNL) is a chemically transformed mouse liver cell line (25) obtained through the American Type Culture Collection. NIH 3T3, BR-lT (21), and BMK cells were routinely cultured in Dulbecco's modified Eagle medium (Sigma) supplemented with 0.375% sodium bicarbonate, 100 U of penicillin/ml, 100 μg of strepyomycin/ml, and 10% calf serum in a 5% CO2 incubator at 37°C. Wild-type polyomavirus strain RA has been described previously (8). The D2N and Δ2-4 mutants were grown on BNL or by transfection of BR-lT cells with the mutant viral DNAs.
Plasmids.
Vector pAS2-1 (Clontech) was used to construct the polyomavirus small T antigen [sT]-Gal4 DNA-binding domain bait (psT-Gal4BD) for yeast two-hybrid screening. The complete wild-type sT sequence was cloned 5′ to the Gal4 DNA-binding domain sequence in the vector to leave the normal amino terminus of the sT antigen exposed. Prokaryotic and eukaryotic glutathione S-transferase (GST) fusion expression vectors pGEX-4T-1 (Amersham Bioscience) and pEBG were used to generate GST-TAZ fusion proteins, respectively. For studies of the effects of viral infection on TAZ function, full-length and mutant forms of TAZ (see below) were inserted into a eukaryotic Gal4 fusion vector (pcDNA3; Invitrogen). The RA strain of polyomavirus was cloned into pBS vector (Stratagene) through the BamHI site to generate plasmid RAHI. To construct cytomegalovirus (CMV) promoter-driven T antigen expression plasmids, lT, middle T antigen (mT), and sT cDNAs were amplified by PCR and subcloned into pcDNA3 vector (Invitrogen). Py-wt Ori contained the polyomavirus origin and the entire noncoding region. Py-ΔOri was derived from Py-wt Ori by deleting the polyomavirus enhancer-binding protein 2α (PEBP-2α)-binding site (ACCGCAG); both were cloned into pGL3Basic vector (Promega). All T antigen and TAZ mutant constructs were generated with the QuikChange site-directed mutagenesis kit (Stratagene) based on wild-type plasmids. TAZ-ΔPDZ has a deletion of the C-terminal 10 amino acids, and TAZ-ΔWW has a deletion of residues 125 to 156. Constructs were verified by sequencing.
Luciferase assay.
For experiments on the effects of viral infection on transactiavation by TAZ, NIH 3T3 cells in 24-well plates were transfected with plasmids as indicated in the text with Lipofectamine 2000 reagent (Invitrogen), the cells were infected with wild-type virus and harvested 36 h later. Total transfected DNA was fixed by adding corresponding amounts of backbone plasmid. Firefly and Renilla luciferase activities were assayed by the dual luciferase assay system (Promega), and firefly luciferase activity was normalized with respect to Renilla luciferase activity.
Antibodies.
Rabbit polyclonal anti-TAZ antibody (Covance) was made with a GST-mTAZ (amino acids 125 to 395) fusion protein as antigen. Rat polyclonal anti-T ascites (31) was used to do immunoprecipitation (IP) assays, and mouse anti-T monoclonal antibody (F4) was used to do Western blot analysis. A rat anti-lT monoclonal antibody (Ab-1; Calbiochem) was used for immunofluorescent staining.
Yeast-two hybrid screening.
A 17-day mouse embryo cDNA library (Clontech) was used for screening with Saccharomyces cerevisiae strain AH109 with psT-Gal4BD as bait (see above). The library contains 3.5 × 106 independent clones, and the titer is 3 × 108 to 4 × 108 CFU/ml. The yeast two-hybrid screening was performed according to instructions in the user manual (Clontech).
Transfection and infection.
Lipofectamine 2000 reagent (Invitrogen) was used in transfections based on the company's instructions. Approximately 30 μl of transfection reagent and 10 μg of plasmids were used for one 10-cm plate transfection. For DNA replication assays, cells were seeded on six-well plates, and 1 μg of plasmid DNA(s) was transfected per well. For IP experiments, cells were seeded onto 10-cm plates and were infected with 0.5 ml of virus suspension (RA and NG59, 5 × 107 to 2 × 108 PFU/ml) for 2 h at 37°C. For the DNA replication assay, cells were seeded on six-well plates and infected with 0.05 ml of virus suspensions per well.
Tetracycline-regulated TAZ expression.
Mouse TAZ full-length cDNA was cloned into a Tet-regulated vector pTet-Splice (consisting of regulatory sequence from the tetracycline resistance operon) to generate construct pTet-mTAZ. pTet-mTAZ, pTet-tTAK (containing the gene for the tetracycline transactivator tTA under the control of TetP), and pcDNA3 were cotransfected into NIH 3T3 cells in the presence of tetracycline. The stable cell lines were selected and established according to the manufacturer's instructions (GIBCO BRL). Expression of TAZ is induced by the withdrawal of tetracycline from the medium.
IP assays and Western blotting.
Lysates of uninfected, infected, or transfected cells were prepared at the indicated times with NP-40 lysis buffer (20 mM Tris [pH 8.0], 135 mM NaCl, 1 mM MgCl2, 0.1 mM CaCl2, 10% glycerol, 1% NP-40, 0.1 mM Na3VO4, 50 mM β-glycerophosphate, 10 mM NaF, and the protease inhibitor Complete Mini [Roche]) and used for Western blotting. For IP assays, cells were lysed in the lysis buffer indicated above, extracts (2 to 3 mg of protein) were incubated with antibody at 4°C for 3 h, and the immune complexes were recovered with protein A-Sepharose CL-4B (Amersham Pharmacia). The proteins were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose membranes. The membranes were blocked with 5% nonfat dry milk in TNET buffer (10 mM Tris, 2.5 mM EDTA, 50 mM NaCl, and 0.1% Tween 20) and incubated with various antibodies for 1 h at room temperature. The blots were washed four times with TNE buffer (10 mM Tris, 2.5 mM EDTA, and 50 mM NaCl), incubated with secondary antibody conjugated to horseradish peroxidase, and then washed four times in TNE buffer. The bands were visualized by chemiluminescence (Perkin-Elmer Life Sciences).
GST pull-down assay.
Lysates of transfected cells were prepared with NP-40 lysis buffer 36 h posttransfection. Extracts (2 to 3 mg of protein) were incubated with Glutathione Sepharose 4B (Amersham Bioscience) overnight at 4°C, and the beads were washed five times with lysis buffer. The complexes were resolved by SDS-PAGE, and Western blotting was performed as described above.
Viral DNA replication assay.
The polyomavirus origin replication assay has been described previously (19). Cells were grown on six-well plates; virus infection or DNA transfection has been described earlier. A total of 5 μg of DNA was subjected to restriction digestion. For virus infection, the viral genome was linearized with EcoRI. For transfection, Py-wt Ori and the mutant were digested with DpnI and Hind III. The newly synthesized, but not the input, DNA is DpnI resistant due to the lack of methylation. The DNA was resolved on a 0.8% agarose gel and probed for Southern analysis with polyomavirus origin-specific or neo gene probes as described in the text.
Immunofluorescent staining.
Immunofluorescent staining was performed as previously described (10). Briefly, BMK cells were plated in complete Dulbecco's modified Eagle medium on coverslips and incubated at 37°C. After infection, infected and uninfected cells were fixed with 4% paraformaldehyde in phosphate-buffered saline for 30 min and stained with rabbit anti-TAZ polyclonal and rat anti-lT monoclonal antibodies. The primary antibodies were detached with Organ Green-conjugated anti-rabbit immunoglobulin G (IgG) secondary antibody and rhodamine red-conjugated anti-rat IgG antibody, respectively. Immunofluorescence results were quantitated by examining single lT-positive cells to determine whether they showed evidence of nuclear accumulation of TAZ compared to neighboring uninfected (lT-negative) cells. Results were obtained for cells infected by wild-type virus and hr-t mutants NG59 and NG18 (3, 13).
RESULTS
A polyomavirus thr mutant leads to the identification of TAZ as a binding partner of the three T antigens.
BNL cells used as a permissive host were originally derived as a methylcholanthrene epoxide-transformed mouse liver cell line. These cells show anchorage-independent growth and are tumorigenic in immunocompetent mice (25). Randomly mutagenized polyomavirus DNA was obtained by cycling a plasmid carrying the PTA wild-type polyomavirus genome through the MutD error-prone strain of Escherichia coli (19). The mutagenized viral DNA was excised and transfected into BNL cells, and the resulting lysate was used for single-plaque isolations on BNL monolayers. Individual plaque suspensions were screened for the absence of cytopathic effect on primary BMK cells. A plaque isolate showing clear differential cytopathic effect—i.e., positive on BNL cells and negative on BMK cells—was molecularly cloned for further study. Sequencing of this isolate showed a substitution at the second position of the common N terminus of the T antigens (D2N). To separate this mutation from others that may have been present in the original isolate, a D2N mutation was introduced into wild-type viral DNA, and the tumor host range of this virus mutant was confirmed (data not shown).
A yeast two-hybrid screen of a 17-day mouse embryo cDNA library (Clontech) was carried out to search for a binding target. The bait was constructed with full-length wild-type polyomavirus sT fused N terminally to the Gal4 DNA-binding domain to expose the region of the T antigens presumed to bind the putative cellular factor (Fig. 1a). The screening yielded several positive clones, one with a novel sequence showing roughly 40% homology to YAP (yes-associated protein), a protein that binds to the SH3 domain of the c-yes proto-oncogene product (32). The sequence of this sT-binding cDNA clone subsequently proved to be the 303 C-terminal amino acids of TAZ, a transcriptional coactivator with the PDZ-binding motif (17). Murine TAZ is a 395-amino-acid protein with a serine at position 89 that constitutes a phosphorylation-dependent 14-3-3-binding site. TAZ also has a WW domain (residues 125 to 156) essential for binding to certain transcription factors through the PPXY motif, a transactivation domain (residues 165 to 395), as well as a PDZ-binding motif at the C terminus important for localization within the nucleus and for transcriptional coactivator function (Fig. 1b) (17).
FIG. 1.
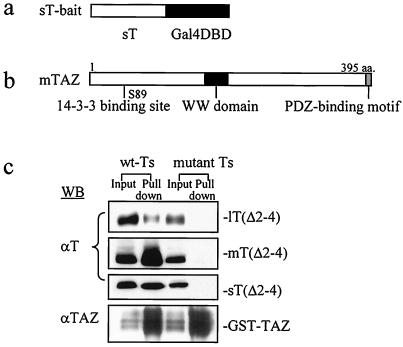
TAZ is identified as a binding partner for all polyomavirus T antigns. (a) A “flipped” bait with wild-type sT fused through its C terminus to the Gal4 DNA-binding domain was used to perform yeast two-hybrid screening. (b) Target TAZ combines a C-terminal PDZ-binding motif, a 14-3-3-binding sequence, and a WW domain. (c) Transfections and pull-down assays with GST-TAZ. Wild-type and mutant D2N and Δ2-4 versions of T antigen cDNAs were cotransfected into NIH 3T3 cells along with GST-TAZ. Extracts of transfected cells were incubated with GST beads. Bound proteins were eluted and analyzed by Western blotting with F4 monoclonal antibody recognizing all three T antigens. Wild-type T antigens are shown to the left of the panels; mutant T antigens are shown to the right in parentheses. D2N sT bound TAZ roughly 60% as well as wild-type sT. The mutation Δ2-4 effectively abolished binding of TAZ by each of the T antigens. A total of 5% of the cell lysate was loaded in the input lanes.
To demonstrate interaction between TAZ and the polyomavirus T antigens, NIH 3T3 cells were transfected with wild-type or mutant T antigen cDNAs along with a GST-TAZ construct. Transfected cell extracts were incubated with GST beads, followed by elution, separation, and Western analysis with an anti-T monoclonal antibody that recognizes sequences common to all three T antigens (Fig. 1c). When individually expressed, each of the wild-type T antigens bound to GST-TAZ. The D2N mutation weakened but did not abolish TAZ binding to sT and mT (data not shown). Several N-terminal deletions were introduced into sT in an attempt to find one with a more complete effect on TAZ binding. A three-amino-acid deletion of residues 2 to 4 was found to be the minimal deletion that effectively prevented TAZ binding to sT. The Δ2-4 mutation in mT and lT also abolished binding to TAZ in these in vitro pull-down experiments (Fig. 1c).
A mutant virus constructed with the Δ2-4 mutation displayed clear differential growth and cytopathic effects when infected onto BNL and BMK monolayers (Fig. 2). The Δ2-4 mutant replicated and induced cytopathology on the permissive BNL cells, although its growth was slower and virus yields were significantly lower (2 to 5%) than those of wild-type virus infecting the same cells. In contrast, the mutant induced no detectible cytopathic effect in the BMK cultures even after prolonged periods of incubation. Growth of the mutant on nonpermissive BMK cells was estimated to be at least 4 orders of magnitude less than that of the wild-type virus. Because the Δ2-4 mutant preserved the thr phenotype of the original (D2N) isolate but showed a more pronounced defect in terms of absence of TAZ binding, further experiments were carried out with Δ2-4 as the thr mutant.
FIG. 2.
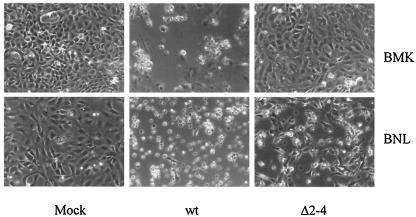
Growth of thr mutant Δ2-4 is restricted to BNL cells. Monolayer cultures of BNL and primary BMK cells were uninfected (mock) or infected by wild-type virus or thr mutant Δ2-4 virus. The photographs, taken 5 days postinfection, show the cytopathic effects indicative of virus growth in all infected cultures, except for BMK infected by the mutant.
T antigen interactions with TAZ.
To confirm and further analyze the binding of T antigens to endogenous TAZ in the context of a normal viral infection, BMK cells were infected by wild-type virus, and extracts were immunoprecipitated with antibody to TAZ. Analysis by Western blotting with anti-T antibody showed that each of the T antigens was brought down by the anti-TAZ antibody, though with different efficiencies (Fig. 3a). Based on the ratios of the amounts of each T antigen recovered in the IP assays to the amounts present in the infected cell extracts, the relative affinities of the T antigens for TAZ were estimated to be roughly 1:7:100 (lT:mT:sT). These estimates do not account for differences in effective concentrations of TAZ and T antigens in different subcellular compartments or for effects of the anti-TAZ polyclonal antibodies which may affect binding and recovery differently among the different T antigen-TAZ complexes.
FIG. 3.
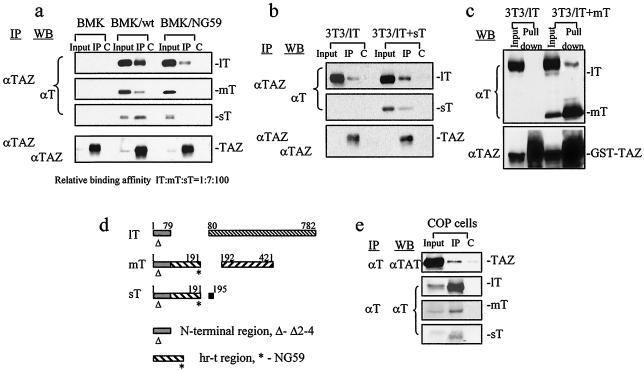
Analyses of T antigen interactions with TAZ. (a) Extracts of uninfected, wild-type, and NG59 mutant virus-infected BMK cells were immunoprecipitated by cross-linked rabbit anti-TAZ polyclonal antibody (IP) or by preimmune antiserum as a control (C). Proteins were separated and blotted with anti-T and anti-TAZ antibodies. (b) Cotransfection of NIH 3T3 cells with TAZ and lT cDNA alone or together with sT cDNA, followed by anti-TAZ IP and blotting for T antigens. (c) Cotransfection with GST-TAZ and lT alone or together with mT cDNAs followed by GST pull-down and Western blotting for T antigens. A total of 5% of cell lysates were loaded in the input lanes (a to c). (d) Schematic representation of the polyomavirus T antigens showing the two regions of interaction with TAZ, the shared amino terminal sequences (Δ) common to all three T antigens, and the hr-t (large T intron) sequences shared by mT and sT (*). (e) A GST-TAZ expression plasmid was transfected into COP cells, and extracts were immunoprecipitated by anti-T antibody or by normal rat IgG as a control (C). SDS-PAGE and Western blotting for TAZ were performed.
The hr-t mutants (1) have defects in the common sT/mT region encoded by the lT intron (Fig. 3d) while encoding a normal lT protein (3, 13). A prototype hr-t mutant, NG59, has a substitution and insertion at residue 179 of sT/mT (179D→NI). These alterations produce functionally defective sT and mT proteins, the former unable to bind PP2A and the latter unable to bind pp60c-src (7, 24, 29, 30). The lT encoded by the NG59 mutation is unaltered. Interestingly, the sT and mT of NG59 fail to bind TAZ despite retention of a normal sequence at the N terminus (Fig. 3a). An unaltered N terminus is thus necessary but not sufficient for TAZ binding to sT and mT; sequences in the hr-t region unique to these two T antigens are also required.
The lT of NG59 binds TAZ, although distinctly less efficiently than the identical lT in a wild-type virus infection. This indicates that a normal N terminus in lT suffices for some TAZ binding and further suggests that sT or mT, or possibly both, may act in some way to enhance binding of TAZ to lT. To test these possibilities directly, NIH 3T3 cells were transfected with lT cDNA alone or with sT or mT cDNAs. Coexpression of sT clearly enhanced binding of lT to TAZ (Fig. 3b). Similarly, coexpression of mT with lT led to increased binding of the latter to GST-TAZ (Fig. 3c). The regions of the polyomavirus T antigens involved in TAZ binding are shown schematically in Fig. 3d. Polyomavirus-transformed mouse cells (34) expressing all three T antigens were used to demonstrate TAZ-T antigen interactions, using anti-T antigen antibody to IP from cell extracts, followed by blotting for TAZ (Fig. 3e).
Alterations of the amino terminus of the Δ2-4 mutant might be expected to alter the efficiency of initiation of translation leading to lower steady-state levels of the mutant T antigens. This is indeed observed following transfection of the T antigen cDNAs where the mutant proteins accumulated to levels roughly 60 to 70% of those of the corresponding wild-type proteins (Fig. 1c). To show that the reduced levels of protein did not affect the ability of the Δ2-4 lT to bind other known targets of lT, the binding of Δ2-4 lT to pRB (9, 18) and p53 was tested by co-IP and blotting. The mutant lT bound to each of these target proteins as efficiently as wild-type lT (data not shown). The effect of the Δ2-4 mutation is thus specific for TAZ binding.
Simian virus 40 (SV40) T antigens show some homology to polyomavirus T antigens in their amino terminal sequences, with two conservative changes in the first six residues. SV40, however, does not encode an mT protein, and the region of sT/mT altered in NG59 and important for TAZ binding shows no homology with SV40 sT. Attempts to show interaction of the SV40 T antigens with TAZ by using GST-TAZ and SV40 sT and lT cDNAs were negative (data not shown).
TAZ recognizes the polyomavirus T antigens through its WW domain.
To determine the region(s) of TAZ essential for binding to the polyomavirus T antigens, GST-TAZ constructs containing either full-length TAZ, TAZ deleted of its WW domain (residues 125 to 156), or TAZ deleted of its PDZ-binding motif (residues 386 to 395) were transfected into NIH 3T3 cells. Transfected cells were then infected by wild-type polyomavirus for 24 h. Extracts were prepared and GST pull-down assays were performed. Full-length TAZ and TAZ-ΔPDZ bound the three T antigens with the same relative efficiencies, while TAZ-ΔWW failed to bind detectible levels of any of the T antigens (Fig. 4). The WW domain of TAZ, essential for binding cellular transcription factors (17), is thus essential for binding to each of the polyomavirus T antigens.
FIG. 4.
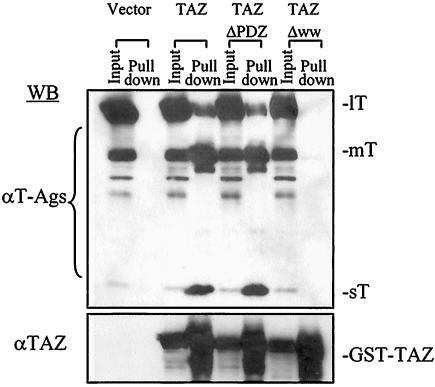
The WW domain but not the PDZ-binding motif in TAZ is essential for binding to polyomavirus T antigens. Wild-type GST-TAZ (TAZ), GST-TAZΔPDZ (lacking residues 386 to 395; TAZ ΔPDZ), and GST-TAZΔWW (lacking residues 125 to 156; TAZ ΔWW) constructs were transfected into NIH 3T3 cells. Transfected cells were then infected by wild-type virus. Cell extracts were prepared, and GST pull-down assays were performed 24 h postinfection. After SDS-PAGE, Western blot analyses for T antigens and TAZ were performed. A total of 5% of cell lysates were loaded in input lanes.
Wild-type polyomavirus infection leads to nuclear accumulation of TAZ.
The distribution of TAZ was examined in infected and uninfected primary BMK cells by double immunofluorescent staining with anti-T and anti-TAZ antibodies (Fig. 5a). TAZ was found in both the nucleus and cytoplasm in uninfected cells, with nuclear staining showing a punctate pattern. In cells infected by wild-type virus, there was a partial redistribution of TAZ from the cytoplasm to the nucleus, evidenced by brighter nuclear than cytoplasmic TAZ staining in wild-type lT-positive cells compared to neighboring uninfected cells (Fig. 5a, top panels). Approximately 80% of large T-positive cells showed a detectible increase in nuclear TAZ staining. NG59, which fails to bind TAZ through its mT and sT (Fig. 3a), was much less efficient in inducing nuclear TAZ accumulation (Fig. 5). Similar results were found for NG18, a deletion hr-t mutant which, unlike NG59, expresses no detectible sT or mT protein (13). Thus, movement of a portion of TAZ from the cytoplasm to the nucleus accompanies wild-type infection but not infection by hr-t mutants defective in the small and middle T antigens.
FIG. 5.
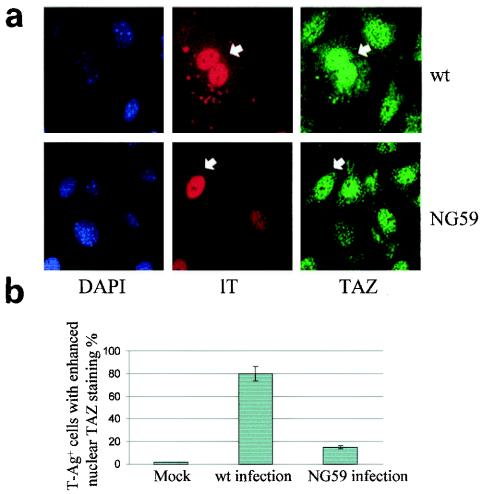
Expression of wild-type sT and mT leads to nuclear accumulation of TAZ. (a) BMK cells were infected by wild-type virus or NG59 mutant virus. After 24 h, cells were fixed, and immunofluorescence staining was performed with anti-lT (rat) and anti-TAZ antibody (rabbit) followed by secondary antibodies, rhodamine red and Oregon green, respectively. (b) Quantitation of results (see Materials and Methods).
Effects of TAZ on viral DNA replication.
The ability of the Δ2-4 mutant to replicate its DNA in nonpermissive BMK cells was greatly diminished relative to wild-type virus (Fig. 6a). TAZ binding to one or more of the T antigens is therefore essential for optimal viral DNA replication. At high levels of expression, TAZ has an inhibitory effect on wild-type virus replication. This was shown using a line of NIH 3T3 cells with Tet-regulated TAZ expression. In the presence of Tet, the cells show undetectable levels of TAZ, and the wild-type virus replicates well. With Tet removed and TAZ highly expressed, viral DNA synthesis was inhibited, as seen by the minimal increase in viral DNA accumulation at 24 and 40 h compared to input DNA at 0 h (Fig. 6b).
FIG. 6.
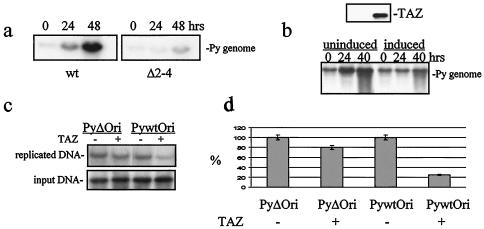
Effects of TAZ on viral DNA replication. (a) BMK cells were infected by wild-type virus or Δ2-4 mutant virus. Low-molecular-weight DNA was isolated, and a Southern blot analysis was performed using 32P-labeled polyomavirus DNA as a probe. The Δ2-4 mutant was sharply reduced in its ability to replicate its DNA.(b) High-level expression of TAZ in a Tet-repressible NIH 3T3 cell line blocks wild-type viral DNA replication. TAZ expression was induced by removing Tet 16 h before infection. TAZ-induced and uninduced cells were infected by wild-type polyomavirus, and low-molecular-weight DNA was extracted at different times. A Southern blot analysis was performed using a polyomavirus plasmid as a probe. Viral DNA replication was strongly inhibited by TAZ as seen from the minimal amplification of input viral DNA at time zero compared to 40 h. (c) The PEBP-2α-binding site is essential for TAZ inhibition of viral DNA synthesis. Wild-type polyomavirus Ori (Py-wt Ori) and an Ori with the PEBP-2α-binding site deleted (Py-Δ Ori) were transfected into TAZ-induced or uninduced cells. TAZ expression was induced by removing Tet 16 h before transfection, and cells were infected by polyomavirus immediately after transfection. Low-molecular-weight DNA was extracted after 24 h, and Southern blotting was performed with the luciferase-encoding region of the ori plasmid as a probe. The band of replicated DNA was compared to input plasmid DNA, and the replication of wild-type ori plasmid in uninduced cells was taken as 100%. (d) Quantitation shows that the inhibition of viral DNA replication by TAZ depends on retention of the PEBP-2α-binding site in the viral enhancer.
The inhibitory effect of TAZ on viral DNA replication may be mediated by PEBP-2α binding to the viral enhancer, since this member of the RUNX family of transcription factors can have negative as well as positive effects on viral DNA synthesis (26, 27). This was tested in an Ori replication assay with a wild-type Ori and an Ori with a deletion of the PEBP-2α-binding site introduced into the NIH 3T3 Tet-TAZ cell line followed by wild-type viral infection. Replication of the wild-type Ori was inhibited roughly fourfold in cells expressing high levels of TAZ. Only slight inhibition of the ori plasmid lacking the PEBP-2α-binding site was seen under the same conditions (Fig. 6c and d).
Virus infection inhibits transactivation by TAZ.
TAZ was previously shown to function as a transcriptional coactivator (17). To examine the effect of the polyomavirus T antigens on the coactivator function of TAZ, NIH 3T3 cells were cotransfected with a plasmid encoding the Gal4 DNA-binding domain by itself or fused to various TAZ constructs, along with a luciferase reporter plasmid driven by Gal4-binding sites linked to a thymidine kinase promoter. Transfected cells were either infected or not by wild-type polyomavirus. In uninfected cells, a 3.8-fold activation by the TAZ fusion was seen compared to Gal4 DNA-binding domain alone (Fig. 7). Infected cells showed a roughly 50% reduction in activation, commensurate with a roughly 60% infection rate based on staining for lT. A similar degree of transactivation was seem using the TAZΔWW construct; however, inhibition by wild-type virus infection was much less efficient, consistent with the importance of the WW domain in binding to the T antigens (Fig. 4). Transactivation by TAZΔPDZ was greatly reduced, as previously reported (17); inhibition by viral infection was still observed.
FIG. 7.
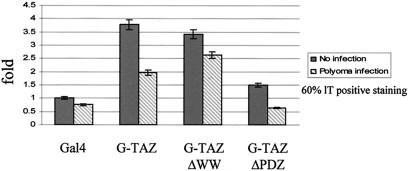
Wild-type polyomavirus infection inhibits transactivation by TAZ. NIH 3T3 cells were cotransfected with 50 ng of tk-LUC, 50 ng of pRL-EF, and 400 ng of CMV-Gal4 vector along with plasmid expressing the indicated GAL4-TAZ fusion proteins. The luciferase activities were normalized by Renilla activities.
DISCUSSION
Randomly mutagenized polyomavirus DNA was cycled through cells derived from a chemical carcinogen-transformed and tumorigenic mouse liver cell line, thus amplifying any virus that could replicate and kill these cells. The resulting virus lysate was used to isolate a thr virus mutant that retained some ability to replicate in the transformed cells but was unable to grow in normal BMK cells. The mutation in this isolate leads to a single-amino-acid substitution (D2N) in the common N terminus of the T antigens. A yeast two-hybrid screen of a mouse embryo cDNA library was carried out to search for a cellular protein that could bind to the wild-type sT protein. The bait was configured with wild-type sT fused through its C terminus to the yeast Gal4 DNA-binding domain to expose the normal N-terminal T antigen sequence. A target representing a C-terminal fragment of the transcriptional coactivator TAZ (17) was identified. Results of pull-down experiments with GST-TAZ and T antigen cDNAs showed that all three wild-type T antigens bound TAZ. The T antigens expressed during a lytic infection of primary BMK cells bound to endogenous TAZ, although with remarkably different apparent efficiencies (sT > mT > lT). The D2N mutation partially reduced TAZ binding, while a site-directed mutant Δ2-4 completely abolished all TAZ-T antigen interactions. A virus mutant constructed with the Δ2-4 mutation behaved as a thr mutant with the same host range as the original isolate.
The WW domain of TAZ is essential for its interaction with the T antigens. This domain is also critical to the coactivator function of TAZ and of the related YAP (32) through interactions with PPXY sequences in their target transcription factors (4, 17, 36). Interestingly, the latter include the RUNX family and, specifically, RUNX1 or AML1 (17), which was originally discovered as PEBP-2α (14, 28). PEBP-2α binds to an element (ACCGCAG) in the A domain of the polyomavirus enhancer and has complex effects in regulating both transcription (16) and replication (26, 27) of viral DNA. The PDZ-binding motif at the C terminus of TAZ is not essential for T antigen binding but is important in regulating the subcellular distribution of TAZ, including association with the plasma membrane and in punctate nuclear structures (17). PDZ domains are found in proteins of diverse function and subcellular localization (12, 23). The PDZ-binding motif of TAZ is essential for coactivator function, suggesting that TAZ may interact with one or more nuclear proteins with a PDZ domain. The 14-3-3-binding site in TAZ serves to negatively regulate its coactivator activity, presumably by sequestering TAZ in the cytoplasm in a phosphorylation-dependent manner (17).
Understanding the full significance of TAZ-T interactions promises to be difficult, in part because of the multiple effects TAZ is expected to have potentially affecting all three T antigens and in part because the functions of TAZ, operating in multiple subcellular compartments, are complex and not fully understood. Some initial insights can nevertheless be derived from the present results. It is clear that two domains of the polyomavirus T antigens are involved in interactions with TAZ, the amino terminus common to all three T proteins and the hr-t region encoding shared portions of sT and mT but not lT (Fig. 3d). Alterations in either domain suffice to abolish TAZ binding. While the Δ2-4 mutation prevents each of the T antigens from binding TAZ, mutation in the sT/mT common region has more complicated effects, as shown by results with the mutant NG59. This mutant encodes T antigens with a normal N terminus; sT and mT are altered in their uniquely shared region, while lT is unchanged. In NG59-infected cells, TAZ fails to bind sT and mT but does bind lT. The mutation in the sT/mT common region abolishes TAZ binding by these T antigens despite their retention of an unaltered N terminus. An intact N terminus is thus sufficient for TAZ binding to lT but not to sT or mT. The amount of TAZ bound to lT in NG59-infected cells is less than the amount bound to the identical lT expressed in wild-type virus-infected cells. Coexpression of either sT or mT can boost the binding of TAZ to lT. The latter observations account for the lower amount of TAZ binding by NG59 lT. Although the T antigens are able to bind TAZ when expressed individually, the functional interaction(s) whereby sT and mT increase the binding of TAZ to lT come into play in virus-infected cells. In a wild-type viral infection, the transactivation function of TAZ was inhibited. This inhibition was partially dependent on interaction of TAZ through its WW domain with the T antigens.
Though lT shows the least amount of binding among the three T antigens, lT-TAZ interaction appears to play an important role in virus replication. The ability of lT to bind TAZ independently of the other T antigens greatly enhances the ability of the virus to replicate in normal BMK mouse cells. Thus, while the thr mutant Δ2-4 fails to bind TAZ through any of its T antigens and is unable to replicate in BMK cells, the hr-t mutant NG59 binds a reduced amount of TAZ and only through its lT and grows to significant titers in the same cells (11). PEBP-2α is a nuclear matrix-associated transcription factor which can act positively on early viral transcription as well as on viral DNA replication (5, 35), events which are also dependent on lT. PEBP-2α also appears to function together with TAZ as a coactivator (17). Under conditions of DNA damage, PEBP-2α may exert a negative effect on polyomavirus DNA replication (26, 27). In our hands, TAZ has an inhibitory effect on viral DNA replication when highly expressed, and this inhibition is not seen when the PEBP-2α site is deleted from the viral enhancer. Thus, lT-TAZ interaction is critical for viral DNA replication, and some effects of TAZ may be mediated through PEBP-2α binding to the viral enhancer.
In a previous study of alterations introduced into the amino terminal portion of mT, a D2N mutation was shown to have a modest effect on transformation, reducing the efficiency of focus formation by rat fibroblasts to roughly 60% of wild-type mT; more drastic changes effectively abolished transformation as well as activation of pp60c-src (6). The original thr isolate carrying the same mutation partially reduced binding to TAZ. This suggests the possibility that TAZ may be important in the assembly of active complexes between mT and pp60c-src. Many PDZ proteins reside at the plasma membrane, and one or more such proteins could conceivably be involved along with TAZ in the assembly of mT signal transduction complexes. However, deletion of the PDZ-binding motif in TAZ has no effect on its binding to mT (Fig. 5). Based on results comparing wild-type virus with NG59, the functions of sT and mT, acting alone or together, result in translocation of TAZ into the nucleus (Fig. 6). Nuclear translocation of TAZ may reflect reduced phosphorylation of TAZ, e.g., on serine-89, leading to loss of interaction with 14-3-3 in the cytoplasm (17). A plausible mechanism currently under investigation is that nuclear accumulation of TAZ in polyomavirus-infected cells may depend on dephosphorylation of TAZ mediated by sT and PP2A. Studies of newborn mice injected with the thr Δ2-4 mutant are under way to determine the effect(s) of the loss of TAZ binding to all three T antigens on virus replication and tumor induction.
Acknowledgments
This work has been supported by grants from the National Cancer Institute (RO1 CA92520 and a project under PO1 CA50661, D. Livingston, principal investigator).
We gratefully acknowledge the advice of Joanna Gilbert on the immunofluorescent staining and helpful discussions with Jeong-Ho Hong and Michael Yaffe.
REFERENCES
- 1.Benjamin, T. L. 1970. Host range mutants of polyoma virus. Proc. Natl. Acad. Sci. USA 67:394-399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Benjamin, T. L. 2001. Polyoma virus: old findings and new challenges. Virology 289:167-173. [DOI] [PubMed] [Google Scholar]
- 3.Carmichael, G. G., and T. L. Benjamin. 1980. Identification of DNA sequence changes leading to loss of transforming ability in polyoma virus. J. Biol. Chem. 255:230-235. [PubMed] [Google Scholar]
- 4.Chen, H. I., and M. Sudol. 1995. The WW domain of Yes-associated protein binds a proline-rich ligand that differs from the consensus established for Src homology 3-binding modules. Proc. Natl. Acad. Sci. USA 92:7819-7823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen, L. F., K. Ito, Y. Murakami, and Y. Ito. 1998. The capacity of polyomavirus enhancer binding protein 2αB (AML1/Cbfa2) to stimulate polyomavirus DNA replication is related to its affinity for the nuclear matrix. Mol. Cell. Biol. 18:4165-4176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cook, D. N., and J. A. Hassell. 1990. The amino terminus of polyomavirus middle T antigen is required for transformation. J. Virol. 64:1879-1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Courtneidge, S. A., and A. E. Smith. 1983. Polyoma virus transforming protein associates with the product of the c-src cellular gene. Nature 303:435-439. [DOI] [PubMed] [Google Scholar]
- 8.Dawe, C. J., R. Freund, G. Mandel, K. Ballmer-Hofer, D. A. Talmage, and T. L. Benjamin. 1987. Variations in polyoma virus genotype in relation to tumor induction in mice. Characterization of wild type strains with widely differing tumor profiles. Am. J. Pathol. 127:243-261. [PMC free article] [PubMed] [Google Scholar]
- 9.Freund, R., R. T. Bronson, and T. L. Benjamin. 1992. Separation of immortalization from tumor induction with polyoma large T mutants that fail to bind the retinoblastoma gene product. Oncogene 7:1979-1987. [PubMed] [Google Scholar]
- 10.Gilbert, J. M., and T. L. Benjamin. 2000. Early steps of polyomavirus entry into cells. J. Virol. 74:8582-8588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goldman, E., and T. L. Benjamin. 1975. Analysis of host range of nontransforming polyoma virus mutants. Virology 66:372-384. [DOI] [PubMed] [Google Scholar]
- 12.Harris, B. Z., and W. A. Lim. 2001. Mechanism and role of PDZ domains in signaling complex assembly. J. Cell Sci. 114:3219-3231. [DOI] [PubMed] [Google Scholar]
- 13.Hattori, J., G. G. Carmichael, and T. L. Benjamin. 1979. DNA sequence alterations in Hr-t deletion mutants of polyoma virus. Cell 16:505-513. [DOI] [PubMed] [Google Scholar]
- 14.Ito, Y. 1989. Signals and transcription factors. Gan To Kagaku Ryoho 16:509-515. (In Japanese.) [PubMed] [Google Scholar]
- 15.Ito, Y. 1996. Structural alterations in the transcription factor PEBP2/CBF linked to four different types of leukemia. J. Cancer Res. Clin. Oncol. 122:266-274. [DOI] [PubMed] [Google Scholar]
- 16.Jackson, M. E., and M. S. Campo. 1995. Both viral E2 protein and the cellular factor PEBP2 regulate transcription via E2 consensus sites within the bovine papillomavirus type 4 long control region. J. Virol. 69:6038-6046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kanai, F., P. A. Marignani, D. Sarbassova, R. Yagi, R. A. Hall, M. Donowitz, A. Hisaminato, T. Fujiwara, Y. Ito, L. C. Cantley, and M. B. Yaffe. 2000. TAZ: a novel transcriptional co-activator regulated by interactions with 14-3-3 and PDZ domain proteins. EMBO J. 19:6778-6791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Larose, A., N. Dyson, M. Sullivan, E. Harlow, and M. Bastin. 1991. Polyomavirus large T mutants affected in retinoblastoma protein binding are defective in immortalization. J. Virol. 65:2308-2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li, D., K. Dower, Y. Ma, Y. Tian, and T. L. Benjamin. 2001. A tumor host range selection procedure identifies p150(sal2) as a target of polyoma virus large T antigen. Proc. Natl. Acad. Sci. USA 98:14619-14624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li, Q. L., K. Ito, C. Sakakura, H. Fukamachi, K. Inoue, X. Z. Chi, K. Y. Lee, S. Nomura, C. W. Lee, S. B. Han, H. M. Kim, W. J. Kim, H. Yamamoto, N. Yamashita, T. Yano, T. Ikeda, S. Itohara, J. Inazawa, T. Abe, A. Hagiwara, H. Yamagishi, A. Ooe, A. Kaneda, T. Sugimura, T. Ushijima, S. C. Bae, and Y. Ito. 2002. Causal relationship between the loss of RUNX3 expression and gastric cancer. Cell 109:113-124. [DOI] [PubMed] [Google Scholar]
- 21.Lukacher, A. E., R. Freund, J. P. Carroll, R. T. Bronson, and T. L. Benjamin. 1993. Pyvs: a dominantly acting gene in C3H/BiDa mice conferring susceptibility to tumor induction by polyoma virus. Virology 196:241-248. [DOI] [PubMed] [Google Scholar]
- 22.Lund, A. H., and M. van Lohuizen. 2002. RUNX: a trilogy of cancer genes. Cancer Cell 1:213-215. [DOI] [PubMed] [Google Scholar]
- 23.Nourry, C., S. G. Grant, and J. P. Borg. 2003. PDZ domain proteins: plug and play! Sci. STKE 2003:RE7. [DOI] [PubMed]
- 24.Pallas, D. C., V. Cherington, W. Morgan, J. DeAnda, D. Kaplan, B. Schaffhausen, and T. M. Roberts. 1988. Cellular proteins that associate with the middle and small T antigens of polyomavirus. J. Virol. 62:3934-3940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Patek, P. Q., J. L. Collins, and M. Cohn. 1978. Transformed cell lines susceptible or resistant to in vivo surveillance against tumorigenesis. Nature 276:510-511. [DOI] [PubMed] [Google Scholar]
- 26.Rutberg, S. E., L. R. Dolan, and Z. Ronai. 1994. PEBP2—a modulator of polyoma DNA replication. DNA Cell Biol. 13:865-874. [DOI] [PubMed] [Google Scholar]
- 27.Rutberg, S. E., S. Y. Fuchs, and Z. Ronai. 1995. Ultraviolet irradiation and c-jun over-expression regulates replication of polyoma sequences in WOP cells through a PEBP2 binding site. Biochim. Biophys. Acta 1261:90-98. [DOI] [PubMed] [Google Scholar]
- 28.Satake, M., T. Ibaraki, Y. Yamaguchi, and Y. Ito. 1989. Loss of responsiveness of an AP1-related factor, PEBP1, to 12-O-tetradecanoylphorbol-13-acetate after transformation of NIH 3T3 cells by the Ha-ras oncogene. J. Virol. 63:3669-3677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schaffhausen, B. S., T. J. Liang, G. G. Carmichael, and T. L. Benjamin. 1985. Residual transforming activity of PY1178T, a mutant lacking the principal in vitro tyrosine phosphorylation site, is not affected by removal of the secondary tyrosine phosphorylation site at residue 322. Virology 143:671-675. [DOI] [PubMed] [Google Scholar]
- 30.Schaffhausen, B. S., J. E. Silver, and T. L. Benjamin. 1978. Tumor antigen(s) in cell productively infected by wild-type polyoma virus and mutant NG-18. Proc. Natl. Acad. Sci. USA 75:79-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Silver, J., B. Schaffhausen, and T. Benjamin. 1978. Tumor antigens induced by nontransforming mutants of polyoma virus. Cell 15:485-496. [DOI] [PubMed] [Google Scholar]
- 32.Sudol, M., P. Bork, A. Einbond, K. Kastury, T. Druck, M. Negrini, K. Huebner, and D. Lehman. 1995. Characterization of the mammalian YAP (Yes-associated protein) gene and its role in defining a novel protein module, the WW domain. J. Biol. Chem. 270:14733-14741. [DOI] [PubMed] [Google Scholar]
- 33.Tenen, D. G. 2003. Disruption of differentiation in human cancer: AML shows the way. Nat. Rev. Cancer 3:89-101. [DOI] [PubMed] [Google Scholar]
- 34.Tyndall, C., G. La Mantia, C. M. Thacker, J. Favaloro, and R. Kamen. 1981. A region of the polyoma virus genome between the replication origin and late protein coding sequences is required in cis for both early gene expression and viral DNA replication. Nucleic Acids Res. 9:6231-6250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Watanabe, S., Y. Ito, A. Miyajima, and K. Arai. 1995. Granulocyte macrophage-colony stimulating factor-dependent replication of polyoma virus replicon in hematopoietic cells. Analyses of receptor signals for replication and transcription. J. Biol. Chem. 270:9615-9621. [DOI] [PubMed] [Google Scholar]
- 36.Yagi, R., L. F. Chen, K. Shigesada, Y. Murakami, and Y. Ito. 1999. A WW domain-containing yes-associated protein (YAP) is a novel transcriptional co-activator. EMBO J. 18:2551-2562. [DOI] [PMC free article] [PubMed] [Google Scholar]