

Abstract
Retroviral genomes consist of two unspliced RNAs linked noncovalently in a dimer. Although these two RNAs are generally identical, two different RNAs can be copackaged when virions are produced by coinfected cells. It has been assumed, but not tested, that copackaging results from random RNA associations in the cytoplasm to yield encapsidated RNA homodimers and heterodimers in Hardy-Weinberg proportions. Here, virion RNA homo- and heterodimerization were examined for Moloney murine leukemia virus (MLV) using nondenaturing Northern blotting and a novel RNA dimer capture assay. The results demonstrated that coexpressed MLV RNAs preferentially self-associated, even when RNAs were identical in known packaging and dimerization sequences or when they differed overall by less than 0.1%. In contrast, HIV-1 RNAs formed homo- and heterodimers in random proportions. We speculate that these species-specific differences in RNA dimer partner selection may at least partially explain the higher frequency of genetic recombination observed for human immunodeficiency virus type 1 than for MLV.
As retroviruses such as Moloney murine leukemia virus (MLV) and human immunodeficiency virus type 1 (HIV-1) bud from infected cells, they coencapsidate two copies of viral genomic RNA. These RNAs are unspliced host RNA polymerase II transcripts of integrated proviruses, and they are identical in primary sequence to the mRNAs that encode the major viral structural proteins and enzymes. How or whether this single molecular species of unspliced viral RNA is partitioned into the RNAs that will be packaged as genomes and those destined to serve as mRNAs is an area of active investigation (6).
Although HIV-1, MLV, and other retroviruses share the requirement for active nuclear export of unspliced RNAs for genome encapsidation, the mechanisms and host machinery that they employ differ. For example, nuclear export of unspliced HIV-1 RNA is dependent on the interaction of the viral Rev protein with a specific HIV-1 RNA structure as well as the human nuclear export factor CRM1 (27, 51, 53). Once in the cytoplasm, HIV-1 unspliced RNAs appear to reside in a single genetic pool from which both mRNAs and genomes can be recruited (7, 12). The host factors that participate in unspliced RNA export for gammaretroviruses such as MLV are unknown. However, observations from infected cells treated with the general transcription inhibitor actinomycin D suggest that MLV genomes and gag-pol mRNAs exist in two functionally distinct pools that have different half-lives and do not equilibrate (12, 24, 25, 31). Thus, gammaretroviruses may use two different nuclear export strategies to traffic the single species of unspliced RNA that is required for their replication.
Unlike other viruses, retroviruses possess two complete genomes per virion. One postulated advantage of copackaging two RNAs is in providing a source of recombinational repair for damaged genomes (9). During viral DNA synthesis, reverse transcriptase (RT) can switch from one copackaged RNA to the other, thereby generating a recombinant provirus that is a chimera of both parental RNA genomes. Crossovers between two identical RNA genomes are not detectable by examining product DNAs; thus, recombination is often monitored experimentally by studying the reverse transcription products of virions harvested from cells coexpressing two genetically distinct RNAs (19). The RNA dimers within such virions are presumed to consist of a heterogeneous population and, thus, recombination frequencies calculated using these approaches have generally involved dividing the number of detectable proviral recombinants by the proportion of virions predicted to contain RNA heterodimers. Because copackaging of RNA from coinfected producer cells is assumed to be random, the frequency of heterozygous virions has been modeled by the Hardy-Weinberg equation (A2 + 2AB + B2 = 1), where A is the fraction of RNA A in total virion RNA, B is the fraction of type B RNA, and 2AB is the proportion of RNA heterodimers (19, 39).
In such single-replication-cycle recombination experiments, genetic markers cosegregate about 10-fold more frequently for HIV-1 than for gammaretroviruses like MLV, suggesting that marker reassortment is more frequent for HIV than MLV (3, 21, 35, 45, 48). Although it was initially suggested that this might reflect differences between HIV and MLV RTs (21), subsequent work determined that template switching occurs at similar frequencies for HIV and MLV when assayed using donor and acceptor sequences that coreside on single RNAs (35). These latter findings seem consistent with the alternate possibility that MLV may preferentially copackage two identical RNAs (9, 18, 32), while HIV may be more likely to copackage two different RNAs (45).
Here, we tested the hypothesis that MLV and HIV-1 differ in randomness of encapsidated RNA dimerization. Using nondenaturing gel electrophoresis as well as a novel RNA capture assay, we examined the extent of MLV RNA homo- and heterodimerization in virions harvested from cells coexpressing two different packagable RNAs. We show that the majority of MLV genomic RNAs had apparently self-associated, resulting in a virus population containing less than half of the RNA heterodimers predicted from random dimerization. In contrast, RNA capture assay results showed that HIV-1 RNAs associated randomly.
MATERIALS AND METHODS
Plasmids.
The MLV helper function plasmid was pMLV Ψ−, which encodes all MLV proteins but contains a packaging sequence deletion between MLV nucleotides (nt) 215 and 368 (41). pMLV is the infectious MLV proviral clone also known as pNCA (10). pMΨ Puro is an MLV-based vector containing a puromycin resistance gene driven by a simian virus 40 (SV40) early promoter (23). pMLV-syn is derived from an infectious proviral plasmid that encodes an RNA identical to that transcribed from pMLV. Using mutagenic primers and overlap extension PCR, seven synonymous substitutions, indicated by the designation “syn,” were introduced into pol (5′-CAAGCCCCACATACAGA-3′ changed to 5′-AAAACCGCATATTCAAC-3′; nucleotide substitutions are in bold type), such that RNA transcribed from this vector could not anneal efficiently to the biotinylated oligonucleotide used in RNA capture assays. pMLV-GPP, previously called pGPP because it encodes MLV gag pol and puromycin resistance (47), is a replication-defective MLV vector in which env has been replaced by an SV40 promoter-driven puromycin resistance gene.
HIV experiments used the helper function plasmid pCMVΔR8.2 (33), which encodes all HIV-1 proteins except envelope. Previously called pHIV LacPuro (2), pHΨ LacPuro is an HIV-1-based retroviral vector containing all required cis-acting HIV-1 sequences as well as a cytomegalovirus (CMV) promoter-driven lacZ followed by an SV40 promoter-driven puromycin resistance gene. pHΨ Puro is a derivative of pHΨ LacPuro in which the region between SnaBI and EcoRI has been replaced with a 1,086-bp MscI (in gag)-Mfe I (in pol) fragment from MLV, thereby removing the CMV promoter and all but 56 bp of lacZ. The incorporated MLV fragment included sequences complementary to the biotinylated oligonucleotide used in RNA capture assays.
RNA capture assay riboprobes were templated by three pBluescript II SK(+) (Stratagene)-derived plasmids. pD 1040-2 contained a 330-bp MscI-BsrBI fragment from MLV gag (47). pJF 813-1 contained a 309-bp MLV pol PCR fragment, and pJF 954-7 contained a 321-bp PCR product templated by pHΨ Puro that included part of the inserted MLV pol sequences.
Virus production and harvest.
For virion production, 80% confluent ET cells (a 293T derivative that constitutively expresses ecotropic envelope [41]) or ΦNXE cells (a 293T-derived MLV-based packaging cell line [40]) were transiently cotransfected with HIV-1 or MLV helper and/or vector plasmids as described below. Note that in order to achieve the ratios of encapsidated HΨ Puro to HΨ LacPuro RNAs for the HIV-1 RNA capture experiment, it was necessary to transfect plasmids at a 1:30 molar ratio. Transfections were performed in 6-cm dishes either by calcium phosphate precipitation (5) or with Fugene 6 (Roche) using a total of 10 or 4 μg of plasmid DNA, respectively. Culture medium was replaced 24 h posttransfection, and a total of 12 ml of virus-containing medium per 6-cm dish was harvested and pooled by collection at 36, 48, and 60 h posttransfection. Residual cells were removed by filtration, and virus-containing medium was stored at −70°C prior to use.
To generate virus produced from integrated proviruses, cells that coexpressed MLV-GPP and MLV-syn RNAs were generated two separate times as follows. ET cells were transiently transfected with either pMLV-syn or pMLV-GPP by calcium phosphate precipitation, and MLV-syn and MLV-GPP virion-containing media were harvested separately. Approximately 50% confluent NIH 3T3 cells were transduced with MLV-GPP by first incubating virus-containing medium with cells for 2 h in the presence of 0.8 μg of Polybrene (hexadimethrine bromide; Sigma)/ml and then, beginning 48 h postinfection, selecting in medium containing 6 μg of puromycin/ml. Pooled puromycin-resistant cells were infected with the replication-competent MLV-syn virion-containing medium. After spread of the MLV-syn virus had been confirmed by increased RT activity, cells were passaged several times and virions were harvested from the medium of coinfected cell pools.
All 293T-derived cell lines were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (Gemini). NIH 3T3 cells chronically infected with MLV and the MLV-GPP/MLV-syn-coexpressing cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% calf serum (Gibco).
Isolation of dimeric viral RNA.
Twelve milliliters of virus-containing medium was centrifuged at 115,000 × g for 90 min at 4°C. Pelleted virus was resuspended in 400 μl of lysis buffer (50 mM Tris [pH 7.5]-10 mM EDTA-1% sodium dodecyl sulfate [SDS]-100 mM NaCl-50 μg of tRNA/ml-100 μg of proteinase K/ml), incubated at 37°C for 30 min, extracted with an equal volume of phenol-chloroform-isoamyl alcohol (25:24:1 [vol/vol/vol]), and ethanol precipitated. Precipitated RNA was incubated for 15 min at 37°C in 100 μl of 25 mM Tris-Cl (pH 8)-50 mM MgCl2-5 mM dithiothreitol-100 U of RNase-free DNase (Roche)/ml-400 U of RNasin (Promega)/ml. DNase treatment was stopped by addition of 25 μl of 50 mM EDTA-1.5 M sodium acetate (pH 5.2)-1% SDS. Samples were phenol-chloroform-isoamyl alcohol (25:24:1 [vol/vol/vol]) extracted, ethanol precipitated, and suspended in 15 μl of 10 mM Tris-1 mM EDTA-100 mM NaCl-1% SDS.
Native Northern blotting.
The protocol for native Northern blotting was adapted from that of Khandjian and Meric (22). Viral RNA was separated on a 0.7% agarose gel in 1× Tris-borate-EDTA at 70 V for 4.5 h, denatured in situ by incubating the gel in 300 ml of a 10% formaldehyde solution at 65°C for 30 min, partially hydrolyzed, subsequently neutralized by soaking in 1 liter of 0.05 M NaOH-1.5 M NaCl for 30 min and 1 liter 0.5 M Tris-Cl (pH 7.4)-1.5 M NaCl for 20 min, respectively, and then passively transferred to a nylon membrane (Hybond N; Amersham Pharmacia Biotech) for 16 h in 10× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate). RNA was cross-linked to the membrane (Stratalinker; Stratagene), and the membrane was prehybridized at 42°C for 5 h in 5× SSC-5× Denhardt's solution-50% formamide-1% SDS-100 μg of denatured salmon sperm DNA/ml before addition of denatured DNA probe. Probes were prepared by random-prime labeling of PCR products containing puroR or MLV env sequences by using a RediPrime II kit (Amersham Pharmacia) and [α-32P]TTP (Perkin-Elmer). Membranes were hybridized at 42°C for 18 h and washed twice each with 2× SSC-0.1% SDS and 0.5× SSC-0.1% SDS for 10 min at 37°C before exposure to film. For subsequent probings, the nylon membrane was stripped by incubation at 80°C in 0.1% SDS and then prehybridized and probed as described above.
RNA capture assay.
Viral RNA purified from 4 ml of transfected cell medium as described above was combined with 200 pmol of a 3′-biotinylated anti-MLV pol oligonucleotide (5′-CTCTGTATGTGGGGCTTG-3′). Note that the same biotinylated oligonucleotide was used in all capture assays in this study because one of each pair of coexpressed RNAs was engineered to contain sequences complementary to this oligonucleotide. This RNA plus oligonucleotide sample was incubated in 100 μl of 2× SSC at 42°C for 15 min and then cooled to 25°C over 30 min. Cooled oligonucleotide-bound RNA was immediately added to streptavidin-coated beads (Promega; prewashed with 0.5× SSC three times and suspended in 100 μl of 0.5× SSC) and gently mixed for 10 min at room temperature. Beads were captured using a magnetic stand, and the supernatant that was removed to a new tube was designated the flowthrough sample. Beads were then washed four times with 0.5× SSC to remove residual unbound RNA. The supernatant from the final wash was retained for analysis (final wash). After the final wash, beads were resuspended in 100 μl of RNase-free water and incubated at 90°C for 5 min to elute the RNA from the bound oligonucleotide. This first elution was pooled with a second 150-μl elution to yield the elution samples analyzed by RNase protection assay. A separate aliquot of viral RNA from 1.5 ml of cell medium from the same transfection was diluted to a final volume of 30 μl in RNase-free water to serve as the input sample. All samples—input, flowthrough, final wash, and elution—were ethanol precipitated with 15 μg of carrier tRNA and resuspended with a molar excess of the appropriate riboprobe for analysis by RNase protection assay.
To generate riboprobes (i) for MLV/MΨ Puro, pD 1040-2 was linearized with EcoRI and transcribed using T3 RNA polymerase (Promega). This created a 400-nt runoff transcript which protected 330 nt of MLV RNA and 289 nt of MΨ Puro RNA. (ii) For MLV/MLV-syn, pJF 813-1 was linearized with XbaI and transcribed using T7 RNA polymerase (Promega), generating a 357-nt runoff transcript that protected 309 nt of MLV RNA and 265 nt of MLV-syn RNA. (iii) For HΨ Puro/HΨ LacPuro, pJF 954-7 was linearized with EcoRI and transcribed using T7 RNA polymerase (Promega). A 377-nt runoff transcript was produced that protected 321 nt of HΨ Puro RNA and 270 nt of HΨ LacPuro RNA. All riboprobes were radiolabeled by including [α-32P]CTP (Perkin-Elmer) in the reaction mixture and were resuspended in hybridization buffer [80% formamide-40 mM piperazine-N,N′-bis(2-ethanesulfonic acid) (pH 6.4)-400 mM NaCl-1 mM EDTA].
For RNase protection assays, samples were incubated in probe-containing hybridization buffer, digested with RNases A and T1, and separated on 5% acrylamide-8 M urea gels by standard techniques (5). Dried gels were exposed to film or phosphorimager screens (Molecular Dynamics) for quantitative analysis using ImageQuant software. To deduce molar ratios, phosphorimager values were normalized for the number of radiolabeled C residues in each protected fragment.
(i) RNA capture assay data analysis.
Elution ratios measured by phosphorimager analysis of RNase protection assays were compared to the ratios predicted for random RNA dimerization as follows. First, the proportion of each coexpressed RNA (A and B) in the virus population was determined from normalized phosphorimager values of the input lane (for example, A = 0.1 and B = 0.9). To deduce the distribution of RNA homo- and heterodimers predicted for random RNA dimerization, these values were used in the Hardy-Weinberg equation (A2 + 2AB + B2 = 1, where A2 and B2 represent the percentage of each homodimer and 2AB denotes the heterodimer population). For this hypothetical sample calculation, AA homodimers made up 1% (0.12) of the viral population, BB homodimers made up 81% (0.92), and AB heterodimers represented the remaining 18% of the population. Assuming there were 100 RNA dimers in the population (made up of 20 RNA A molecules and 180 RNA B molecules) and that only RNA A bound the biotinylated oligonucleotide, there would be 1 AA homodimer and 18 AB heterodimers retained by the beads and 81 BB homodimers in the flowthrough sample. Thus, if dimerization followed Hardy-Weinberg equilibrium, the ratio of RNA A to RNA B predicted to be in the elution would be 20:18 ([2 × 1 + 18]:18), or 1.1:1. In the experiments below, the calculated expected elution ratios were compared to actual elution ratios measured by phosphorimager to determine the extent of RNA heterodimerization.
(ii) Calculating differences between expected and observed MLV RNA heterodimerization.
If experimentally measured elution ratios differed from the expected value, indicating that the virion RNA homo- and heterodimer frequencies had diverged from Hardy-Weinberg equilibrium, then the extent of heterodimerization was determined as follows, using values from the hypothetical example above. First, because RNA A was the oligonucleotide-bound RNA, all 20 RNA A molecules would be retained on the beads and be present in the elution. RNA B, on the other hand, was retained on the beads via its association with RNA A, and its presence in the elution represented the heterodimer population. With a hypothetical elution ratio measured by phosphorimager to be 2.5:1, there would be only 8 RNA Bs (20/2.5) in the elution and, therefore, only 8 heterodimers in the population: 44% of the number (18 heterodimers) predicted above for random dimerization.
RESULTS
Visualizing RNA dimers separated on nondenaturing gels.
Length differences in 3′-truncated in vitro-transcribed retroviral RNAs have previously been used to differentiate homo- and heterodimers on nondenaturing gels, with RNA heterodimers displaying mobilities intermediate to those of long and short homodimers (26, 36, 42). Here, in the initial experiments to examine RNA dimerization, two different-length MLV Ψ+ packagable RNAs were coexpressed in virion producer cells, purified from viral particles, and examined using nondenaturing Northern blotting. A large size difference between coexpressed RNA species (8.3 versus 2.7 kb) was selected with the intention of facilitating RNA heterodimer detection based on mobility differences from either homodimer on native agarose gels. One coexpressed RNA was the native 8.3-kb Moloney MLV genome (Fig. 1A), while the second, significantly shorter RNA was MΨ Puro (Fig. 1A). The 5′ 1 kb of wild-type MLV sequences shared by these RNAs included all sequences known to be required for RNA packaging and initiation of dimerization (1, 4, 36, 43).
FIG. 1.
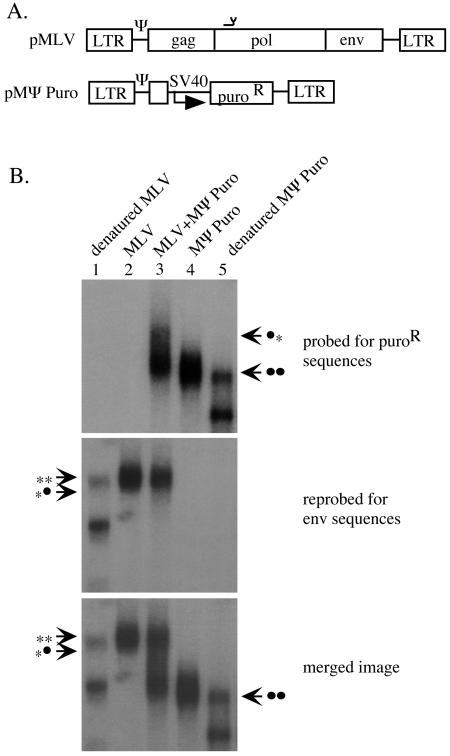
Native agarose gel electrophoresis of MLV Ψ+ RNAs. (A) Schematic representation of MLV-derived vectors used for this experiment. pMLV encodes an 8.3-kb full-length native MLV RNA. pMΨ Puro encodes a 2.7-kb RNA, of which the first 1,000 nt are identical to that of MLV RNA. The region of pMLV complementary to the anti-MLV pol oligo used for RNA capture experiments is indicated by the symbol above pol in the pMLV vector. Ψ, cis-acting packaging signal; SV40, SV40 promoter. (B) Nondenaturing Northern blot depicting migration of MLV Ψ+ dimeric RNAs. Viral RNA was harvested from the medium of packaging cells transfected with pMLV (lanes 1 and 2), pMΨ Puro (lanes 4 and 5), or an equimolar ratio of pMLV and pMΨ Puro (lane 3). Each panel is labeled with the DNA probe used. To create the merged image, film exposures of the top and middle panels were physically overlaid and scanned together. **, migration of MLV RNA homodimers; *•, migration of putative MLV/MΨ Puro heterodimers; ••, migration of MΨ Puro homodimers.
To generate virions containing either one or both of the Ψ+ RNAs, 293T-derived packaging cells were transiently transfected with infectious pMLV alone, pMΨ Puro, or an equimolar ratio of pMLV and pMΨ Puro. We have previously determined that under the transfection conditions used here, more than 20 plasmids are coexpressed by each transfected 293T cell (47). RNA from virions produced by these transfected cells was purified under conditions that maintained dimer linkages, separated on 0.7% native agarose gels, and transferred to nylon membranes. Radiolabeled DNA probes that detected the puromycin resistance gene or MLV envelope sequences were used sequentially to probe a single membrane, and exposures of each probing were examined separately or overlaid to form a merged image (Fig. 1B). Mobility standards indicating the migration of monomeric MLV and MΨ Puro RNAs (Fig. 1B, lanes 1 and 5, respectively) were generated from RNA aliquots denatured at 65°C. Because cells were cotransfected with equimolar amounts of pMLV and pMΨ Puro, random dimerization, as modeled by the Hardy-Weinberg equation, predicted roughly 25% of each RNA homodimer and 50% of the heterodimer in virions from vector-cotransfected cells. RNase protection assays were used to quantify the ratio of coexpressed RNAs in virions, which was 1.2:1 for this experiment and would be predicted to yield 49.6% RNA heterodimers.
As seen in the panels probed for either puroR or envelope sequences alone, homodimers of short and long vector RNAs were visualized in the single vector expression lanes (Fig. 1B, lane 4 of top panel and lane 2 of middle panel, respectively). However, contrary to what would be predicted if the RNAs dimerized at random, there was an apparent lack of heterodimers with intermediate mobility among viral RNAs from vector-coexpressing cells (Fig. 1B, lane 3 in all three panels). Some upward smearing was visible among cotransfection products visualized with the puroR probe alone (Fig. 1B, lane 3, top panel), possibly reflecting the presence of some heterodimer product with a mobility similar to that of the long vector homodimer. However, the radioactive signal of this shifted product was significantly lower than that in the shorter RNA homodimer product. These results suggest that MLV RNA homodimers predominated in virions produced by transfected cells coexpressing two different-length Ψ+ RNAs.
RNA capture assays to assess associations in MLV RNA dimers.
Intact retroviral RNAs are notoriously difficult to isolate (9). Native agarose gel electrophoresis of these lengthy and fragile RNAs yields diffuse bands that do not allow ready quantification. An additional limitation of the gel-based assays used above is that two different-length RNAs must be used to detect heterodimerization. Thus, to establish a more quantitative and experimentally tractable way of examining dimerization and to allow study of dimerization properties of same-length RNAs, a novel RNA capture assay was developed (Fig. 2A).
FIG. 2.
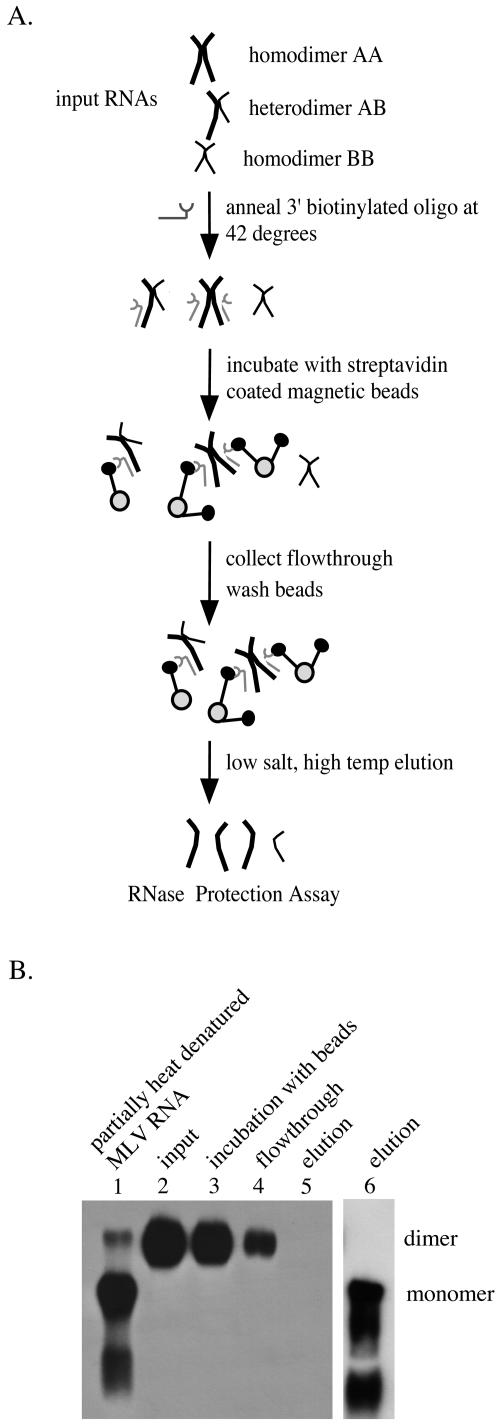
RNA capture assay. (A) Flow chart of the RNA capture assay. Genetically distinct RNA molecules are represented as A (thick line) and B (thin line). RNA dimers were annealed to a biotinylated oligonucleotide before incubation on streptavidin-coated beads. Captured RNA was eluted, and RNA samples were subjected to analysis by RNase protection assay. (B) RNA dimers remained intact through steps of the RNA capture assay. Viral RNA was isolated from the medium of NIH 3T3 cells chronically infected with MLV, annealed to the anti-MLV pol oligonucleotide (lanes 3 to 5) or the biotinylated anti-MLV pol oligonucleotide (lane 6), carried through the RNA capture assay to the indicated step, and separated on 0.7% native agarose. RNA was transferred to a nylon membrane and probed for MLV env sequences. To indicate migration of MLV RNA monomers and dimers, respectively, viral RNA was partially denatured by incubation at 65°C for 10 min (lane 1) or left on ice to retain dimer linkages (lane 2). Note that RNA was present in the elution only when the biotinylated version of the anti-MLV pol oligonucleotide was used (compare lanes 5 and 6).
For these experiments, a 3′-biotinylated oligonucleotide (anti-MLV pol) complementary to a segment of the MLV RT coding sequence was annealed to a population of viral RNAs under reaction conditions determined to maintain dimer linkages. The same biotinylated oligonucleotide was used in all of the RNA capture assays described in this study, and vectors were designed to ensure that the oligonucleotide annealed to sequences present on only one of two coexpressed RNAs (for example, RNA A but not RNA B [Fig. 2A]). After annealing the oligonucleotide to virion RNAs, the mixture was incubated with streptavidin-coated magnetic beads, allowing oligonucleotide-bound RNAs, including both AA homodimers and AB heterodimers, to bind. The streptavidin beads were then washed four times to remove unbound RNAs. RNAs retained on the beads were subsequently eluted with a low-salt, high-temperature incubation that caused the biotinylated oligonucleotide to dissociate from the bound RNA. Controls were performed to determine that viral RNA remained dimeric under the conditions of all the processing steps, except elution (Fig. 2B).
To monitor RNA retention on the beads, the ratio of the coexpressed RNAs in the input sample, the flowthrough material, the final wash, and the elution step were quantified by RNase protection assay. The input was an aliquot of virion RNA that was not incubated with biotinylated oligonucleotide. The flowthrough consisted of the RNAs from the annealed mixture that were not captured by the streptavidin-coated beads, and the elution was comprised of those RNAs that were retained by the beads. The final wash served as a control to demonstrate that all detectable nonspecifically bound RNA had been removed before the captured RNAs were eluted.
RNase protection assay data were quantified by phosphorimager, and the proportion of each RNA in the input sample was used to calculate the percentage of heterodimers expected from random dimerization. This percentage was then used to determine the expected ratio of eluted RNAs if dimerization were random (see Materials and Methods). The expected ratio was compared to the experimentally determined elution ratio for each experiment. The difference between predicted and observed ratios was taken as an indication of the randomness of RNA dimerization. The farther the measured elution ratio was from that predicted, the less random (more biased) the dimerization between coexpressed RNAs.
Initial RNA capture assays tested dimerization between infectious MLV and MΨ Puro RNAs, the two MLV RNAs whose dimerization was examined on nondenaturing gels above. To allow greater sensitivity for heterodimers, a molar excess of pMΨ Puro was cotransfected with pMLV. Under these conditions, where the less-abundant RNA was the one bound by oligonucleotide, random dimerization modeled by the Hardy-Weinberg equation predicts that almost every MLV RNA would be dimerized to a MΨ Puro RNA. Therefore, the elution ratio for the two RNAs would be nearly 1:1. RNase protection controls were performed to demonstrate that the ratio of intracellular packagable RNAs was nearly the same as the RNAs' ratio in virions (within a factor of 1.1-fold [data not shown]).
A representative RNase protection assay is illustrated in Fig. 3. For the experiment shown in this figure, the measured RNA input ratio was 1:3.6 (MLV to MΨ Puro). Lanes 1 to 4 and lanes 5 to 8 present control RNA capture assay results analyzing viral RNAs produced by packaging cells transfected with infectious pMLV or pMΨ Puro alone, respectively. MΨ Puro RNA, which could not anneal to the biotinylated oligonucleotide and bind the streptavidin beads, was present exclusively in the flowthrough and absent from the final elution (lanes 5 to 8). In contrast, much of the MLV RNA was retained on the beads, although a significant amount of MLV RNA was consistently present in the flowthrough (lanes 1 to 4). This unintended flowthrough by some of the MLV RNA may have been due to inefficient annealing of the oligonucleotide to virion RNA at the relatively low (42°C) annealing temperature used to ensure that dimer linkage structures were retained, or inaccessibility of some biotin moieties when oligonucleotides were bound to RNAs. Control experiments in which a vast excess of radiolabeled biotinylated oligonucleotide was incubated alone with streptavidin beads demonstrated that >99% of the oligonucleotide was bound and, thus, the binding capacity of the beads was not exceeded in any experiments performed here (data not shown).
FIG. 3.
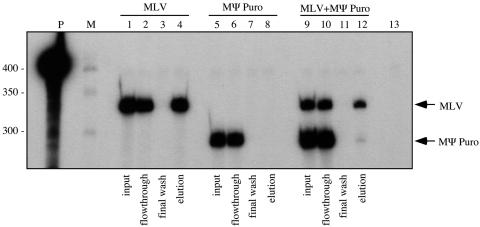
MLV-MΨ Puro RNA capture assay. Viral RNA was isolated from the medium of packaging cells transfected with pMLV (lanes 1 to 4), pMΨ Puro (lanes 5 to 8), or a 1:10 molar ratio of pMLV to pMΨ Puro (lanes 9 to 12) and was subjected to the RNA capture assay. RNase protection assays of input samples (lanes 1, 5, and 9), flowthroughs (lanes 2, 6, and 10), final washes (lanes 3, 7, and 11), and elutions (lanes 4, 8, and 12) are represented. Lane 13 is a digested riboprobe control. Migration of protected fragments representing each RNA is indicated at the right. Note that all phosphorimager values were normalized for the number of radiolabeled CTPs in each protected fragment. M, RNA marker; P, undigested riboprobe.
In Fig. 3, lanes 9 to 12, coexpressed MLV and MΨ Puro RNAs were examined. The observed elution ratio of MLV to MΨ Puro was not roughly 1.3:1, as would be predicted if virion RNAs with the determined 1:3.6 input ratio were present in random dimer proportions (lane 12), but was instead 5.3:1 (MLV to MΨPuro). Similar extents of biased homodimerization were observed in each of four independent repetitions of this experiment, as summarized by the first three bars (labeled MLV/MΨ Puro) in Fig. 4. Shown in this figure are the elution ratio of coexpressed RNAs predicted from random dimerization, normalized to 1, and the experimentally determined excess of oligonucleotide-bound RNA, calculated as described in Materials and Methods. Because all RNAs complementary to the oligonucleotide were not retained on the streptavidin beads, the same data sets were reevaluated based on calculations that assumed MLV homodimers, which have two oligo-complementary sites, were retained on beads twice as well as MLV/MΨ Puro heterodimers, which have only one. Regardless of whether it was assumed that all oligonucleotide-bound dimers were retained equally well or that oligo-bound homodimers bound streptavidin twice as well as heterodimers, the findings suggested a marked excess of RNA homodimers. Similar biases for homodimerization were observed when the expression ratios of MLV and MΨ Puro RNAs were varied (data not shown).
FIG. 4.
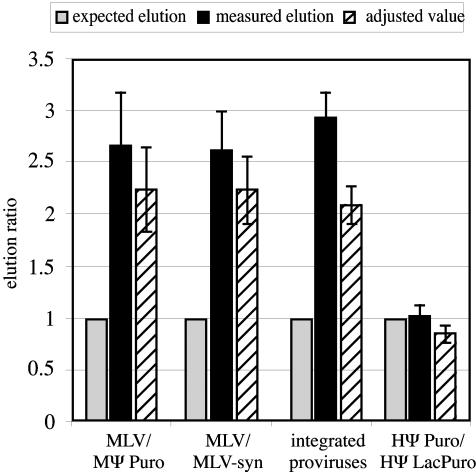
Summarized elution data from RNA capture experiments. Open bars, average expected elution ratio; filled bars, average measured elution ratio; hatched bars, average elution ratios corrected for the assumption that twice as many oligonucleotide-bound homodimers as heterodimers are retained on the beads. Standard errors are indicated. For comparison among experiments, expected elution ratios were set to a value of 1 (1 oligonucleotide-bound RNA/1 unbound RNA), and measured elution ratios were normalized accordingly. Thus, the higher the value on the y axis, the lower the amount of unbound RNA relative to values predicted based on random dimerization, and the higher the experimentally determined bias toward preferential RNA homodimerization. Input RNA ratios for the experiments used to generate these data were as follows: 1:6.5,1:4.9, 1:3.6, and 1:3.1 MLV to MΨ Puro for the four repetitions averaged in MLV-MΨ Puro experiments; 1:3.2,1:5.4, 1:5.6, and 1:8.5 MLV to MLV-syn for the four repetitions used to generate the MLV-MLV-syn data; 1:1.8, 1:5.6, 1:4.8, and 1:7.1 HΨ Puro to HΨ LacPuro for the four repetitions averaged in HΨ Puro-HΨ LacPuro experiments; and 1:1.3, 1:1.6, 1:1.5, 1:1.4, and 1:1.5 MLV-GPP to MLV-syn for the five repetitions used to generate the data from MLV-GPP-MLV-syn-coexpressing cells.
Testing whether or not sequence homology outside known packaging and dimerization regions affected RNA associations.
Although MLV and MΨ Puro RNAs share all sequences believed to be critical for dimerization, it remained possible that sequence homology outside of canonical dimerization regions might affect virion RNA dimer partner selection during viral replication. It has been suggested that viral RNA dimers make several stabilizing contacts outside of the dimer linkage region (28, 37), and the higher-order structures of full-length viral RNAs may differ from those of the shorter transcripts that have been used to study dimerization in vitro (11, 14-16, 29). Additionally, all sequences involved in dimerization during virus replication may not yet have been elucidated. Thus, to address whether or not differences between MLV and MΨ Puro RNAs contributed to the preferential self-association observed, the RNA capture assay was next used to study dimerization of nearly identical full-length MLV RNAs.
First, seven silent point mutations were introduced far from known dimerization sequences over a span of 17 nt within the RT coding sequences of an infectious MLV proviral plasmid. These synonymous substitutions, indicated by the designation syn in Fig. 5A, were the only differences between the RNA expressed by this new vector plasmid, pMLV-syn, and pMLV. The seven changes did not alter the protein coding sequence but were intended to prevent the biotinylated oligonucleotide from annealing to MLV-syn RNA.
FIG. 5.
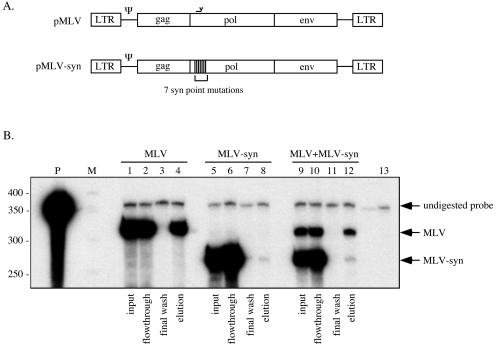
MLV-MLV-syn RNA capture assay. (A) Schematic representation of plasmids transfected into 293T-based cells for virus production. Both vectors encode an 8.3-kb RNA. However, because of the seven point mutations indicated (not to scale), MLV-syn RNA was unable to efficiently anneal to the biotinylated oligonucleotide. The region of pMLV complementary to the anti-MLV pol oligonucleotide is indicated above pol, as in Fig. 1A. (B) Viral RNA was isolated from the medium of cells transfected with pMLV (lanes 1 to 4), pMLV-syn (lanes 5 to 8), or a 1:10 molar ratio of pMLV to pMLV-syn (lanes 9 to 12) and subjected to an RNA capture assay. RNase protection assay results for inputs, flowthroughs, final washes, and elutions are illustrated. Migration of protected fragments representing each RNA is indicated at the right. For the experiment whose data are shown in this figure, the MLV/MLV-syn input ratio was 1:5.6. The elution ratio expected from random dimerization was 1.2:1 (MLV to MLV-syn), and the elution ratio measured by phosphorimager was 4.2:1 (MLV to MLV-syn). M, RNA marker; P, undigested riboprobe.
For these experiments, a molar excess of pMLV-syn was transiently cotransfected with pMLV into 293T-derived cells (Fig. 5A). Virion RNAs were harvested and analyzed using the RNA dimer capture assay. Again, the measured elution ratios for coexpressed RNAs diverged from the elution ratios predicted if dimerization were random (Fig. 5B, lane 12). In four repetitions of this experiment, measured elution ratios were, on average, 2.6-fold higher than the expected ratios (Fig. 4), demonstrating that MLV RNA self-association was observed even for two coexpressed full-length RNAs that differed by less than 0.1% overall. Note that when expressed alone, a small but quantifiable amount of MLV-syn RNA (in different experimental repetitions, from 0.1 to 1% of the total) was detectably retained on the biotinylated beads (Fig. 5B, lane 8). This unintended retention of small amounts of MLV-syn RNA suggest that calculations based on wild type plus MLV-syn coexpression may slightly underestimate biases toward MLV RNA homodimerization.
Dimerization of MLV RNAs produced by integrated proviruses in coinfected murine cells.
The contexts and amounts of viral RNA expression in transiently transfected cells and in naturally coinfected cells differ significantly. An additional artificial aspect of the experiments performed above is that they examined MLV RNAs expressed in human cells. If any host factors affect RNA dimerization, or if transcripts of nonintegrated DNAs differ from those of integrated proviruses, the properties of MLV RNAs harvested from transiently transfected human cells may differ from those of RNAs produced from the natural murine host. To address whether or not these parameters affected MLV RNA dimerization, the dimer capture assay was used to examine virion RNAs from MLV-coinfected murine cells.
Murine NIH 3T3 cells that coexpressed two different MLV RNAs were established two separate times by first transducing them with MLV-GPP, a replication-defective vector that expresses Gag and Pol but contains a puromycin resistance gene in place of env coding sequences (47). These 3T3 cells expressing MLV-GPP were subsequently infected with replication-competent MLV-syn, the infectious virus containing synonymous substitutions in RT described above. Virions were harvested from cells in which both MLV-GPP and MLV-syn had integrated, and virion RNA was purified and analyzed by the dimer capture assay. Predicted elution ratios based on RNA proportions in the input samples were compared to those measured by phosphorimager. In all repetitions of these experiments, RNA homodimers were again observed in large excess (2.9-fold, on average) over values that would be predicted if dimerization were random (Fig. 4, integrated proviruses).
HIV-1 RNA analyzed by dimer capture.
The high rates of recombination reported for HIV-1 suggest it must readily copackage two different RNAs (44, 48, 50, 52). Thus, dimerization of the HIV-1-derived vectors, pHΨ Puro and pHΨ LacPuro, was also examined using the RNA capture assay (Fig. 6A). pHΨ Puro contained approximately 1 kb of MLV pol-derived sequences which included, near their center, the region complementary to the biotinylated oligonucleotide used in the RNA capture assays. HΨ LacPuro RNA shared 2.3 kb of HIV-1-derived sequence at its 5′ end with HΨ Puro but did not contain the oligonucleotide-complementary sequences. Thus, only RNA dimers that contained at least one HΨ Puro RNA molecule would be retained on streptavidin beads in the RNA capture assay.
FIG. 6.
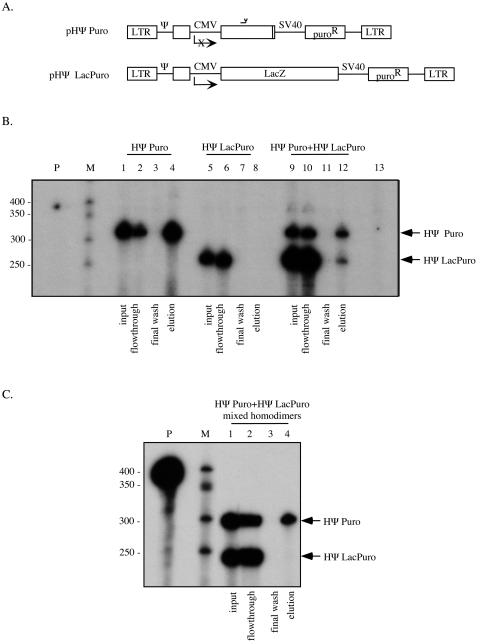
HΨ Puro-HΨ LacPuro RNA capture assay. (A) Schematic representation of HIV-1 Ψ+ vectors used to transfect 293T-based cells for virus production. pHΨ Puro encodes a 4.7-kb RNA containing 1 kb of MLV pol sequences (the anti-MLV pol complementary region is indicated). pHΨ LacPuro templates transcription of a 6.9-kb RNA. Ψ, packaging signal; CMV, CMV immediate early promoter; SV40, SV40 promoter. X indicates inactivation of the promoter. (B) Viral RNA isolated from the medium of cells transfected with HIV-1 Ψ− helper and pHΨ Puro (lanes 1 to 4), pHΨ LacPuro (lanes 5 to 8), or a ratio of plasmids resulting in approximately a 1:10 ratio of encapsidated HΨ Puro to HΨ LacPuro RNAs (lanes 9 to 12) was analyzed in the RNA capture assay. Lane 13 is a digested probe control. Inputs (lanes 1, 5, and 9), flowthroughs (lanes 2, 6, and 10), final washes (lanes 3, 7, and 11), and elutions (lanes 4, 8, and 12) are illustrated. Migration of protected fragments representing each RNA is indicated to the right. For this example, the measured HΨ Puro/HΨ LacPuro input ratio was 1:7.1. The elution ratio expected from random dimerization was 1.1:1 (HΨ Puro to HΨ LacPuro), and the elution ratio measured by phosphorimager was 1.4:1 (HΨ Puro to HΨ LacPuro). M, RNA marker; P, undigested riboprobe. (C) Medium from cells transfected with HΨ Puro or HΨ Lac Puro was mixed, virus was pelleted, and viral RNA dimers were isolated and analyzed in an RNA capture assay. Input, flowthrough, final wash, and elution are illustrated (lanes 1 to 4, respectively). Migration of protected fragments representing each RNA is indicated on the right. M, RNA marker; P, undigested riboprobe.
293T-based cells were cotransfected with the Ψ− HIV-1 helper, pCMVΔR8.2, and either of the two HIV-based vectors separately or together, using a molar excess of pHΨ LacPuro over pHΨ Puro. A representative RNase protection assay is presented in Fig. 6B. Single vector controls demonstrated that HΨ LacPuro RNA was present only in the flowthrough and not in the eluted fraction, as intended (Fig. 6B, lanes 6 and 8). A significant amount of HΨPuro RNA was retained on the beads via its association with the biotinylated oligonucleotide and was thereby detectable in the final eluted fraction (Fig. 6B, lane 4).
Dimer capture experiments were then used to analyze coexpressed HIV-1-derived viral RNAs (Fig. 6B, lanes 9 to 12). Dimerization of coexpressed HΨPuro and HΨLacPuro was more random than that observed for MLV. In fact, measured elution ratios from four independent experiments averaged only 1.03-fold higher than the expected 1:1 ratios (Fig. 6B, lane 12, and Fig. 4). These values were remarkably similar to the elution ratios predicted for random RNA dimerization. In controls where single RNA species virions were mixed prior to RNA purification, there was no detectable HΨ LacPuro RNA in the elution (Fig. 6C, lane 4). This control demonstrated that the RNA heterodimers detected in virions harvested from HIV-derived vector-coexpressing cells could not be ascribed to postreplication RNA associations that arose during RNA processing steps. Taken together, these results suggest that when two genetically distinct HIV-1-derived vector RNAs are coexpressed, they dimerize at random.
DISCUSSION
In this report, the RNA populations in MLV and HIV-1 particles produced by vector coexpressing cells were compared. MLV RNA dimers were examined both by native agarose gel electrophoresis and using a newly developed RNA capture assay. The results of both experimental approaches were consistent and suggested that coexpressed MLV RNAs displayed a marked preference for self-association, with significantly more homodimers observed among virion RNAs than would be expected if RNA dimerization were random. Similar biases toward homodimerization were observed for MLV Ψ+ RNAs that shared only the first 1 kb of MLV RNA and for two nearly identical full-length MLV genomes. In contrast, HIV-1 formed RNA homo- and heterodimers in proportions remarkably similar to those predicted if coexpressed RNAs were joined in dimers at random. Although MLV RNA capture elution ratios suggested that the virion heterodimer populations consistently averaged less than half (approximately 40%) of that expected if dimerization were random, a significant amount of MLV heterodimers was readily detectable.
Nonrandom dimerization may have significant implications for the frequency of retroviral recombination and suggests that MLV recombination rates have been underestimated. It has previously been suggested that preferential RNA homodimerization might contribute to retroviral “negative interference” (9, 18, 35). The term negative interference refers to a genetic phenomenon of simple retroviruses in which the minority of all reverse transcription products that are phenotypic recombinants are determined to contain a higher number of genetic crossovers than would be predicted if each recombination event were independent. We speculated that differences in copackaging might be responsible for at least some part of the reported 6- to 20-fold differences in HIV-1 and MLV recombination frequencies and may contribute to negative interference. Our results are largely consistent with this hypothesis; however, the measured decreases in RNA heterodimer populations were not great enough to account for the entire difference in recombination frequencies that have been reported for HIV and MLV. Thus, either MLV virions containing RNA homodimers are more likely to complete reverse transcription of integration-competent DNAs than those with heterodimeric RNAs, or virus species-specific differences, in addition to nonrandom dimerization, likely contribute to differences in HIV and MLV genetic marker segregation rates. Because even minor differences in selective advantage can have a major impact on viral populations (8), these differences between HIV and MLV likely have minimal impact on virus evolution, but they do have important implications for previously noted differences in single replication cycle recombination frequencies (35).
Although not explicitly tested by the work presented here, these new findings, combined with what is known about retroviral RNA trafficking, suggest the following speculative model for the mechanism of biased RNA dimerization seen for MLV (Fig. 7). Several lines of evidence suggest that full-length retroviral RNAs adopt different structures than do shorter transcripts (11, 14-16, 29). In reconstituted reactions, shorter (<1.5 kb) in vitro-transcribed 3′-truncated RNAs can dimerize, but RNAs derived from MLV's 5′ end that are longer than about 1.5 kb cannot (N. D. Robson and A. Telesnitsky, unpublished data). Therefore, it seems possible that MLV RNA may be in a conformation more favorable for dimerization before it has been fully transcribed. This model postulates that while RNA polymerase is transcribing MLV proviruses, some of the incomplete transcripts may adopt a fold conducive for initiating dimer-linkage interactions. Because cotranscribed RNAs would be present at a higher local concentration than other RNAs capable of dimerization, immature homodimers of cotranscribed RNAs would form preferentially. The resulting MLV RNAs that commit to dimerization early and those that remain monomeric likely adopt different RNA structures that may bind different sets of RNA nuclear export or adaptor proteins. Thus, depending on which alternate RNA structure—monomeric or dimeric 5′ leader region—was adopted, individual unspliced MLV RNAs would be trafficked into the different functional cytoplasmic pools of genomes and mRNAs. Recent studies suggest that adaptor and other proteins involved in specifying RNA export fates are loaded cotranscriptionally (49). Evidence that some retroviral Gag precursors enter the nucleus during late replication phases, and that this somehow affects RNA dimerization, suggests that one factor that affects unspliced RNAs' fates may be Gag (13, 46). The mechanism by which identical full-length MLV RNAs segregate into nonequilibrating, functionally distinct populations within the cytoplasm of infected cells has long remained unresolved, and we hypothesize that transcriptional commitment to dimerization may be part of the answer. Examples in the literature, which describe how two RNAs with identical sequences can have distinct extranuclear fates depending on their mode of nuclear export, provide precedence for this model (34).
FIG. 7.
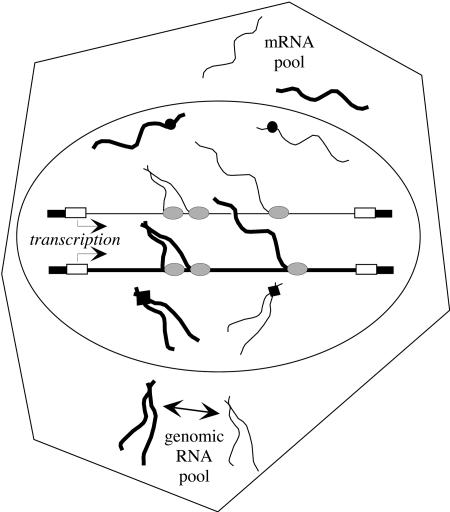
Model of MLV RNA self-association, suggesting MLV RNAs may commit to dimerization cotranscriptionally. The figure represents a cell containing two integrated proviruses: one is represented by a heavy line, and the other is shown by a light line. RNAs committed to dimerization and those that remain as monomers are postulated to adopt different higher-order structures and thus bind different sets of nuclear export factors (represented by a filled diamond versus a filled circle) that initiate trafficking to distinct cytoplasmic pools (mRNA pool or genomic RNA pool). Due to the weakness of molecular interactions in early dimer linkages, a limited exchange of RNA molecules within the genomic RNA pool, indicated by the double-sided arrow, is postulated to occur, giving rise to encapsidated heterodimers. Light gray ovals, RNA polymerase II; open rectangles, LTRs; large oval, nucleus; nuclear arrowheads, direction of provirus transcription.
This model does not clearly account for how the observed, albeit reduced, level of MLV heterodimers might form. Even under conditions where genomic RNA is limiting, MLV RNAs are packaged into virions as dimers (24, 30). Although these observations support the notion that RNAs have acquired a dimer molecular identity before they are encapsidated, the dimer interactions among Moloney MLV RNAs in assembling virions are much weaker than in mature virus (17). Thus, to explain the subset of MLV RNA heterodimers observed here and those described previously within RNA-coexpressing cells (38), we postulate that the weak dimer linkages that dictate trafficking of MLV genomes to sites of assembly may remain capable of RNA exchange until postassembly RNA maturation (Fig. 7).
Unlike MLV RNAs, the unspliced HIV-1 transcripts which serve genome and mRNA functions appear to reside in a single cytoplasmic pool (7, 12). The 5′ ends of these HIV-1 RNAs exist in equilibrium between two primary conformations, the long-distance interaction and the branched multiple hairpin (20). The first of these two conformers is the more stable thermodynamically, but it is incapable of dimer formation. Coating of the long-distance interaction conformer with nucleocapsid protein in vitro aids in transition to the dimer-competent branched multiple hairpin structure, thereby suggesting a role for the nucleocapsid domain of the Gag polyprotein as a regulatory switch between gene expression and dimerization of unspliced RNAs during HIV replication (20). This suggests that HIV-1 commitment to dimerization occurs within a single cytoplasmic pool where the Gag polyprotein can serve as an RNA chaperone to induce structural rearrangements that facilitate random dimerization among coexpressed HIV-1 RNAs.
Acknowledgments
We thank Nam-Hee Shin for preliminary observations, Sharon Marr for assistance designing experimental approaches, Jennifer Masterson for excellent technical contributions, and Mike Imperiale and John Moran for helpful comments on the manuscript.
This work was supported by NIH grant CA69300 to A.T. J.A.F. was supported in part by NIH training grant T32-GM08353.
REFERENCES
- 1.Adam, M. A., and A. D. Miller. 1988. Identification of a signal in a murine retrovirus that is sufficient for packaging of nonretroviral RNA into virions. J. Virol. 62:3802-3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.An, W., and A. Telesnitsky. 2001. Frequency of direct repeat deletion in a human immunodeficiency virus type 1 vector during reverse transcription in human cells. Virology 286:475-482. [DOI] [PubMed] [Google Scholar]
- 3.Anderson, J. A., V. K. Pathak, and W. S. Hu. 2000. Effect of the murine leukemia virus extended packaging signal on the rates and locations of retroviral recombination. J. Virol. 74:6953-6963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Armentano, D., S. F. Yu, P. W. Kantoff, T. von Ruden, W. F. Anderson, and E. Gilboa. 1987. Effect of internal viral sequences on the utility of retroviral vectors. J. Virol. 61:1647-1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ausubel, F. M. 1994. Current protocols in molecular biology. John Wiley & Sons, New York, N.Y.
- 6.Butsch, M., and K. Boris-Lawrie. 2002. Destiny of unspliced retroviral RNA: ribosome and/or virion? J. Virol. 76:3089-3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Butsch, M., and K. Boris-Lawrie. 2000. Translation is not required to generate virion precursor RNA in human immunodeficiency virus type 1-infected T cells. J. Virol. 74:11531-11537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coffin, J. M. 1995. HIV population dynamics in vivo: implications for genetic variation, pathogenesis and therapy. Science 267:483-489. [DOI] [PubMed] [Google Scholar]
- 9.Coffin, J. M. 1979. Structure, replication, and recombination of retrovirus genomes: some unifying hypotheses. J. Gen. Virol. 42:1-26. [DOI] [PubMed] [Google Scholar]
- 10.Colicelli, J., and S. P. Goff. 1988. Sequence and spacing requirements of a retrovirus integration site. J. Mol. Biol. 199:47-59. [DOI] [PubMed] [Google Scholar]
- 11.De Tapia, M., V. Metzler, M. Mougel, B. Ehresmann, and C. Ehresmann. 1998. Dimerization of MoMuLV genomic RNA: redefinition of the role of the palindromic stem-loop H1 (278-303) and new roles for stem-loops H2 (310-352) and H3 (355-374). Biochemistry 37:6077-6085. [DOI] [PubMed] [Google Scholar]
- 12.Dorman, N., and A. Lever. 2000. Comparison of viral genomic RNA sorting mechanisms in human immunodeficiency virus type 1 (HIV-1), HIV-2, and Moloney murine leukemia virus. J. Virol. 74:11413-11417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dupont, S., N. Sharova, C. DeHoratius, C. A. Virbasius, X. Zhu, A. G. Bukrinskaya, M. Stevenson, and M. R. Green. 1999. A novel nuclear export activity in HIV-1 matrix protein required for viral replication. Nature 402:681-685. [DOI] [PubMed] [Google Scholar]
- 14.Feng, Y. X., W. Fu, A. J. Winter, J. G. Levin, and A. Rein. 1995. Multiple regions of Harvey sarcoma virus RNA can dimerize in vitro. J. Virol. 69:2486-2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fosse, P., N. Motte, A. Roumier, C. Gabus, D. Muriaux, J. L. Darlix, and J. Paoletti. 1996. A short autocomplementary sequence plays an essential role in avian sarcoma-leukosis virus RNA dimerization. Biochemistry 35:16601-16609. [DOI] [PubMed] [Google Scholar]
- 16.Fu, W., R. J. Gorelick, and A. Rein. 1994. Characterization of human immunodeficiency virus type 1 dimeric RNA from wild-type and protease-defective virions. J. Virol. 68:5013-5018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fu, W., and A. Rein. 1993. Maturation of dimeric viral RNA of Moloney murine leukemia virus. J. Virol. 67:5443-5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu, W.-S., E. H. Bowman, K. A. Delviks, and V. K. Pathak. 1997. Homologous recombination occurs in a distinct retroviral subpopulation and exhibits high negative interference. J. Virol. 71:6028-6036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu, W. S., and H. M. Temin. 1990. Genetic consequences of packaging two RNA genomes in one retroviral particle: pseudodiploidy and high rate of genetic recombination. Proc. Natl. Acad. Sci. USA 87:1556-1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huthoff, H., and B. Berkhout. 2001. Two alternating structures of the HIV-1 leader RNA. RNA 7:143-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jetzt, A. E., H. Yu, G. J. Klarmann, Y. Ron, B. D. Preston, and J. P. Dougherty. 2000. High rate of recombination throughout the human immunodeficiency virus type 1 genome. J. Virol. 74:1234-1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khandjian, E. W., and C. Meric. 1986. A procedure for Northern blot analysis of native RNA. Anal. Biochem. 159:227-232. [DOI] [PubMed] [Google Scholar]
- 23.Kulpa, D., R. Topping, and A. Telesnitsky. 1997. Determination of the site of first strand transfer during Moloney murine leukemia virus reverse transcription and identification of strand transfer-associated reverse transcriptase errors. EMBO J. 16:856-865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Levin, J. G., P. M. Grimley, J. M. Ramseur, and I. K. Berezesky. 1974. Deficiency of 60 to 70S RNA in murine leukemia virus particles assembled in cells treated with actinomycin D. J. Virol. 14:152-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Levin, J. G., and M. J. Rosenak. 1976. Synthesis of murine leukemia virus proteins associated with virions assembled in actinomycin D-treated cells: evidence for persistence of viral messenger RNA. Proc. Natl. Acad. Sci. USA 73:1154-1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ly, H., and T. G. Parslow. 2002. Bipartite signal for genomic RNA dimerization in Moloney murine leukemia virus. J. Virol. 76:3135-3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Malim, M. H., J. Hauber, S. Y. Le, J. V. Maizel, and B. R. Cullen. 1989. The HIV-1 rev trans-activator acts through a structured target sequence to activate nuclear export of unspliced viral mRNA. Nature 338:254-257. [DOI] [PubMed] [Google Scholar]
- 28.Mangel, W. F., H. Delius, and P. H. Duesberg. 1974. Structure and molecular weight of the 60-70S RNA and the 30-40S RNA of the Rous sarcoma virus. Proc. Natl. Acad. Sci. USA 71:4541-4545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marquet, R., J. C. Paillart, E. Skripkin, C. Ehresmann, and B. Ehresmann. 1994. Dimerization of human immunodeficiency virus type 1 RNA involves sequences located upstream of the splice donor site. Nucleic Acids Res. 22:145-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meric, C., and S. P. Goff. 1989. Characterization of Moloney murine leukemia virus mutants with single-amino-acid substitutions in the Cys-His box of the nucleocapsid protein. J. Virol. 63:1558-1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Messer, L. I., J. G. Levin, and S. K. Chattopadhyay. 1981. Metabolism of viral RNA in murine leukemia virus-infected cells: evidence for differential stability of viral message and virion precursor RNA. J. Virol. 40:683-690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mikkelsen, J. G., and F. S. Pedersen. 2000. Genetic reassortment and patch repair by recombination in retroviruses. J. Biomed. Sci. 7:77-99. [DOI] [PubMed] [Google Scholar]
- 33.Naldini, L., U. Blomer, P. Gallay, D. Ory, R. Mulligan, F. H. Gage, I. M. Verma, and D. Trono. 1996. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 272:263-267. [DOI] [PubMed] [Google Scholar]
- 34.Ohno, M., S. Segref, S. Kuersten, and I. W. Mattaj. 2002. Identity elements used in export of mRNAs. Mol. Cell 9:659-671. [DOI] [PubMed] [Google Scholar]
- 35.Onafuwa, A., W. An, N. D. Robson, and A. Telesnitsky. 2003. Human immunodeficiency virus type 1 genetic recombination is more frequent than that of Moloney murine leukemia virus despite similar template switching rates. J. Virol. 77:4577-4587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oroudjev, E. M., P. C. Kang, and L. A. Kohlstaedt. 1999. An additional dimer linkage structure in Moloney murine leukemia virus RNA. J. Mol. Biol. 291:603-613. [DOI] [PubMed] [Google Scholar]
- 37.Ortiz-Conde, B. A., and S. H. Hughes. 1999. Studies of the genomic RNA of leukosis viruses: implications for RNA dimerization. J. Virol. 73:7165-7174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pal, B. K., L. Scherer, L. Zelby, E. Bertrand, and J. J. Rossi. 1998. Monitoring retroviral RNA dimerization in vivo via hammerhead ribozyme cleavage. J. Virol. 72:8349-8353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Panganiban, A. T., and D. Fiore. 1988. Ordered interstrand and intrastrand DNA transfer during reverse transcription. Science 241:1064-1069. [DOI] [PubMed] [Google Scholar]
- 40.Pear, W. S., M. L. Scott, and G. P. Nolan. 1996. Generation of high titer, helper-free retroviruses by transient transfection, p. 41-57. In P. Robbins (ed.), Methods in molecular medicine: gene therapy protocols. Humana Press, Totowa, N.J. [DOI] [PubMed]
- 41.Pfeiffer, J. K., R. S. Topping, N. H. Shin, and A. Telesnitsky. 1999. Altering the intracellular environment increases the frequency of tandem repeat deletion during Moloney murine leukemia virus reverse transcription. J. Virol. 73:8441-8447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Polge, E., J. L. Darlix, J. Paoletti, and P. Fosse. 2000. Characterization of loose and tight dimer forms of avian leukosis virus RNA. J. Mol. Biol. 300:41-56. [DOI] [PubMed] [Google Scholar]
- 43.Prats, A. C., C. Roy, P. A. Wang, M. Erard, V. Housset, C. Gabus, C. Paoletti, and J. L. Darlix. 1990. cis elements and trans-acting factors involved in dimer formation of murine leukemia virus RNA. J. Virol. 64:774-783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Quinones-Mateu, M. E., Y. Gao, S. C. Ball, A. J. Marozsan, A. Abraha, and E. J. Arts. 2002. In vitro intersubtype recombinants of human immunodeficiency virus type 1: comparison to recent and circulating in vivo recombinant forms. J. Virol. 76:9600-9613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rhodes, T., H. Wargo, and W. S. Hu. 2003. High rates of human immunodeficiency virus type 1 recombination: near-random segregation of markers one kilobase apart in one round of viral replication. J. Virol. 77:11193-11200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Scheifele, L. Z., R. A. Garbitt, J. D. Rhoads, and L. J. Parent. 2002. Nuclear entry and CRM1-dependent nuclear export of the Rous sarcoma virus Gag polyprotein. Proc. Natl. Acad. Sci. USA 99:3944-3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shin, N. H., D. Hartigan-O'Connor, J. K. Pfeiffer, and A. Telesnitsky. 2000. Replication of lengthened Moloney murine leukemia virus genomes is impaired at multiple stages. J. Virol. 74:2694-2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.St. Louis, D. C., D. Gotte, E. Sanders-Buell, D. W. Ritchey, M. O. Salminen, J. K. Carr, and F. E. McCutchan. 1998. Infectious molecular clones with the nonhomologous dimer initiation sequences found in different subtypes of human immunodeficiency virus type 1 can recombine and initiate a spreading infection in vitro. J. Virol. 72:3991-3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stutz, F., and E. Isaurralde. 2003. The interplay of nuclear mRNP assembly, mRNA surveillance and export. Trends Cell Biol. 13:319-327. [DOI] [PubMed] [Google Scholar]
- 50.van Wamel, J. L. B., and B. Berkhout. 1998. The first strand transfer during HIV-1 reverse transcription can occur either intramolecularly or intermolecularly. Virology 244:245-251. [DOI] [PubMed] [Google Scholar]
- 51.Wolff, B., J. J. Sanglier, and Y. Wang. 1997. Leptomycin B is an inhibitor of nuclear export: inhibition of nucleo-cytoplasmic translocation of the human immunodeficiency virus type 1 (HIV-1) Rev protein and Rev-dependent mRNA. Chem. Biol. 4:139-147. [DOI] [PubMed] [Google Scholar]
- 52.Yu, H., A. E. Jetzt, Y. Ron, B. D. Preston, and J. P. Dougherty. 1998. The nature of human immunodeficiency virus type 1 strand transfers. J. Biol. Chem. 273:28384-28391. [DOI] [PubMed] [Google Scholar]
- 53.Zapp, M. L., and M. R. Green. 1989. Sequence-specific RNA binding by the HIV-1 Rev protein. Nature 342:714-716. [DOI] [PubMed] [Google Scholar]