

Abstract
Recent events have raised concern over the use of pathogens, including variola virus, as biological weapons. Vaccination with Dryvax is associated with serious side effects and is contraindicated for many people, and the development of a safer effective smallpox vaccine is necessary. We evaluated an attenuated vaccinia virus, modified vaccinia virus Ankara (MVA), by use of a murine model to determine its efficacy against an intradermal (i.d.) or intranasal (i.n.) challenge with vaccinia virus (vSC8) or a recombinant vaccinia virus expressing murine interleukin-4 that exhibits enhanced virulence (vSC8-mIL4). After an i.d. challenge, 15 of 16 mice who were inoculated with phosphate-buffered saline developed lesions, one dose of intramuscularly administered MVA was partially protective (3 of 16 mice developed lesions), and the administration of two or three doses of MVA was completely protective (0 of 16 mice developed lesions). In unimmunized mice, an i.n. challenge with vSC8 caused a significant but self-limited illness, while vSC8-mIL4 resulted in lethal infections. Immunization with one or two doses of MVA prevented illness and reduced virus titers in mice who were challenged with either vSC8 or vSC8-mIL4. MVA induced a dose-related neutralizing antibody and vaccinia virus-specific CD8+-T-cell response. Mice immunized with MVA were fully protected from a low-dose vSC8-mIL4 challenge despite a depletion of CD4+ cells, CD8+ cells, or both T-cell subsets or an antibody deficiency. CD4+- or CD8+-T-cell depletion reduced the protection against a high-dose vSC8-mIL4 challenge, and the depletion of both T-cell subsets was associated with severe illness and higher vaccinia virus titers. Thus, MVA induces broad humoral and cellular immune responses that can independently protect against a molecularly modified lethal poxvirus challenge in mice. These data support the continued development of MVA as an alternative candidate vaccine for smallpox.
The continued threat of bioterrorism has led to concern over the reemergence of smallpox. Global immunity against poxviruses has declined over the last 20 years, as routine immunization against smallpox ceased in the early 1980s following the declaration of smallpox eradication by the World Health Organization. The consequences of a deliberate smallpox release would be great, as mortality for nonimmune persons has been reported to be near 30% (10).
While historical experience supports the efficacy of replication-competent vaccinia virus immunization against natural smallpox, the current Food and Drug Administration (FDA)-licensed vaccine Dryvax presents some safety concerns. Previous studies have described a significant rate of serious complications following Dryvax administration, including death, in 1 per 1 million vaccinees (26, 27). This rate could be even higher if mass vaccination were instituted today because of the large and growing number of persons for whom Dryvax is contraindicated. This has led to hesitation within the medical community for the use of widespread vaccination (9, 33). As a result, there have been renewed investigations into the development of a safer second-generation smallpox vaccine.
Previous strategies for the development of safe smallpox vaccines have focused on less virulent vaccinia virus strains, including recombinant vaccinia viruses with selected deletions of virulence genes or insertions of proinflammatory cytokines (13, 14, 34, 40) and empirically attenuated live vaccinia virus strains (21, 25, 29, 30, 37). The modified vaccinia virus Ankara (MVA) was attenuated by >570 passages in chicken embryo fibroblasts, resulting in the loss of approximately 15% of its parent genome, including several host range genes (2, 29, 31). MVA is consequently unable to replicate effectively in mammalian cells, which reduces the risk of dissemination and transmission (8, 35). In addition, MVA no longer encodes many of the soluble inhibitors of cytokine and chemokine function as well as other factors that play a role in immune evasion (1, 6, 39). However, epitopes that are known to elicit neutralizing antibodies are conserved (16, 32, 44), and recently three human CD8+ cytotoxic T lymphocyte (CTL) epitopes restricted to HLA-A*0201 have been identified that are present in MVA, the Copenhagen strain of vaccinia virus, and variola virus (11, 41). Thus, MVA can induce significant vaccinia virus-specific immune responses that are unmodified by normal vaccinia virus immune evasion mechanisms.
Earlier work with MVA demonstrated its safety and its ability to protect against the development of poxvirus infections in several animal models (22, 30, 43). Recently, MVA immunization has been shown to provide protection against a pulmonary vaccinia virus challenge (4, 11). With the threat of bioterrorism and the potential for exposure to genetically manipulated weaponized smallpox, the ability of a new vaccine to protect against pathogens with enhanced virulence may be necessary. Type 2 cytokines have been shown to diminish CTL activity in vivo and to inhibit viral clearance (2, 12, 24, 42). The coexpression of interleukin-4 (IL-4) in the presence of vaccinia virus infection results in the downregulation of type 1 cytokines, reduces cytolytic activity, and delays viral clearance (2, 3, 24, 36). The demonstration of potent immunity and in vivo protection by novel second-generation vaccines against vaccinia virus strains with enhanced virulence would lend further support to the development of a new approach to smallpox immunization.
We sought to evaluate the comparative efficacies of MVA and a replication-competent vaccinia virus strain, vSC8, against both intradermal and pulmonary challenges of vaccinia virus in a mouse model. MVA immunization elicited both humoral and cellular immunity equivalent to that elicited by replication-competent vaccinia virus and protected mice from the illness associated with poxvirus challenge, including a lethal intranasal challenge with a recombinant vaccinia virus, vSC8-mIL4. MVA immunization reduced viral replication in the lung tissue and reduced the pathology associated with vaccinia virus pulmonary infections. After the selective depletion of B- and T-cell subsets, mice immunized with MVA retained sufficient immunity for protection, suggesting that the immunologic correlate of protection from vaccinia virus is complex and redundant.
MATERIALS AND METHODS
Viruses and cells.
The vaccinia virus strain vSC8 expressing β-galactosidase was provided by Bernie Moss (National Institutes of Health [NIH], Bethesda, Md.). Viral stocks were grown in BSC40 cells and purified as previously described (23). A vaccinia virus expressing murine IL-4 (vSC8-mIL4) was constructed from wild-type vSC8 as described previously (24). Modified vaccinia virus Ankara was provided by Therion Biologics (Cambridge, Mass.). BSC40 cells were obtained from Shiu-Lok Hu (University of Washington, Seattle) and were maintained in minimum essential medium (MEM) supplemented with 10% fetal calf serum, glutamine, and antibiotics. P815 cells were obtained from Jonathan Yewdell (NIH) and were maintained in RPMI medium supplemented with 10% fetal calf serum, glutamine, and antibiotics.
Mice.
Six- to 8-week-old pathogen-free BALB/c mice were purchased from Charles River Laboratories (Raleigh, N.C.). Six- to 8-week-old mice in a C57BL/6 genetic background that lacked mature B cells (B6.12952-Igh-6tmlCgn mice) were purchased from Jackson Laboratory (Bar Harbor, Maine). Age-specific C57BL/6 mice purchased from Jackson Laboratory served as a control strain for the B-cell-deficient mice. All mice were housed and cared for in accordance with the Guide for the Care and Use of Laboratory Animals prepared by the Committee on Care and Use of Laboratory Animals of the Institute of Laboratory Animal Resources, National Research Council.
Mouse immunization and challenge.
Mice were immunized intramuscularly with a 0.1-ml volume in each thigh with MVA, the vSC8 vaccinia virus, or phosphate-buffered saline (PBS). MVA and vSC8 were administered at a dose of 2 × 106 PFU per vaccination. As indicated for each experiment, groups of mice received single or multiple doses of MVA, with each dose separated by 2 weeks. Intradermal and intranasal challenges were performed 4 weeks after the last immunization. Intradermal challenges were performed by the administration of a 50-μl volume of vSC8 intradermally at the base of the tail (2 × 106 PFU). Mice were observed daily for lesion formation at the site of challenge for 14 days.
For intranasal challenges, mice were anesthetized intramuscularly with xylazine-ketamine and then infected with vSC8 or vSC8-mIL4 in a 100-μl volume. The mice were weighed and observed for illness daily, as previously described (23).
In vivo replication of vaccinia virus.
Subgroups of mice were sacrificed on days 4, 8, and 12. The right lung tissue was removed, weighed, and immediately quick-frozen in minimum essential medium supplemented with 10% fetal bovine serum (10% MEM). Briefly, the samples were quick-thawed at 37°C and maintained at 4°C prior to being individually ground with a mortar and pestle. Serial 10-fold dilutions of clarified supernatants were used to infect subconfluent monolayers of BSC40 cells in triplicate in 12-well plates. After 1 h, the plates were covered in 0.75% methylcellulose in 10% MEM and incubated at 37°C. The cells were fixed with formalin 2 days after infection and stained with 2% crystal violet-40% methanol, and plaques were counted under a dissecting microscope. Data are presented as geometric mean log10 PFU per gram of lung at dilutions that produced more than three plaques per well.
Vaccinia virus neutralization assay.
Retro-orbital bleeding of mice was performed on days 14 and 28 postimmunization. Blood samples were centrifuged at 5,000 rpm for 5 min in a Sorvall Fresco Biofuge microcentrifuge, and the sera were collected. Serum samples were diluted 1:10 in PBS and stored at −20°C. Each sample was thawed and diluted two- or fourfold in a 96-well plate. An equal volume of vSC8 vaccinia virus (2 × 105 PFU/well) was added to each well. A negative control and standard vaccinia immune globulin positive control (obtained from Hana Golding, FDA, Bethesda, Md.) were used in each assay. The plates were incubated overnight at 37°C, and then 50 μl of each sample was added to a subconfluent monolayer of BSC40 cells, with each sample tested in triplicate. After 1 h at room temperature, the cells were overlaid with 10% MEM containing 0.75% methylcellulose. The plates were incubated at 37°C for 48 h and then fixed with 3.7% formaldehyde prior to staining with 0.2% crystal violet-40% methanol. Each well was then evaluated for the number of plaques under a dissecting microscope, and the 60% plaque reduction value was determined and expressed as the log2 reciprocal neutralization titer. Cumulative data are given as mean titers ± standard deviations.
Intracellular cytokine assay.
Mice were sacrificed 14 and 28 days after their last immunization, and their spleens were harvested. Lymphocytes were isolated by centrifugation on a Ficoll-Hypaque cushion (specific gravity, 1.09) at room temperature, washed twice, and resuspended in RPMI containing 10% fetal bovine serum. P815 cells were infected for 6 h with vSC8 at a multiplicity of infection of 3 to 5 in serum-free RPMI at 37°C. After incubation, vaccinia virus-infected and uninfected P815 cells were added to 2 × 106 lymphocytes at a 1:10 ratio. The cells were costimulated with monoclonal anti-mouse CD28 and anti-mouse CD49d antibodies. Phorbol myristate acetate (0.2 μg/5 ml) and ionomycin (2 μg/5 ml) served as positive controls. After 1 h, brefeldin A (Pharmingen, San Diego, Calif.) was added to each sample at 3.5 μg/5 ml. The samples were incubated for 12 to 16 h at 37°C. The cells were then fixed, permeabilized (Cytofix/Cytoperm; Pharmingen), and stained with anti-gamma interferon (IFN-γ) (clone XMG1.2), anti-CD3e (clone 145-2C11), anti-CD4a (clone RM4-5), and anti-CD8a (clone 53-6.7) monoclonal antibodies. After being washed, the cells were resuspended in staining buffer and analyzed in a FACSCalibur instrument (BD Biosciences, San Jose, Calif.). Analyses of samples were performed with FlowJo software (TreeStar Inc., Ashland, Oreg.). All antibodies were purchased from Pharmingen.
Antibody depletion.
Mice were injected intraperitoneally with an antibody specific to either CD4 (clone GK 1.5) or CD8 (clone 2.43), with both of these antibodies, or with an isotype control antibody (clone HB 151), as previously described (19). On days −2 and −1 and on the day of challenge, mice received 200 μg of the indicated antibody intraperitoneally. On day 7, the remaining mice received an additional injection of 500 μg of antibody. Mice from each depletion group were sacrificed on day 11 postchallenge to measure the frequencies of CD3-, CD4-, and CD8-positive splenocytes by flow cytometry. Relative to isotype control-treated mice, the depleted mice had only 3.2% of their original CD4+ T cells and 2.8% of the CD8+ T cells remaining.
Statistical analysis.
Data were maintained in a Paradox database. Determinations of neutralization antibody titers were performed with SAS statistical software (SAS Institute Inc., Cary, N.C.) as previously described (24). Weight loss and neutralizing antibody titers were analyzed by analyses of variance with Kruskal-Wallis and Wilcoxon rank sum tests. Comparisons were made between individual experiments by statistical modeling, and trend analysis was performed by the general linear model method of the SAS software. Virus titers and T-cell responses were compared by Tukey's t test analysis in Sigma Stat 2.0 (SPSS Inc., Chicago, Ill.).
RESULTS
MVA immunization protects against intradermal challenge with vaccinia virus.
The intradermal inoculation of vaccinia virus results in a characteristic development of lesions at the site of the inoculation. After vaccinia virus immunization of humans, the lesions progress from papules to pustules or vesicles prior to the formation of scabs. BALB/c mice were immunized intramuscularly with 2 × 106 PFU of MVA or vSC8 given as one, two, or three doses at 2-week intervals. Four weeks after immunization, the mice were challenged with vSC8 intradermally at the base of the tail, and the development of lesion formation was compared with that in PBS-inoculated controls. For control mice, single or multiple lesions were noted at the site of inoculation beginning on day 6. Although the numbers and sizes of the lesions varied, pox lesions were noted for nearly all animals (94%) (Table 1). A single immunization with MVA or vSC8 provided a partial yet significant reduction in the numbers of lesions compared to the control animals (P < 0.05) (Table 1). After two or three doses of MVA, complete protection from the formation of pox lesions was noted. These data suggest that a single dose of MVA elicits an immune response equivalent to that against a single dose of replication-competent vaccinia virus when given intramuscularly and that immunity improves with multiple injections.
TABLE 1.
MVA immunization protects against intradermal vSC8 challengea
| Vaccination regimen | Frequency of lesion formation (no. of mice with lesions/total no. of mice [%])b |
|---|---|
| PBS | 15/16 (94) |
| MVA | 3/16 (19)* |
| MVA-MVA | 0/16 (0)* |
| MVA-MVA-MVA | 0/16 (0)* |
| VSC8 | 2/8 (25)* |
Mice were immunized with PBS, vSC8, or single or multiple doses of MVA and then challenged intradermally at the base of the tail 4 weeks after the last immunization with vSC8. Mice were observed daily for pox lesion formation. Data are a combination of two experiments and demonstrate a significant decrease in lesion formation in all immunized mice. There was partial protection in mice that received a single injection of MVA, and complete protection was observed for mice immunized with two or three doses of MVA.
*, P < 0.05.
MVA immunization protects against illness and reduces viral burden in mice challenged intranasally with vaccinia virus.
After demonstrating the protection from intradermal challenge of MVA immunization, we next immunized mice with MVA or vSC8 and challenged them intranasally with vSC8 at 107 PFU. The challenge volume of 100 μl is known to deposit at least 70% of the inoculum directly in the lung (18). Mice were immunized with either one or two doses of MVA prior to the challenge, as two doses demonstrated complete protection in the intradermal challenge model. After the challenge, the mice were observed daily for illness and weight loss. PBS-inoculated mice developed weight loss beginning on day 4 (Fig. 1A). On day 6, control mice suffered a peak weight loss of nearly 25% and then gradually recovered by day 12. In sharp contrast, all immunized mice were protected from significant weight loss. Lungs were harvested on days 4, 8, and 12 after the challenge, and vaccinia virus titers were determined (Fig. 1B). The viral titers reflected the illness of the PBS-inoculated mice: the vaccinia virus titers per gram of lung were 7.52 ± 0.50 and 6.99 ± 0.26 log10 on days 4 and 8, respectively. The vaccinia virus titers were significantly reduced on day 4 for all groups of immunized mice compared to those in PBS-inoculated mice (P < 0.05), but there were detectable virus titers on day 4 in mice that had received a single MVA immunization. By day 8, there was no detectable vaccinia virus in any immunized mouse. Thus, clinical protection was achieved after a single-dose immunization with MVA and correlated directly with a reduction in the overall viral burden and the rapid clearance of vaccinia virus.
FIG. 1.
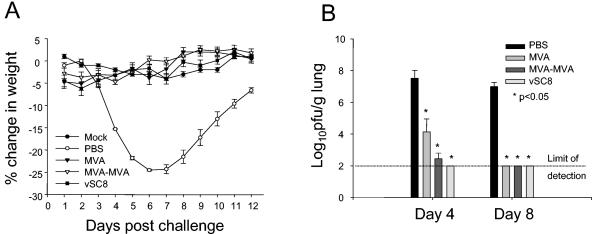
Mice immunized with MVA demonstrate absence of weight loss and reduced vaccinia virus titers in the lungs after an intranasal challenge with vSC8. Mice were immunized with vSC8 or one or two doses of MVA or were inoculated with PBS and then were challenged 4 weeks later with intranasal vSC8. (A) PBS-inoculated mice demonstrated weight loss that peaked on days 6 and 7. MVA- and vSC8-immunized mice were protected from weight loss and clinical illness. (B) Lungs were harvested on days 4 and 8, and vaccinia virus titers were determined and expressed as log10 PFU per gram of lung tissue. The limit of detection for this assay was 2.0 log10 PFU/g. On day 4, there was a significant decline in vaccinia virus titer in MVA-immunized mice compared to PBS-inoculated mice. By day 8, there was a complete clearance of vaccinia virus from the lungs of mice that were immunized with MVA.
MVA induces both humoral and cellular immune responses.
Vaccinia virus neutralization was performed by use of a plaque reduction assay with samples collected 2 and 4 weeks after immunization. A dose-dependent humoral response was elicited by MVA immunization (Fig. 2A). The peak response was seen on day 14 and persisted to at least day 28. A single dose of MVA elicited a response equivalent to that seen with intramuscular vSC8. The administration of multiple doses of MVA boosted the antibody response, with a significant enhancement of neutralization seen after three doses on days 14 and 28 compared to that with a single dose (P < 0.05).
FIG. 2.
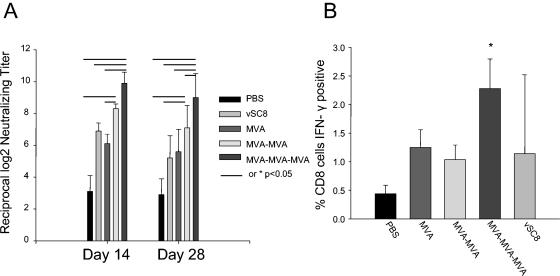
MVA elicits dose-dependent humoral and cellular immune responses. After immunization with PBS, vSC8, or MVA, mice were assessed for an immune response by plaque neutralization and intracellular IFN-γ production. (A) On days 14 and 28 after the last immunization, neutralizing antibody titers were determined and expressed as reciprocal log2 serum dilutions resulting in 60% plaque reduction. Data represent means from two independent experiments, resulting in data for 10 mice per group. The lines between bars indicate values that are significantly different (P < 0.05). (B) Twenty-eight days after immunization, splenocytes were harvested, and intracellular IFN-γ production in vaccinia virus-specific T cells was measured by flow cytometry. The data are for five mice in each group from one representative experiment of four total experiments.
Similarly, the magnitude of the cellular response was dependent on the number of MVA immunizations. Twenty-eight days after immunization, splenocytes were isolated, stimulated with vaccinia virus, and evaluated for intracellular IFN-γ production by flow cytometry. The percentage of vaccinia virus-specific CD8+ CD3+ T cells was determined for mice receiving PBS, vSC8, or MVA (Fig. 2B). A higher frequency of vaccinia virus-specific CD8+ T cells was elicited by the three-dose regimen of MVA than by either one or two doses of MVA (P < 0.05).
MVA protects against lethal vSC8-mIL4 intranasal challenge.
In order to determine the efficacy of MVA immunization against a vaccinia virus strain with enhanced virulence, we challenged mice intranasally with a recombinant vaccinia virus encoding murine IL-4, vSC8-mIL4. As with the vSC8 parent strain, mice immunized with one or two doses of MVA were protected from the development of clinical illness, as measured by weight loss (Fig. 3A). Nonimmunized mice lost weight rapidly, and all mice died or were euthanized because of extreme illness by day 8 after the challenge. Nonimmunized mice demonstrated significantly higher vaccinia virus titers in the lungs than did MVA-immunized mice (Fig. 3B). Lung histopathologies were evaluated to compare the inflammatory processes in immunized versus nonimmunized mice. MVA-immunized mice showed only a minimal infiltrate on day 8, while there was a profound alteration of the lung architecture in nonimmunized mice, with severe mononuclear cell peribronchiolar and interstitial infiltrates, alveolar edema and exudate, and epithelial necrosis (Fig. 4).
FIG. 3.
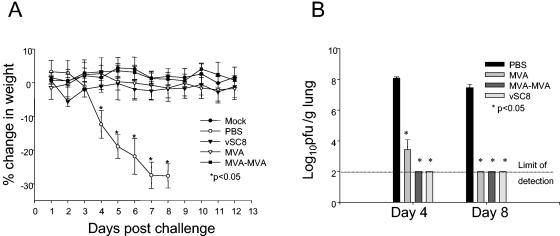
MVA immunization provides clinical and virologic protection after an intranasal challenge with vSC8-mIL4. Mice were immunized with PBS, vSC8, or one or two doses of MVA and then challenged 4 weeks after immunization with intranasal vSC8-mIL4. (A) PBS-inoculated mice demonstrated a rapid decline in weight loss that resulted in death. MVA-immunized mice were protected from weight loss and clinical illness. (B) Lungs were harvested on days 4 and 8, and vaccinia virus titers in the lungs were determined and expressed as log10 PFU per gram of lung tissue. The limit of detection for this assay was 2.0 log10 PFU/g. Similar to the case for the vSC8 challenge, there was a significant decline in viral titer in MVA-immunized mice compared to PBS-inoculated mice on day 4, with a complete clearance of vaccinia virus by day 8.
FIG. 4.
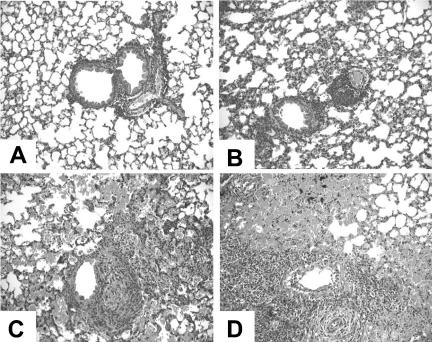
Lung histopathology in mice challenged with vSC8-mIL4. Mice were sacrificed on day 8 after the vSC8-mIL4 challenge, lungs were fixed in 10% formalin phosphate and embedded in paraffin, and thin sections were stained with hematoxylin and eosin. The images were obtained with a Zeiss Axioplan microscope using a 20× objective. (A) Lung from a mouse immunized intradermally with Dryvax prior to vSC8-mIL4 challenge; (B) lung from a mouse immunized with one dose of MVA prior to vSC8-mIL4 challenge; (C and D) lungs from mice challenged with vSC8-mIL4 subsequent to PBS inoculation. Both Dryvax and MVA immunization provided significant protection from the extensive peribronchiolar and interstitial inflammation, alveolar exudates and edema, and epithelial necrosis seen in the lungs of unimmunized mice.
T-cell depletion reduces efficacy of MVA immunization.
Groups of mice were depleted of CD4+ cells, CD8+ cells, or both by the use of specific antibodies prior to an intranasal challenge with 107 PFU of vSC8-mIL4. PBS-inoculated mice developed significant illness and mortality by day 8 after the challenge (Fig. 5A). MVA-immunized mice that received an isotype control antibody or mice that were depleted of CD4+ cells, CD8+ cells, or both had no significant weight loss. In mice that were depleted of both CD4+ and CD8+ cells, there was a higher vaccinia virus titer in the lungs on day 4 after the challenge than those for the other groups of MVA-immunized mice (P < 0.05) (Fig. 5B). On day 8, vaccinia virus was still detectable in the lungs of CD4- and CD8-depleted mice, while all other MVA-immunized mice had cleared the vaccinia virus. To better define the roles of the CD4+ and CD8+ T cells in this setting, we challenged other groups of mice that had been immunized with two doses of MVA with a higher dose (108 PFU) of vSC8-mIL-4 intranasally after differential T-cell subset depletion. With the higher-titer vaccinia virus challenge, the MVA vaccine-induced immunity was partially mitigated by either CD4+- or CD8+-T-cell depletion alone (Fig. 6B), but illness was most severe when both T-cell subsets were depleted prior to the challenge (Fig. 6A). The most severe illness was associated with higher residual vaccinia virus titers on both days 4 and 8 in mice that were depleted of both T-cell subsets. These data indicate that both CD4+ and CD8+ vaccine-induced T-cell responses are important for controlling vaccinia virus replication and illness.
FIG. 5.
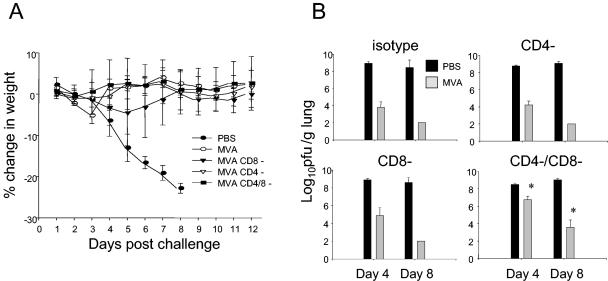
T-cell-depleted mice that are immunized with MVA are partially protected from a vaccinia virus intranasal challenge. Mice were immunized with two doses of MVA and then challenged 4 weeks after immunization with 107 PFU of intranasal vSC8-mIL4. Prior to challenge, the mice were depleted of CD4+, CD8+, or both CD4+ and CD8+ T cells on days −2, −1, and 0, and repeat depletion was done on day 7. (A) Changes in baseline weight were determined daily. The depletion of either CD4+ or CD8+ T cells did not result in weight loss in mice receiving the lower-titer challenge. Compared to PBS-inoculated controls, all MVA-immunized mice recovered to their baseline weight by day 8. (B) Vaccinia virus titers in the lungs were determined on days 4 and 8 after the challenge. Clinical illness corresponded to vaccinia virus titers in MVA-immunized mice. MVA-immunized double-depleted mice had significantly higher vaccinia virus titers on days 4 and 8 than did MVA-immunized mice treated with the isotype control or an antibody to either CD4 or CD8.
FIG. 6.
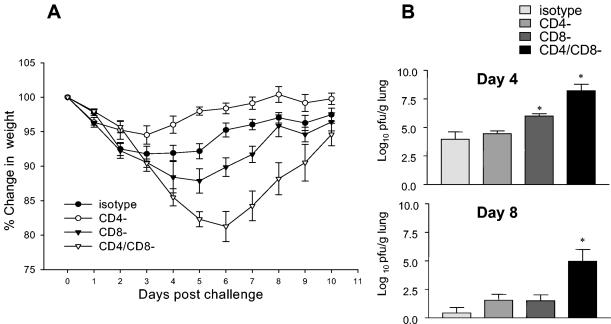
Mice were immunized with two doses of MVA and then challenged intranasally 4 weeks after immunization with 108 PFU of vSC8-mIL4. Prior to challenge, the mice were depleted of CD4+, CD8+, or both CD4+ and CD8+ T cells on days −2, −1, and 0, and repeat depletion was done on day 7. (A) Changes in baseline weight were determined daily. After the high-titer challenge, the depletion of either CD4+ or CD8+ T cells resulted in modest weight loss, despite the prior immunization with MVA. The depletion of both CD4+ and CD8+ T cells resulted in a more severe illness, suggesting that both T-cell subsets contribute to immunity. (B) The vaccinia virus titers in the lungs were determined on days 4 and 8 after challenge and demonstrated the same patterns as those seen in the weight loss curves and were also consistent with the patterns seen after the lower-dose vSC8-mIL4 challenge. Mice depleted of both CD4+ and CD8+ T cells had more residual virus on days 4 and 8 than the other groups.
Mice lacking mature B cells.
In order to assess the contribution of the humoral immune response to vaccine-induced immunity, we immunized mice lacking mature B cells, B6.12952-Igh-6tmlCgn (B6.12952) mice, with one or two doses of MVA prior to a challenge with vSC8-mIL4. Neutralization assays were performed on day 28 postimmunization with B6.12952 and C57BL/6 mice. C57BL/6 mice immunized with MVA demonstrated the presence of neutralizing antibodies, while there were no detectable neutralizing antibodies in the B6.12952 mice (Fig. 7A). MVA-immunized mice showed no decline in weight after the challenge, while PBS-inoculated B6.12952 and C57BL/6 mice showed significant weight reductions that peaked on day 7 (Fig. 7B). The vaccinia virus lung titers measured on day 4 postinfection revealed a significant decline in titer in the B6.12952 mice that received one or two doses of MVA compared to those in PBS-inoculated B6.12952 mice (Fig. 7C). However, when compared to that in C57BL/6 MVA-immunized mice, the degree of vaccinia virus inhibition was significantly lower (P < 0.05). Both B6.12952 and C57BL/6 mice that were immunized with MVA cleared the virus from the lungs by day 8, while PBS-inoculated mice that survived still had high titers of vaccinia virus. These data support a significant role for the humoral immune response in protection against vaccinia virus, particularly in controlling the peak of vaccinia virus replication, but mice deficient in neutralizing antibodies were eventually able to clear the virus without significant clinical illness.
FIG. 7.
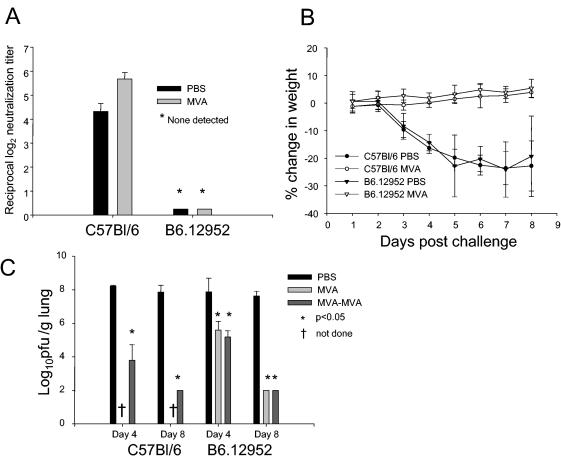
B-cell-deficient mice challenged with vSC8-mIL4 after MVA immunization are protected from weight loss but demonstrate higher vaccinia virus titers in the lungs than MVA-immunized C57BL/6 mice. B6.12952 mice lacking mature B cells were immunized with PBS or one or two doses of MVA and then challenged intranasally 4 weeks after immunization with vSC8-mIL4. C57BL/6 mice served as controls. (A) C57BL/6 mice had significant neutralizing antibody responses postchallenge, while there was no detectable antibody for B6.12952 mice. (B) After the challenge with vSC8-mIL4, PBS-inoculated mice demonstrated a rapid decline in weight while MVA-immunized mice lacked clinical illness despite the absence of neutralizing antibodies. (C) Vaccinia virus titers in the lungs were determined and expressed as log10 PFU per gram of lung tissue. The viral burden in MVA-immunized B6.12952 mice was significantly less than that in PBS-inoculated mice, but they had a higher vaccinia virus titer on day 4 than that in C57BL/6 mice with normal humoral immunity.
DISCUSSION
Following the anthrax attacks in the fall of 2001, discussions about the potential use of other virulent pathogens as biological weapons have increased. The threat of a smallpox outbreak elicits great concerns because of its acute and significant morbidity and mortality rates and the lack of significant immunity against poxviruses among civilians born after 1970, despite a large vaccination campaign effort among the military and medical first responders (17). Discussions about the widespread use of the present FDA-approved vaccine Dryvax have resulted in much debate in the medical community. While the vaccine is efficacious, the complications associated with Dryvax make the development of a safer second-generation smallpox vaccine a medical priority.
For the present study, we evaluated the efficacy and immune response of a highly attenuated vaccinia virus, MVA, by use of a mouse model. Investigations of this vaccinia virus strain began in the late 1950s after attenuation of the parent strain CVA in chicken embryo fibroblasts. Since that time, MVA has been evaluated in several animal models as well as in humans (22, 30, 43). During the 1970s, more than 120,000 people were immunized with MVA (37). Reports reflect a good safety profile for MVA, including safe administration to immunocompromised animals (38, 43). However, the specific determinants of MVA-induced immunity compared with those induced by replication-competent vaccinia virus have only recently been studied (5, 45).
We demonstrated the ability of a single dose of MVA to protect against both intradermal and intranasal challenges with vaccinia virus. In particular, we demonstrated that MVA immunization can protect against a lethal respiratory challenge with a molecularly modified recombinant vaccinia virus expressing murine IL-4. While previous studies have shown a dose-related effect following single-dose escalation (4, 11), we showed that a multidose schedule of MVA may enhance the overall immune response and provide added protection against vaccinia virus replication. For both the intradermal and intranasal models, virologic protection, as judged by the presence or absence of pox lesions, and vaccinia virus titers in the lungs improved in mice that received two or three immunizations (Table 1 and Fig. 1). While clinical illness was reduced in all MVA-immunized mice after an intranasal challenge, the vaccinia virus titers in the lungs were higher for mice that received a single immunization. This enhanced protection correlated with the magnitude of both the humoral and cellular immune responses, as judged by the neutralization activity and intracellular IFN-γ production in vaccinia virus-specific T cells, respectively (Fig. 2). In mice receiving multiple immunizations, the immune responses were enhanced, suggesting that strategies to optimize the protection of humans against poxviruses such as variola virus and monkeypox virus may necessitate a multidose immunization regimen. Although it was not evaluated in these studies, the route of immunization is also important. For example, MVA was administered intramuscularly in our studies, and the responses compared favorably to those induced by replication-competent vaccinia virus given by the intramuscular or intradermal route. However, when vaccinia virus is given by intraperitoneal injection, much higher frequencies of CD4+- and CD8+-T-cell responses can be achieved in splenocytes (20). Therefore, additional exploration of the route of poxvirus immunization may be warranted.
With the threat of bioterrorism, there is concern not only about the potential release of virulent pathogens, but also about the purposeful manipulation of pathogens in an effort to augment their virulence. Importantly, we have demonstrated that MVA immunization can protect against a lethal pulmonary challenge with a molecularly modified recombinant vaccinia virus expressing murine IL-4. Prior studies have demonstrated the importance of cytokine balance in the pathogenesis of viral, bacterial, and parasitic diseases. The Th1 cytokine IFN-γ plays a key role in the control of vaccinia virus infection (24). Disruption of the Th1-Th2 balance has been shown to adversely affect viral clearance and protection from dissemination. As previously described (24, 36), we demonstrated that the virulence of vaccinia virus is enhanced in the setting of excess IL-4 production, with a prolonged elevation of lung viral titers and death in mice infected with vSC8-mIL4 (Fig. 3). Despite this enhanced virulence, MVA immunization protected mice from a lethal pulmonary challenge with vSC8-mIL4 and eliminated clinical illness and weight loss. The ability to protect against a lethal molecularly modified vaccinia virus lends further support to MVA as a candidate vaccine against smallpox.
While they are effective against vaccinia virus challenges, the determinants of immunity elicited by MVA and other vaccinia viruses have not been clearly defined. The immunization of human subjects with Dryvax has been shown to elicit both humoral and cellular immunity (15). Historical reports suggest that both arms of the immune system are relevant to protection from smallpox. Investigations of villages during outbreaks of smallpox correlated the vaccine take and antibody response with immunity (28). More recent reports point to the importance of the cellular immune response for containing disseminated vaccinia virus (7). It becomes important in the evaluation of novel vaccines to dissect the role of each component of the immune response in order to demonstrate similar patterns of response to both replication-competent and attenuated vaccines.
Recent work suggested that selected human CD8+ CTL epitopes are conserved between various poxviruses, including MVA and variola virus (11, 41). The selective depletion of either CD4+ or CD8+ T cells alone prior to vSC8-mIL4 challenge resulted in minimal weight loss and viral replication, which suggests that neither cell type is required for the containment of vaccinia virus replication (Fig. 4). The depletion of both CD4+ and CD8+ T cells resulted in weight loss, which corresponds to enhanced viral replication and delayed clearance, but the weight loss was minimal. While this demonstrates the importance of cellular immunity for vaccinia virus clearance, it suggests that T cells are not the sole factor in the defense against poxvirus infection and that the immune system has evolved redundant mechanisms to control this important class of pathogens.
Contributions from the humoral immune response following immunization are also likely to be important for protection against vaccinia virus infection. We demonstrated a dose-dependent increase in the antibody response to MVA immunization, which correlated with protection. However, in the absence of neutralizing antibodies there was minimal illness in this murine model from an intranasal vaccinia virus challenge (Fig. 5). While MVA-immunized B-cell-deficient mice demonstrated significant viral replication in the lungs on day 4 after the intranasal challenge, they lacked signs of clinical illness and cleared the vaccinia virus by day 8. As for many other virus infections, we believe that the data support an important role for vaccine-induced antibodies in protection from infection, but effective T-cell responses also appear to be important for protection against severe poxvirus-induced disease. These data are consistent with recent studies that evaluated the immune determinants of protection against the WR strain of vaccinia virus (5) and that indicated that vaccine-induced protection from poxviral challenge should not be restricted to a single arm of the immune response, but should include a combination of both cellular and humoral immune responses.
We demonstrated a robust immune response following MVA immunization in a murine model. MVA not only protected mice from a standard vaccinia virus challenge, but it was also able to prevent illness and limit viral replication after the administration of a lethal molecularly modified strain of vaccinia virus. These data support the further development of MVA as a stand-alone vaccine against smallpox and infections caused by other orthopoxviruses.
REFERENCES
- 1.Alcami, A., and G. L. Smith. 1995. Vaccinia, cowpox and camelpox viruses encode soluble interferon receptors with novel broad species specificity. J. Virol. 69:4633-4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Antoine, G., F. Scheiflinger, F. Dorner, and F. G. Falkner. 1998. The complete genomic sequence of the modified vaccinia Ankara strain: comparison with other orthopoxviruses. Virology 24:365-396. [DOI] [PubMed] [Google Scholar]
- 3.Aung, S., Y. W. Tang, and B. S. Graham. 1999. Interleukin-4 inhibits induction of cytotoxic T lymphocyte activity in mice infected with recombinant vaccinia virus expressing respiratory syncytial virus M2 protein. J. Virol. 73:8944-8949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aung, S., and B. S. Graham. 2000. Differential regulation of perforin- and FasL-mediated cytotoxicity by IL-4. J. Immunol. 164:3487-3493. [DOI] [PubMed] [Google Scholar]
- 5.Belyakov, I. M., P. Earl, A. Dzutsev, V. A. Kuznetsov, M. Lemon, L. S. Wyatt, J. T. Snyder, J. D. Ahlers, G. Franchini, B. Moss, and J. A. Berzofsky. 2003. Shared modes of protection against poxvirus infection by attenuated and conventional smallpox vaccine viruses. Proc. Natl. Acad. Sci. USA 100:9458-9463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blanchard, T. J., A. Alcami, P. Andrea, and G. L. Smith. 1998. Modified vaccinia virus Ankara undergoes limited replication in human cells and lacks several immunomodulatory proteins: implications for use as a human vaccine. J. Gen. Virol. 79:1159-1167. [DOI] [PubMed] [Google Scholar]
- 7.Bray, M., and M. E. Wright. 2003. Progressive vaccinia. Clin. Infect. Dis. 36:766-774. [DOI] [PubMed] [Google Scholar]
- 8.Carroll, M. W., and B. Moss. 1997. Host range and cytopathogenicity of the highly attenuated MVA strain of vaccinia virus: propagation and generation of recombinant viruses in a nonhuman mammalian cell line. Virology 238:198-211. [DOI] [PubMed] [Google Scholar]
- 9.CDC. 2001. Vaccinia (smallpox) vaccine. Recommendations of the Advisory Committee on Immunization Practices (ACIP). Morb. Mortal. Wkly. Rep. Recommun. Rep. 50(RR-10):1-25. [PubMed] [Google Scholar]
- 10.Cohen, J. 2001. Smallpox vaccinations: how much protection remains? Science 294:985. [DOI] [PubMed] [Google Scholar]
- 11.Drexler, I., C. Staib, W. Kastenmuller, S. Stevanovic, B. Schmidt, F. Lemonnier, H. Rammensee, D. Busch, D. H. Bernhard, H. Bernhard, and V. Erfle. 2003. Identification of vaccinia virus epitope-specific HLA-A*0201-restricted T cells and comparative analysis of smallpox vaccines. Proc. Natl. Acad. Sci. USA 100:217-222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fischer, J. E., J. E. Johnson, R. K. Kuli-Zade, T. R. Johnson, S. Aung, R. A. Parker, and B. S Graham. 1997. Overexpression of interleukin-4 delays virus clearance in mice infected with respiratory syncytial virus. J. Virol. 71:8672-8677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Flexner, C., A. Hugin, and B. Moss. 1987. Prevention of vaccinia virus infection in immunodeficient mice by vector-directed IL-2 expression. Nature 330:259-262. [DOI] [PubMed] [Google Scholar]
- 14.Flexner, C., B. Moss, W. T. London, and B. R. Murphy. 1990. Attenuation and immunogenicity in primates of vaccinia virus recombinants expressing human interleukin-2. Vaccine 8:17-21. [DOI] [PubMed] [Google Scholar]
- 15.Frey, S. E., F. K. Newman, J. Cruz, W. B. Shelton, J. M. Tennant, T. Polach, A. L. Rothman, J. S. Kennedy, M. Wolff, R. B. Belshe, and F. A. Ennis. 2002. Dose-related effects of smallpox vaccine. N. Engl. J. Med. 346:1275-1280. [DOI] [PubMed] [Google Scholar]
- 16.Galmiche, M. C., J. Goenaga, R. Wittek, and L. Rindisbacher. 1999. Neutralizing and protective antibodies directed against vaccinia virus envelope antigens. Virology 254:71-80. [DOI] [PubMed] [Google Scholar]
- 17.Grabenstein, J. D., and W. Winkenwerder. 2003. US military smallpox vaccination program experience. JAMA 289:3278-3282. [DOI] [PubMed] [Google Scholar]
- 18.Graham, B. S., M. D. Perkins, P. F. Wright, and D. T. Karzon. 1988. Primary respiratory syncytial virus infection in mice. J. Med. Virol. 26:153-162. [DOI] [PubMed] [Google Scholar]
- 19.Graham, B. S., L. A. Bunton, P. F. Wright, and D. T. Karzon. 1991. Role of T lymphocyte subsets in the pathogenesis of primary infection and rechallenge with respiratory syncytial virus in mice. J. Clin. Investig. 88:1026-1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harrington, L. E., R. van der Most, J. L. Whitton, and R. Ahmed. 2002. Recombinant vaccinia virus-induced T-cell immunity: quantitation of the response to the virus vector and the foreign epitope. J. Virol. 76:3329-3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hashizume, S. 1975. A new attenuated strain Lc16m8 of vaccinia virus for safer smallpox vaccination. Clin. Virol. 3:229-235. [Google Scholar]
- 22.Hochstein-Mintzel, V., T. Hanichen, H. C. Huber, and H. Stickl. 1975. Vaccine- and variola-protective effect of the modified vaccinia strain MVA in intramuscular immunization. Zentbl. Bakteriol. Hyg. I. Abt. Orig. A 975:283-297. [PubMed] [Google Scholar]
- 23.Johnson, T. R., J. E. Johnson, S. R. Roberts, G. W. Wertz, R. A. Parker, and B. S. Graham. 1998. Priming with secreted glycoprotein G of respiratory syncytial virus (RSV) augments interleukin-5 production and tissue eosinophilia after RSV challenge. J. Virol. 72:2871-2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnson, T. R., J. E. Fischer, and B. S. Graham. 2001. Construction and characterization of recombinant vaccinia viruses co-expressing a respiratory syncytial virus protein and a cytokine. J. Gen. Virol. 82:2107-2116. [DOI] [PubMed] [Google Scholar]
- 25.Kempe, C. H., V. Fulginiti, M. Minamitani, and H. Shinefield. 1968. Smallpox vaccination of eczema patients with a strain of attenuated live vaccinia (CVI-78). Pediatrics 42:980-985. [PubMed] [Google Scholar]
- 26.Lane, J. M., F. L. Ruben, J. M. Neff, and J. D. Millar. 1969. Complications of a smallpox vaccination, 1968. N. Engl. J. Med. 281:1201-1208. [DOI] [PubMed] [Google Scholar]
- 27.Lane, J. M., F. L. Ruben, J. M. Neff, and J. D. Millar. 1970. Complications of smallpox vaccination, 1968: results of ten statewide surveys. J. Infect. Dis. 122:303-309. [DOI] [PubMed] [Google Scholar]
- 28.Mack, T. M., J. Noble, Jr., and D. B. Thomas. 1972. A prospective study of serum antibody and protection against smallpox. Am. J. Trop. Med. Hyg. 21:214-218. [DOI] [PubMed] [Google Scholar]
- 29.Mayr, A., V. Hochstein-Mintzel, and H. Stickl. 1975. Passage history, properties, and applicability of the attenuated vaccinia virus strain MVA. Infection 3:6-14. [Google Scholar]
- 30.Mayr, A., H. Stickl, H. K. Muller, K. Danner, and H. Singer. 1978. The smallpox vaccination strain MVA: marker, genetic structure, experience gained with the parenteral vaccination and behavior in organisms with a debilitated defense mechanism. Zentbl. Bakteriol. 167:375-390. [PubMed] [Google Scholar]
- 31.Meyer, H., G. Sutter, and A. Mayr. 1991. Mapping of deletions in the genome of the highly attenuated vaccinia virus MVA and their influence on virulence. J. Gen. Virol. 72:1031-1038. [DOI] [PubMed] [Google Scholar]
- 32.Moss, B. 2001. Poxviridae: the viruses and their replication, p. 2849-2883. In D. M. Knipe et al. (ed.), Fields virology, 4th ed. Lippincott Williams and Wilkins, Philadelphia, Pa.
- 33.Neff, J. M., J. M. Lane, V. A. Fulginiti, and D. A. Henderson. 2002. Contact vaccinia: transmission of vaccinia from smallpox vaccination. JAMA 288:1901-1905. [DOI] [PubMed] [Google Scholar]
- 34.Perera, L. P., C. K. Goldman, and T. A. Waldmann. 2001. Comparative assessment of virulence of recombinant vaccinia viruses expressing IL-2 and IL-15 in immunodeficient mice. Proc. Natl. Acad. Sci. USA 98:5146-5151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sancho, M. C., S. Schleich, G. Griffiths, and K. Krijnse-Locker. 2002. The block in assembly of modified vaccinia virus Ankara in HeLa cells reveals new insights into vaccinia virus morphogenesis. J. Virol. 76:8318-8334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sharma, D. P., A. J. Ramsay, D. J. Maguire, M. S. Rolph, and I. A. Ramshaw. 1996. Interleukin-4 mediates down regulation of antiviral cytokine expression and cytotoxic T-lymphocyte responses and exacerbates vaccinia virus infection in vivo. J. Virol. 70:7103-7107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stickl, H., V. Hochstein-Mintzel, A. Mayr, H. C. Huber, H. Schafer, and A. Holzner. 1974. MVA vaccination against smallpox: clinical tests with an attenuated live vaccinia virus strain (MVA). Dtsch. Med. Wochenschr. 99:2386-2392. [DOI] [PubMed] [Google Scholar]
- 38.Stittelaar, K. J., T. Kuiken, R. L. de Swart, G. Amerongen, H. W. Vos, H. G. Niesters, P. van Schalkwijk, T. van der Kwast, L. S. Wyatt, B. Moss, and A. D. Osterhaus. 2001. Safety of modified vaccinia virus Ankara (MVA) in immune-suppressed macaques. Vaccine 19:3700-3709. [DOI] [PubMed] [Google Scholar]
- 39.Symons, J. A., A. Alcami, and G. L. Smith. 1995. Vaccinia virus encodes a soluble type I interferon receptor of novel structure and broad species specificity. Cell 81:551-560. [DOI] [PubMed] [Google Scholar]
- 40.Tartaglia, J., M. E. Perkus, J. Taylor, E. K. Norton, J. C. Audonnet, W. I. Cox, S. W. Davis, J. van der Hoeven, B. Meignier, M. Riviere, B. Languet, and E. Paoletti. 1992. NYVAC: a highly attenuated strain of vaccinia virus. Virology 188:217-232. [DOI] [PubMed] [Google Scholar]
- 41.Terajima, M., J. Cruz, G. Raines, E. D. Kilpatrick, J. S. Kennedy, A. L. Rothman, and F. A. Ennis. 2003. Quantitation of CD8+ T cell responses to newly identified HLA A*0201 restricted T cell epitopes conserved among vaccinia and variola (smallpox) viruses. J. Exp. Med. 197:927-932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van Den Broek, M., M. F. Bachmann, G. Kohler, M. Barner, R. Escher, R. Zinkernagel, and M. Kopf. 2000. IL-4 and IL-10 antagonize IL-12-mediated protection against acute vaccinia virus infection with a limited role of IFN-gamma and nitric oxide synthetase 2. J. Immunol. 164:371-378. [DOI] [PubMed] [Google Scholar]
- 43.Werner, G. T., U. Jentzsch, E. Metzger, and J. Simon. 1980. Studies on poxvirus infections in irradiated animals. Arch. Virol. 64:247-256. [DOI] [PubMed] [Google Scholar]
- 44.Wolffe, E. J., S. Vijaya, and B. Moss. 1995. A myristylated membrane protein encoded by the vaccinia virus L1R open reading frame is the target of potent neutralizing monoclonal antibodies. Virology 211:53-63. [DOI] [PubMed] [Google Scholar]
- 45.Xu, R., A. J. Johnson, D. Liggitt, and M. J. Bevan. 2004. Cellular and humoral immunity against vaccinia virus infection of mice. J. Immunol. 172:6265-6271. [DOI] [PubMed] [Google Scholar]