

Abstract
The viral genome of Kaposi's sarcoma-associated herpesvirus (KSHV) persists as an extrachromosomal plasmid in latently infected cells. The KSHV latency-associated nuclear antigen (LANA) stimulates plasmid maintenance and DNA replication by binding to an ∼150-bp region within the viral terminal repeats (TR). We have used chromatin immunoprecipitation assays to demonstrate that LANA binds specifically to the replication origin sequence within the KSHV TR in latently infected cells. The latent replication origin within the TR was also bound by LANA-associated proteins CBP, double-bromodomain-containing protein 2 (BRD2), and the origin recognition complex 2 protein (ORC2) and was enriched in hyperacetylated histones H3 and H4 relative to other regions of the latent genome. Cell cycle analysis indicated that the minichromosome maintenance complex protein, MCM3, bound TR in late-G1/S-arrested cells, which coincided with the loss of histone H3 K4 methylation. Micrococcal nuclease studies revealed that TRs are embedded in a highly ordered nucleosome array that becomes disorganized in late G1/S phase. ORC binding to TR was LANA dependent when reconstituted in transfected plasmids. DNA affinity purification confirmed that LANA, CBP, BRD2, and ORC2 bound TR specifically and identified the histone acetyltransferase HBO1 (histone acetyltransferase binding to ORC1) as a potential TR binding protein. Disruption of ORC2, MCM5, and HBO1 expression by small interfering RNA reduced LANA-dependent DNA replication of TR-containing plasmids. These findings are the first demonstration that cellular replication and origin licensing factors are required for KSHV latent cycle replication. These results also suggest that the KSHV latent origin of replication is a unique chromatin environment containing histone H3 hyperacetylation within heterochromatic tandem repeats.
Kaposi's sarcoma (KS)-associated herpesvirus (KSHV) (also referred to as human herpesvirus 8) has been causally linked to KS, primary effusion lymphoma (PEL), and multicentric Castleman's disease (4, 9, 10, 28, 59). Similar to Epstein-Barr virus (EBV), KSHV genomes can be maintained as multicopy chromatin-associated episomes in latently infected lymphocytes. During latent infection in PEL cells, KSHV transcription is restricted to a few viral genes, including the latency-associated nuclear antigen (LANA), viral FLICE-inhibitory protein (v-FLIP), v-CycD, kaposin, and viral interferon regulatory factor 2 protein (vIRF-2) (7, 23, 36, 48, 51, 55). The latency-associated viral products have been shown to have growth-transforming and cell cycle-deregulating properties likely to contribute to KSHV-associated malignancies (8, 16, 38). In addition to its growth-transforming activity, LANA is required for stable episomal maintenance and DNA replication of the latent viral genome (1, 13-15, 25, 33).
LANA, like EBNA1 from EBV, is a DNA binding protein that stimulates plasmid-based DNA replication (1, 2, 25, 26, 41). Deletion of the LANA-related protein from herpesvirus saimiri caused a complete loss of episomal genomes and failure to establish latent infection (13, 14). KSHV and herpesvirus saimiri LANAs bind to an ∼60-bp sequence within the ∼800-bp G+C-rich terminal repeats (TR) (1, 17, 24, 25). At least two repeats were required for stable episomal maintenance (2, 41), but a single LANA binding site (LBS) was sufficient to support transient DNA replication of plasmids (25). This resembles some of the properties of EBV OriP, which can replicate transiently with a minimum of two EBNA1 sites (65), but requires at least seven additional binding sites in the family of repeats to support stable plasmid maintenance (12, 37, 64). Most viral genomes have from 2 to 20 copies of the TR, and it is not clear how these tandem repeats influence LBS conformations to support plasmid maintenance of the viral genome during latency.
Stimulation of DNA replication by LANA has been presumed to function in a manner similar to that of EBNA1. Like EBNA1, LANA can coimmunoprecipitate from soluble nuclear extracts with members of the origin recognition complex (ORC) (21, 41). ORC binding is thought be an essential step in the establishment of a cellular replication origin (19, 20). ORC binding allows the cell cycle-regulated recruitment of the minichromosome maintenance complex (MCM), which is thought to be the replicative helicase and a target of the licensing mechanism that restricts replication firing to once per cell cycle (3, 61). EBNA1 has been shown to recruit the ORC to OriP in EBV (11, 21, 58), but it has not been formally shown that LANA can recruit ORC and MCM to the replication origin of KSHV.
Epigenetic constraints imposed by chromatin structure may regulate aspects of DNA replication and origin selection (27, 47). Posttranslational modification of histone tails may distinguish chromosomal boundaries and establish higher-order chromatin structures that can regulate protein access to DNA replication origins (35, 60). The KSHV latent cycle replication origin is embedded in the G+C-rich sequence of the TR. G+C-rich tandem repeats have been shown to have a high tendency to form repressive heterochromatin that may exclude binding of cellular factors required for replication preinitiation complex assembly (45, 52). The latent KSHV genome is associated with nucleosomes that can be positioned and modified to repress transcription of key regulatory genes, like the immediate-early open reading frame 50 (ORF50) transcript responsible for activation of the lytic cycle replication pathway (44). LANA has been shown to interact with numerous proteins associated with chromatin functions, including the histone acetyltransferase CBP (40), the double-bromodomain-containing protein BRD2 (also known as RING3) (46, 50), and mSin3 (39). A potential regulatory function of chromatin on KSHV viral replication origins has not been investigated.
Here, we show that the KSHV TR is organized into nucleosome arrays that are highly enriched in acetyl histone H3 at the LBSs. We present evidence that histone acetyltransferases and the double-bromodomain protein BRD2 are localized with LANA at TR. We also demonstrate that the cellular replication factors ORC and MCM associate with TR and function in LANA-mediated DNA replication of TR-containing plasmids.
MATERIALS AND METHODS
Cells and plasmids.
BCBL1 cells, a PEL-derived B-cell line infected with KSHV, were maintained in RPMI medium containing 10% fetal bovine serum and penicillin-streptomycin (50 U/ml). Cell cycle studies were performed, using asynchronous cells; cells were arrested with 400 μM mimosine (Sigma); and cells arrested with 1 μM colchicine (Sigma) overnight and collected at a density of 2 × 105 to 5 × 105 cells/ml of RPMI with 10% fetal bovine serum and antibiotics. Fluorescence-activated cell sorter analysis was performed subsequently to verify the cell cycle status of each condition. 293 cells were maintained in Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum containing penicillin-streptomycin (50 U/ml). The 2 × TR plasmid (N700) contains two contiguous copies of the KSHV TR region inserted into the NotI site of the pCRII vector (Invitrogen) (courtesy of R. Renne). The BKS plasmid (N103) is a pBSII-KS vector purchased from Stratagene. The LANA expression plasmid (N811) was constructed by cloning the LANA gene into the HindIII and XbaI sites of the p3XFlag-myc-CMV-24 vector (Sigma). LANA was also expressed without any epitope tag from pA3 M-LANA (a gift of Erle Robertson, University of Pennsylvania).
ChIP assay.
Chromatin immunoprecipitation (ChIP) was based on the protocol by Upstate Biotechnology, Inc., and was performed as described previously (18, 44). ChIP assays were each performed with ∼1 × 106 BCBL1 cells or 293 cells transfected with the 2 × TR and LANA expression plasmids. ChIP DNA was analyzed by real-time PCR (ABI Prism 7000 sequence detection system; Applied Biosystems) and primers designed with computer software (Primer Express, version 2.0; Applied Biosystems) as previously described (30, 31, 63). Briefly, 5 μl of ChIP DNA and SYBR Green PCR master mix (Applied Biosystems) were combined with amplification primers in a 25-μl reaction mixture. Standard curves for each primer set were constructed by 5 twofold serial dilutions of the input DNA for each IP assay. No-template controls incorporated 5 μl of water in place of DNA. Amplification levels were compared to a preamplified baseline value. The threshold cycle (CT) line was set above the amplification noise in the linear part of the logarithmic growth curve, and all derived amplification values were normalized to the baseline. Standard curves, based on the starting copy number of the target sequence versus the CT at which the fluorescence is first detected above the baseline value, were only considered valid if the slopes were between the values of −3.32 and −3.90. Dissociation curve analysis was simultaneously performed to determine melting temperatures (Tm) of nucleic acid sequences, to verify that amplification signals resulted only from the target DNA species. Result values were calculated using the CT value for the sample and the standard curve. Reported results were normalized to the values of the input controls that were quantitated in parallel for each experiment. The percentage of input partly reflects antibody efficiency for precipitation at each amplified sequence. ChIP assays included rabbit polyclonal antibodies to MeH3K9 (Abcam), immunoglobulin G (IgG) (Santa Cruz Biotechnology), MCM3 (Abcam), HBO1 (for histone acetyltransferase binding to ORC1) (Zymed), ORC2 (PharMingen), CBP (Santa Cruz Biotechnology), and AcH3, AcH3K9, AcH4, MeH3K4, and MCM5 (Upstate Biotechnology). A rat monoclonal antibody for orf73 (LANA) was purchased from Advanced Biotechnology, Inc., while a goat polyclonal antibody for BRD2 was purchased from Bethyl, Inc. A complete list of primers is described in Fig. S1 in the supplemental material.
DNA affinity purification assay.
Plasmids 2 ×TR (N700) and N103 (pBSIIKS vector) were linearized with XhoI and biotinylated by a fill-in reaction with the Klenow fragment of DNA polymerase and 0.2 mM deoxynucleoside triphosphates (lacking dCTP) and brotinylated dCTP (Invitrogen). The labeled plasmid was then phenol-chloroform extracted, precipitated with ethanol, and digested with XbaI/EcoV for N700 or with Asp718/PvuI for N103 to generate a 1.6-kb linear fragment biotinylated at one end. The resulting fragments were purified with the Qiaquick PCR purification kit (QIAGEN) to remove unincorporated nucleotides and then bound to Streptavidin M-280 dynabeads (Dynal) according to the manufacturer's instructions (Dynal). The DNA dynabeads were then incubated with dialyzed nuclear extract (22) from BCBL1 cells for 1 h. Samples were centrifuged and washed three times for 5 to 10 min each time in 500 μl of D150 (150 mM KCl, 20 mM HEPES [pH 7.9], 0.2 mM EDTA, 20% glycerol) with 0.05% NP-40, 5 mM beta-mercaptoethanol, and protease inhibitor cocktail (Sigma). The protein-bead complexes were then boiled in sodium dodecyl sulfate lysis buffer and analyzed by Western blotting with the appropriate antibody. Western blots were visualized with the ECL chemiluminescence reagent (Amersham).
Micrococcal nuclease laddering assay.
BCBL1 cells were either untreated or treated with mimosine and colchicine and then subjected to nuclear extraction as previously described (43, 44). Briefly, the cells were harvested and disrupted with a Dounce homogenizer in lysis buffer (0.3 M sucrose, 2 mM magnesium acetate, 3 mM CaCl2, 1% Triton X-100, and 10 mM HEPES [pH 7.9]). The lysate was then spun through a pad (25% glycerol, 5 mM magnesium acetate, 0.1 mM EDTA, and 10 mM HEPES [pH 7.4]) at 1,000 × g for 15 min at 4°C. The nuclei (2 × 107) were then partially digested with a range of micrococcal nuclease (MNase; 30 U/μl) at 37°C for 10 min in 100 μl of digestion buffer (25 mM KCl, 4 mM MgCl2, 1 mM CaCl2, 50 mM Tris [pH 7.4], and 12.5% glycerol). MNase concentrations ranged from 15 to 120 U for each 100 μl of reaction mixture, as indicated for each experiment. The reaction was stopped by adding an equal volume of stop buffer (2% sodium dodecyl sulfate, 0.2 M NaCl, 10 mM EDTA, 10 mM EGTA, 50 mM Tris [pH 8.0], and 100 μg of proteinase K/ml) for 2 h at 50°C. The resulting DNA was extracted and purified by phenol-chloroform extraction, followed by ethanol precipitation. The purified DNA was electrophoresed on a 1.5% agarose gel, transferred to Zeta-Probe blotting membranes (Bio-Rad), and hybridized with radiolabeled probe (57). The TR probe was made by digesting plasmid N700 with NotI restriction enzyme, followed by gel purification of the TR fragment. The LBS probe was constructed by digestion of the pGC-LBS (pJH10 was obtained courtesy of R. Renne) vector with BamHI and EcoRI, containing the LBS (nucleotides [nt] 509 to 644 of the KSHV TR), followed by gel purification. The ORF50 probe was constructed by amplifying a 298-bp region from the ORF50 promoter by conventional PCR (18, 44). All probe DNA was labeled with the Random Primed DNA labeling kit (Roche) with α-32P-labeled dATP nucleotides and incubated for 30 min at 37°C. The membrane was developed with a PhosphorImager (Molecular Dynamics) and analysis software (ImageQuant).
Nucleosome mapping by indirect end labeling.
Indirect end labeling was performed essentially as described previously (29, 54). Nuclei (1.5 ×107 per reaction mixture) from asynchronous growing BCBL1 cell cultures were isolated as described above and digested with MNase I (300, 1,500, and 7,500 U/ml) at 37°C for 2 min followed by the addition of stop buffer and proteinase K for 2 h at 50°C. DNA was extracted with phenol-chloroform, ethanol precipitated, and subjected to restriction digestion with 500 U of NotI/ml or 1,000 U of DraIII/ml at 37°C for 16 h. Addition of EcoRI (250 U/ml), which does not cleave within the TR, was included to help solubilize genomic DNA. Control DNA from nuclei not treated with MNase I was purified as described above and subjected to identical restriction digestion. Some of the control DNA was subject to MNase treatment with 6 or 60 U of MNase I/ml at 37°C for 5 min as indicated (see Fig. 4). DNA was further purified by phenol-chloroform extraction and ethanol precipitation and analyzed by Southern blotting of 1.7% agarose gels. Restricted DNA was then indirectly end labeled with single-stranded oligonucleotides complementary to the corresponding 5′ or 3′ ends of the NotI or DraIII restriction fragments of the KSHV TR. The probes for the TR region consisted of 5′-NotI (nt 1 to 55) (GCGGCCGCATCCCCCCCCCCTGTTTTACCCCCCCGGGGGGGCGCGCGCGGGCCGC), 3′-NotI (nt 809 to 754) (AAAAGAGGCGGCGCCCGCGGCGGCGGCGGCGCTTGCTTTCGTTTTCTCCCGCGGCC), 5′-DraIII (nt 394 to 448) (TCCAGGGCTCCACGTAGCAAGCACTGAGGAGGCTCCCCCAAACAGGCTCACACAC), and 3′-DraIII (nt 378 to 324) (GAGGCGAGCGGGGGGAGGGGGAGGGGCGGCGCGGGGTGGGGGGCGCGGGGCGCGG). Oligonucleotides were labeled with the DIG Oligonucleotide 3′-end labeling kit, second generation (Roche), according to the manufacturer's specifications. Southern blotting and hybridization were performed as described above.
FIG. 4.
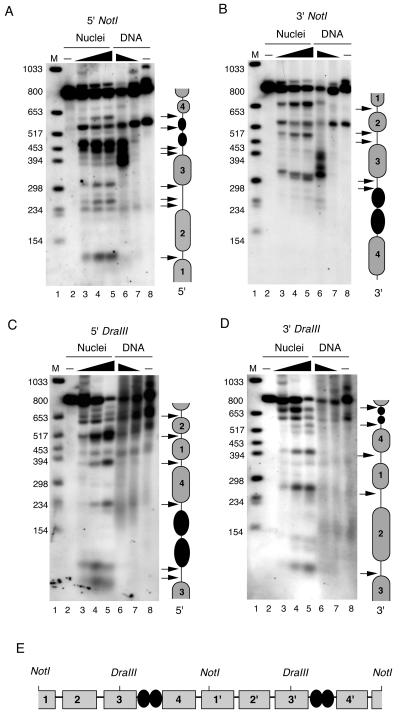
Nucleosome mapping by indirect end labeling. BCBL1 cell nuclei (lanes 2 to 5) or purified DNA (lanes 6 to 8) were treated with various concentrations of MNase I nuclease and subject to indirect end-labeling analysis. The number of MNase U/100 μl of reaction mixture was as follows: 30 (lane 3), 150 (lane 4), 300 (lane 5), 6 (lane 6), 0.6 (lane 7), and 0 (lanes 2 and 8). MNase I-treated DNA was purified and subjected to restriction digestion with NotI (A and B) or DraIII (C and D). Restricted DNA was then subjected to Southern blot analysis and probed with 55-bp oligonucleotides specific for the termini generated at 5′ NotI (A), 3′ NotI (B), 5′ DraIII (C), or 3′ DraIII (D). Interpretations of data are indicated to the right of each panel. Presumed nucleosomes are indicated by grey spheres, and LANA is indicated by black spheres. Arrows indicate major MNase I cleavage sites. (E) Summary of predicted nucleosome positions for two tandem TRs.
DNA replication assay.
The DNA replication assay was performed as described previously (18, 44). Briefly, 293 cells were transfected with small interfering RNA (siRNA) control or siRNA against ORC2, MCM5, or HBO1. After 6 h, transfected cells were replated and cultured for another 16 h. Cells were transfected again with TR plasmid (2 μg), a plasmid expressing Flag-LANA (2 μg), and comparable siRNA (100 nM). At 24 h posttransfection, cells were detached by trypsin, washed twice with phosphate-buffered saline, replated onto 10-cm plates, and cultured for another 2 days. Plasmid DNA was extracted by a modified Hirt method (Invitrogen). The nucleic acid pellets were dissolved in 30 μl of 10 mM Tris (pH 7.8)-1 mM EDTA, and DNA was subjected to restriction digestion with DpnI and BamHI or with BamHI alone. DNA samples were analyzed on 0.7% agarose gels in the absence of ethidium bromide and transferred to Zeta-Probe blotting membranes (Bio-Rad) prior to Southern blotting analysis. Radiographic images were quantified by PhosphorImager analysis. Replication was measured as a function of DpnI-resistant DNA relative to total DNA recovered after BamHI digestion. MCM5 and ORC2 siRNA smart pools were available from Dharmacon, Inc. The siRNA for HBO1 was synthesized by Dharmacon and contained the targeting sequence AAUCUCGAGCACACAGACAGU.
RESULTS
Chromatin organization of the KSHV genome.
To better understand the nucleoprotein and chromatin organization of the KSHV genome in vivo, we used the ChIP assay to determine the distribution of histone modifications and key DNA-protein-interactions (Fig. 1). Real time PCR was used to assay 11 regions of the KSHV genome in BCBL1 PEL cells (Fig. 1A). We first analyzed the KSHV genome for histone H3 and H4 acetylation and for H3 K4 and K9 methlyation (Fig. 1B). Remarkably, we found that acetyl histone H3 was highly enriched at the TR (Fig. 1B, lanes K). Histone H4 acetylation was also enriched at TR, as well as at a region upstream of the K11 promoter and adjacent to a 145-bp tandem repeat. Most of the KSHV genome was subject to histone H3 K4 methylation, with the exception of regions upstream of ORF22 (lanes D) and upstream of the K12 (kaposin) promoter (lanes I). We did not detect significant levels of H3 K9 methylation in these experiments. Together, these data suggest that the KSHV latent genome is organized into distinct chromatin domains marked by variations in histone tail modifications. The most marked distinction was that of histone H3 hyperacetylation at the TR (lanes K).
FIG. 1.

Chromatin organization of the KSHV genomes. (A) Eleven primer pairs were designed to sample regions of the KSHV genome indicated by capital letters above each locus. The amplified regions are delineated by grey bars and KSHV coordinates (53). (B) Real-time PCR analysis of ChIP assays with antibodies to AcH3, AcH3K9, AcH4, MeH3K4, or MeH3K9 as indicated in each graph. Values were reported as percent input and reflect the average of at least three independent experiments. (C) The same analyses as appear in panel B, except with antibodies specific for LANA, ORC2, BRD2, CBP, or control IgG, as indicated for each graph. Letters below each bar correspond to the KSHV loci depicted in panel A.
The ChIP assay was also used to investigate the association of nonhistone proteins with the KSHV genome (Fig. 1C). LANA was found to be associated exclusively with the TR (Fig. 1C, lanes K). Cellular replication factor ORC2 had a distribution pattern identical to that of LANA and associated exclusively with the TR. We also found LANA-associated proteins, CBP and BRD2, were enriched at TR but could also be found at the ORF50 promoter (Fig. 1C, lanes G). Identical results were found in latently infected JSC1 cells, indicating that these findings were not peculiar to one cell type (see Fig. S2 in the supplemental material). These results confirm previous findings that LANA can interact with ORC2, CBP, and BRD2. These results confirm earlier studies implicating LANA binding to TR in vivo (24), and further demonstrates that CBP, BRD1, and ORC2 localize to the latent replication origin within the KSHV TR in latently infected cells.
Cell cycle-associated chromatin modifications at TR.
Cellular replication origins are subject to cell cycle-dependent changes in protein complex assembly and chromatin structure. To determine if such changes occur at the KSHV latent replication origin, we compared ChIP profiles of asynchronous PEL cells with cultures arrested in late G1 with mimosine or G2/M with colchicine (Fig. 2). We found that histone acetylation did not change significantly at these different stages of the cell cycle. Interestingly, we found that histone H3 K4 methylation was significantly reduced (∼7-fold) at the TR in cells arrested in late G1/S by mimosine (Fig. 2B). Mimosine treatment did not produce a similar reduction in H3 K4 methylation at the two other regions of the KSHV genome examined, suggesting the changes at TR were locus specific.
FIG. 2.

Cell cycle-dependent histone modifications and MCM protein binding at the KSHV TR. (A) Fluorescence-activated cell sorter analysis of propidium iodide-treated BCBL1 cells grown as asynchronous cultures or arrested in late G1 with mimosine or in G2/M phase with colchicine. (B) Real-time PCR analysis of ChIP assays with antibodies to AcH4, AcH3, MeH3K4, LANA, ORC2, MCM3, or IgG at KSHV loci A, G, and K as depicted in Fig. 1A. Asynchronous (black), mimosine-arrested (grey), and colchicine-arrested (hatched) cultures are indicated in the bar graph. (C) Western blot analysis of total cellular proteins derived from asynchronous (ASYN), mimosine-arrested (G1), or colchicine-arrested (M) cells probed with antibodies to LANA or β-actin, as indicated.
EBNA1 has been shown to bind to the EBV OriP throughout the cell cycle (32), and it was anticipated that LANA would also remain bound to TR throughout the cell cycle. To our surprise, we found that LANA binding to TR was substantially reduced (∼3-fold) in both mimosine- and colchicine-arrested cells. LANA protein levels were also slightly reduced in colchicine-arrested cells (Fig. 2C), suggesting this may account for some of the reduced LANA binding to TR in M-phase-arrested cells. ORC2 binding did not change, and MCM3 binding was significantly elevated (∼4-fold) in mimosine-arrested cells. The binding of LANA, ORC2, and MCM3 was specific for TR relative to other regions of the genome and with respect to low levels of control IgG binding. These results suggest that the KSHV TR replication origin is subject to cell cycle regulation, similar to cellular DNA replication origins and similar to OriP of EBV.
Nucleosome organization at the KSHV TRs.
The TR region of KSHV consists of ∼809-bp tandem repeats that are G+C rich. Repetitive cellular DNA sequences are often subject to formation of higher-ordered chromatin structure, like heterochromatin, that silences gene expression and restricts DNA binding protein accessibility. The chromatin structure of the KSHV replication origin was examined by micrococcal nuclease (MNase I) digestion patterns (Fig. 3A). We found that the MNase digestion of the TR produced a highly organized ladder reflective of tandem arrays of phased nucleosomes. A similar pattern was found when MNase digested DNA was probed with the 150-bp LBS found within the 809-bp TR. Interestingly, the LBS probe did not detect mononucleosomal bands at ∼146 bp, and the highest concentration of MNase produced a band of ∼350 bp. This suggests that a 350-bp MNase-resistant nucleoprotein complex protects the LBS, consistent with the binding of LANA and other cellular replication proteins. In contrast, mononucleosomal bands were found with the TR and the ORF50 promoter region, which has been shown to have a single positioned nucleosome protecting the transcriptional initiation site (44). These results suggest that chromatin structure at the TR consists of regularly spaced nucleosomes disrupted at the LBS region within each TR segment.
FIG. 3.
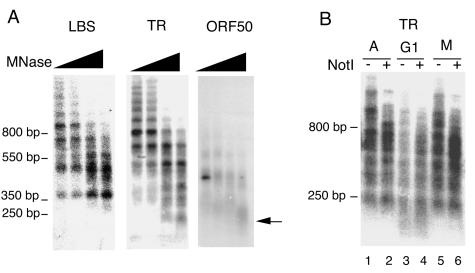
Nucleosome organization at TR. (A) Southern blots of MNase I-digested BCBL1 nuclei. Nuclei were digested with a twofold serial increase in MNase I, ranging from 15 to 120 U/100 μl of reaction mixture as indicated above. Extracted DNA was probed with sequences specific for the LBS, TR, or ORF50 region as indicated above each blot. The arrow indicates mononucleosome fragments of ∼147 bp. (B) MNase I digestions of asynchronous (A), mimosine-arrested (G1), or colchicine-arrested (M) BCBL1 cells were probed with sequences specific for the TR. MNase I-digested samples were cut with NotI (even-numbered lanes) or not restricted (odd-numbered lanes) prior to electrophoresis.
We next examined whether the ordered chromatin structure at the TR was subject to change in cell cycle-arrested PEL cells (Fig. 3B). We found that the MNase pattern in G1-arrested cells (mimosine treatment) led to a decreased intensity of the MNase pattern, suggesting that DNA became more accessible to enzymatic digestion. These results suggest that the stable chromatin pattern at TR is altered during late G1.
To more precisely map nucleosome positions at TR, we used the indirect end-labeling method (Fig. 4) (29, 54). BCBL1 cell nuclei were analyzed by MNase I digestion (Fig. 4A to D, lanes 3 to 5) and compared to the MNase I digestion of purified genomic DNA (lanes 6 and 7). MNase I digestion patterns over TR were first analyzed by NotI restriction digest and indirect end labeling with a 55-bp probe to the 5′ end of the NotI site (Fig. 4A). MNase I protection was observed at the regions 300 to 450 bp from the NotI 5′ end (Fig. 4A, lanes 3, 5, and 6). This ∼150-bp protected region most likely corresponds to a nucleosome (designated nucleosome 3). Strong MNase I-hypersensitive sites in nuclei, but not naked genomic DNA, were indicated by the arrows and most likely correspond to linker regions between nucleosomes. Analysis with the NotI 3′ probe showed clear MNase I protection occurring ∼350 to 520 bp from the 3′ end of the NotI site (Fig. 4B). This ∼170-bp protected area corresponds to nucleosome 3, which lies immediately 5′ of the LBSs at nt 560 to 620 relative to the NotI 5′ end (25).
Since MNase I protection was not immediately obvious for the other presumed nucleosomes, we performed indirect end labeling with DraIII restriction digestion, which cuts at position 378 relative to the 5′ NotI site (Fig. 4C and D). MNase I protection was observed over sequences between ∼234 to 394 and to a lesser extent over sequences between ∼500 to 650 relative to the 5′ DraIII site (Fig. 4C). These protected regions most likely correspond to nucleosomes (designated nucleosomes 4 and 2, respetively). Similar protection could be observed for the regions ∼100 to 270 nt and to a lesser extent from ∼270 to 420 nt relative to the 3′ DraIII site (Fig. 4D). These protected regions most likely correspond to nucleosomes (designated nucleosomes 2 and 1, respectively). A summary of this nucleosome-positioning analysis is shown with a schematic of two tandem TR elements (Fig. 4E). Our data are consistent with the interpretation that a single TR consists of four nucleosomes in addition to the two LBSs.
LANA recruits ORC and MCM components to TR in vivo.
LANA is required for DNA replication and episomal maintenance of plasmids containing at least two TRs. We next determined whether LANA expression was required for the recruitment of ORC components to the TR (Fig. 5). ChIP was performed on 293 cells transiently transfected with 2 × TR (Fig. 5) and a LANA expression vector. We found LANA bound to the TR region of the plasmid but did not bind to the plasmid AMP gene. We also found that ORC2 bound to TR only in cells transfected with LANA but not with cells transfected with vector alone. ORC2 bound with similar sequence specificity as LANA, suggesting that it was recruited to TR in a LANA-dependent manner. Similarly, we found that MCM3 was specifically associated with TR in LANA-transfected cells, suggesting that LANA recruits both ORC and MCM complex components to the TR element. In contrast to ORC and MCM components, histone modifications observed at the viral genome TR were not recapitulated in transient transfection assays with plasmid-based TR sequence. Histone H3 acetylation was not enriched at TR relative to AMP sequences in the absence or presence of LANA coexpression (Fig. 5). These findings suggest that transiently transfected plasmids can assemble ORC and MCM complexes in a LANA-dependent manner but do not recapitulate the same pattern of histone H3 hyperacetylation observed at genomic TR in PEL cells latently infected with KSHV.
FIG. 5.

LANA recruits ORC to TR. 293 cells transfected with 2 × TR plasmid and either LANA expression vector (black bars) or empty vector (grey bars) were then assayed by ChIP assay and real-time PCR. PCR primers specific for the LBS region of TR or the AMP gene of the plasmid were analyzed by ChIP assay with antibodies specific for LANA, ORC2, MCM3, AcH3, and IgG. Data from at least three independent ChIP experiments were normalized as the percentage of total input DNA recovered for each antibody.
ORC and histone acetylases HBO1 and CBP bind TR in vitro.
As a complementary approach to ChIP, we analyzed cellular protein assembly on 2 × TR DNA fragments by DNA affinity purification from nuclear extracts (Fig. 6). BCBL1 cell nuclear extract was incubated with DNA from 2 × TR or with a 1.6-kb control fragment from pBKSII plasmid DNA (BKS) and analyzed by Western blotting with various antibodies. As expected, we found that LANA bound specifically to 2 × TR but not to BKS. We also found evidence that ORC2, BRD2, and CBP bound specifically to 2 × TR but not to BKS by this method. Interestingly, MCM3 protein did not associate with 2 × TR by this method, suggesting that additional licensing events must take place to recapitulate cell cycle-dependent binding in vitro. Telomere repeat binding factor 2, which binds the EBV origin of replication, did not bind to 2 × TR, suggesting that these two replication origins associate with different cellular factors.
FIG. 6.

HBO1 binds TR in vitro and in transfected cells. (A) DNA affinity purification assays with BCBL1-derived nuclear extracts were compared for binding to 2 × TR or BKS DNA. Eluted proteins were analyzed by Western blotting with antibodies specific for LANA, HBO1, ORC2, BRD2, CBP, MCM3, telomere repeat binding factor 2, or nonspecific control rabbit IgG as indicated and visualized by enhanced chemiluminescence. (B) ChIP assay of transiently transfected 293 cells with FLAG-HBO1 and untagged LANA expression vectors. Primer amplifications for TR and AMP genes were quantitated and presented as the increase over IgG for each IP reaction.
The DNA affinity assay was also used to demonstrate that other cellular factors can bind 2 × TR with high specificity. We reasoned that additional histone acetyltransferases are likely to be involved in establishing histone H3 hyperacetylation at the TR in PEL cells. The HBO1 histone acetyltransferase is a Myst family member that interacts with both ORC and MCM complexes and is therefore a likely candidate to associate with the KSHV replication origin in the TR (6, 34). Using the DNA affinity purification procedure, we found that the HBO1 was highly enriched in 2 × TR affinity-purified fractions but not with BKS (Fig. 6A). This robust association of HBO1 with 2 × TR suggests that HBO1 is partly responsible for histone H3 hyperacetylation at TR. To verify the interaction of HBO1 with TR in vivo, we attempted unsuccessfully to assay by ChIP with HBO1-specific monoclonal antibodies in KSHV-infected PEL cell lines. As an alternative, we expressed pCMV-FLAG-HBO1 and 293 cells cotransfected with 2 × TR in the presence or absence of LANA. We found that FLAG-HBO1 associated specifically with the TR plasmid sequence in cells coexpressing LANA (Fig. 6B). These results indicate that HBO1 can associate with TR sequence in vivo and that LANA facilitates this interaction.
Functional requirement for ORC, MCM, and HBO1 in LANA-dependent DNA replication.
The functional contribution of cellular TR binding proteins was determined by DNA replication assays and siRNA depletion experiments (Fig. 7). ORC2, MCM5, and HBO1 were targeted for degradation with siRNA (Fig. 7C). These siRNA had no detectable effect on PCNA or FLAG-LANA expression (Fig. 7C). LANA-dependent DNA replication of 2 × TR was assayed in 293 cells (Fig. 7A and B). We found that siRNA bound to MCM5 produced the most significant reduction in TR replication to 30% of wild-type levels, while siRNA directed against ORC2 and HBO1 inhibited replication ∼2.5-fold 72 h posttransfection. These results suggest that MCM5, ORC2, and HBO1 contribute to LANA-mediated DNA replication of 2 × TR and further suggest that these cellular factors regulate KSHV latent cycle replication.
FIG. 7.
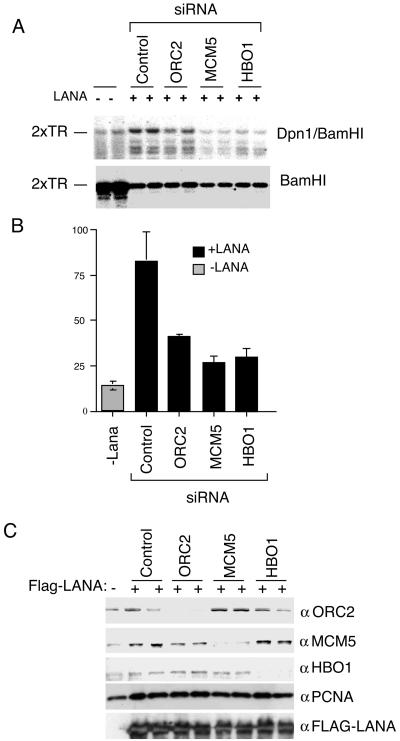
ORC, MCM, and HBO1 contribute to LANA-dependent DNA replication. (A) Transient DNA replication assay of 2 × TR-containing plasmid in 293 cells transfected with LANA expression vectors (+) or with control vector (−). Transfections also contained siRNA directed against ORC2, MCM5, HBO1, or control luciferase as indicated above. Replicated plasmid DNA was analyzed by DpnI resistance and Southern blotting. (B) Quantification of three independent replication assays is indicated in the bar graph. (C) Western blot analysis of ORC2, MCM5, and HBO1 siRNA knockdown experiments. Transfected cell extracts were analyzed with antibodies specific for siRNA-targeted proteins ORC2, MCM5, and HBO1 or control proteins PCNA and FLAG-LANA, as indicated to the right of each panel.
DISCUSSION
Chromatin organization of the KSHV genome.
Packaging of eukaryotic chromosomes into higher-order structures is critical for safeguarding the genome and regulating genetic processes like transcription, DNA replication and repair, chromosome condensation, and segregation (52). The contribution of chromatin structure to gene regulation and plasmid maintenance of the KSHV latent genome is poorly understood. In this work, we investigated the chromatin structure, histone modifications, and regulatory protein interactions with the KSHV genome in latently infected PEL cells. We found that KSHV latent cycle DNA replication origin in the viral TR was enriched in acetylated histone H3. This hyperacetylation at histone H3 was exclusively restricted to the LANA binding region within TR (locus K), since a region 20 bp external to the TR (locus A) was not enriched in acetylated histones. MNase I digestion patterns of TR revealed that nucleosomes were organized in ordered arrays throughout most of the TR, with the exception of the region covering the LBS (Fig. 2C). Indirect end-labeling experiments suggested that four nucleosomes are positioned at TR in addition to the LBSs (Fig. 4). Based on these findings, we propose that the 809-bp TR is organized with four nucleosomes separating the LBS from each tandem repeat. Genetic evidence indicates that at least two TR are required for plasmid maintenance function, and it seems likely that nucleosome packing may facilitate interaction between LBS sites positioned every 809 bp (Fig. 8). While tandem repeats tend to form repressive heterochromatin, the central region of the KSHV TR is maintained in a hyperacetylated state, which presumably facilitates DNA replication factor assembly.
FIG. 8.
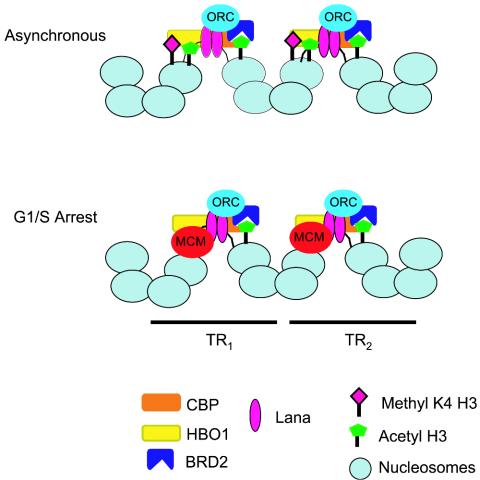
Model of the KSHV latent replication origin at TR. Four nucleosomes are predicted to bind between tandem LBS. The nucleosomes adjacent to LBS have high levels of histone H3 acetylation, maintained by HBO1 and CBP acetyltranferases and the acetyl lysine binding activity of BRD2. G1/S-arrested cells correlate with a disruption of nucleosome structure at the LBS, MCM2 binding, and the loss of H3MeK4.
Cellular origin binding factors assemble at TR and are required for LANA-dependent replication.
Recent studies with EBV revealed that ORC and MCM proteins assemble at OriP. We now show that components of the ORC and MCM complex assemble at or near the LBS of the TR in cells latently infected with KSHV. The ORC complex bound to TR consistently throughout the cell cycle. In contrast, the MCM complex was enriched at the TR in cells arrested in late G1 with mimosine. Knockdown experiments with siRNA revealed that ORC2 and MCM5 were critical for LANA-dependent DNA replication of 2 × TR (Fig. 7). MCM binding and dissociation are hallmarks of replication origin licensing, and our data suggest that KSHV latent replication is subject to the same or similar regulation as most cellular origins (3).
Surprisingly, we also found that LANA binding to the TR was reduced in mimosine- and colchicine-arrested cells (Fig. 2B). LANA protein levels also decreased during M phase, suggesting that LANA expression and activity may be under cell cycle control. Since LANA is thought to be essential for tethering the viral genome to the cellular chromosome during mitosis, it is unlikely that LANA can be completely dissociated from the LBS at these critical periods in the cell cycle. It is also possible that the decrease in ChIP-detected LANA at TR may be a consequence of changes in protein complex with LANA at TR during these stages of the cell cycle. These changes may mask epitopes essential for IP and therefore inhibit detection by ChIP assay.
ORC was found to associate with 2 × TR in vivo (Fig. 1, 2, and 5) and in vitro (Fig. 6). Others have shown that ORC components can interact with LANA (41) and have shown by ChIP assay that LANA can bind to TR in vivo (24). Our results indicate that ORC and LANA bind throughout most of the cell cycle to the TR in PEL cells latently infected with KSHV. LANA and ORC binding was relatively specific for TR, since none of the other 10 amplified regions showed significant interactions with these two factors (Fig. 1). We also found that ORC bound specifically to TR in vitro (Fig. 6). These results are in contrast to other studies that suggest that human ORC possesses nonspecific DNA binding activity (62). Several significant differences in experimental design may explain these seemingly contradictory findings. In particular, Vashee et al. (62) found that purified, recombinant ORC could bind DNA nonspecifically, while we found that endogenous ORC from nuclear extracts bound specifically to 2 × TR in reaction mixtures with a gross excess of competitor DNA. We suggest that ORC is recruited to templates in vivo based primarily on the DNA binding protein composition of the templates. LANA and other TR-specific binding proteins are likely to contribute to the specific recruitment of ORC in these in vitro binding assays. Future experiments with recombinant proteins can resolve these apparent differences.
Maintenance of histone acetylation at the LBS.
In addition to the binding of ORC and MCM at the LBS, we also present evidence that LANA-associated proteins BRD2 and CBP interact with the LBS in vivo. CBP is a histone acetyltransferase that may contribute to the high level of acetylated histone H3 near the LBS. BRD2 is a double-bromodomain-containing protein that is likely to bind acetylated lysines and may contribute to the maintenance and stability of acetylated histones adjacent to the LBS. We also found evidence from in vitro DNA binding assays that HBO1 can be recruited to 2 × TR (Fig. 6). HBO1 is a Myst family histone acetyltransferase that was isolated in two-hybrid screens with ORC1 (34) and in an independent screen with MCM2 (6). A functional role for HBO1 in TR replication was established by the use of siRNA in transient replication assays (Fig. 7). HBO1 is a likely candidate for regulation of KSHV TR replication, since it is known to associate with cellular origin binding factors like ORC and MCM and since our data revealed that TR is hyperacetylated throughout most of the cell cycle. We suggest that HBO1 is the more abundant histone acetyltransferase at TR, based on the DNA affinity experiments, and is more likely responsible for maintaining the high constitutive levels of H3 hyperacetylation near the LBS than the weaker associated CBP.
The histone acetylation at the LBS did not spread 358 bp to the K1 ORF (locus A), suggesting that the hyperacetylation is highly restricted to the region adjacent to the LBS. We were not able to generate primers that would amplify the G+C-rich sequence outside the LBS in the TR, and therefore we could not readily determine the precise nucleosomal boundary of the H3 acetylation. A recent study demonstrated that heterochromatin protein 1 and histone H3 K9 methylation can also be detected at the KSHV TR and the K1 ORF neighboring regions (56). These findings are consistent with our observation that nucleosomes are highly organized in the TR and likely to form heterochromatin. We suspect that chromatin boundary proteins may restrict the hyperaceylated histone H3 to nucleosomes adjacent to LBS and facilitate higher-order chromatin structure necessary for the formation of an active replication origin in the TR. The combination of these origin binding factors, histone acetyltransferases and bromodomain proteins, are likely to create a region of histone H3 hyperacetylation in an otherwise barren region of condensed heterochromatin. These histone modifications and binding proteins may also facilitate interactions between the LBS of each tandem repeat in the TR (Fig. 8).
Replication origins and chromatin regulation.
Cell cycle changes in chromatin structure and modification may regulate replication at the TR. We found that MCM3 binding was enriched in cells arrested in late G1 with mimosine. This correlated with a specific loss of histone H3 K4 methlyation at the LBS (Fig. 2). Loss of H3 K4 methylation was not seen at other regions of the KSHV genome, and no other histone modification was reduced at TR under these conditions. This suggests that histones with H3 K4 methylation are exchanged with unmethylated histones through some remodeling process or that histone H3 K4 is demethylated at G1/S in the vicinity of the LBS. To date, there is no evidence for a histone H3 K4 demethylase or cell cycle-coordinated loss of histone H3 K4 methylation. Future experiments will attempt to distinguish between histone demethylation and histone exchange at the KSHV origin in late G1/S and whether this contributes directly to MCM loading on TR in late G1.
The chromatin architecture of the KSHV latent origin may be a suitable model of origin site selection in cellular heterochromatin or highly repetitive regions of the genome. The KSHV TR origins have some similarity to the origins found in the ribosomal DNA clusters of budding yeast. Origin selection in the yeast ribosomal DNA clusters is subject to SIR2-based epigenetic regulation that limits utilization to ∼20% of the potential origins (5, 42, 49). It is not known whether KSHV TRs utilize more than one of the potential origins within each TR in the viral genome. In contrast to KSHV, the latent replication origin of EBV is within a short unique region of the viral genome adjacent to an active RNA polymerase III transcript that is likely to maintain an open chromatin structure in most latent infections. While KSHV and EBV share the common use of cellular replication origin binding and licensing factors, it seems likely that these two viral origins employ different chromatin modifying and regulatory activities. These findings also suggest that cellular origins may be equally diverse and utilize different regulatory strategies, depending on cell type, genome location, and chromatin environments. These findings also have implications for our understanding of the establishment and maintenance of KSHV latent infection.
Supplementary Material
Acknowledgments
We thank Rolf Renne and Erle Robertson for the generous gift of plasmids and antibodies and the Wistar Institute Cancer Center Core Facilities for Cell Sorting.
This work was funded in part from grants from the NIH (CA 93606 and CA 85678 to P.M.L.) and from the Leukemia-Lymphoma Society (to Z.D.).
Footnotes
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Ballestas, M. E., P. A. Chatis, and K. M. Kaye. 1999. Efficient persistence of extrachromosomal KSHV DNA mediated by latency-associated nuclear antigen. Science 284:641-644. [DOI] [PubMed] [Google Scholar]
- 2.Ballestas, M. E., and K. M. Kaye. 2001. Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen 1 mediates episome persistence through cis-acting terminal repeat (TR) sequence and specifically binds TR DNA. J. Virol. 75:3250-3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blow, J. J., and B. Hodgson. 2002. Replication licensing—defining the proliferative state? Trends Cell Biol. 12:72-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boshoff, C., T. F. Schulz, M. M. Kennedy, A. K. Graham, C. Fisher, A. Thomas, J. O. McGee, R. A. Weiss, and J. J. O'Leary. 1995. Kaposi's sarcoma-asociated herpesvirus infects endothelial and spindle cells. Nat. Med. 1:1274-1278. [DOI] [PubMed] [Google Scholar]
- 5.Brewer, B. J., and W. L. Fangman. 1991. Mapping replication origins in yeast chromosomes. Bioessays 13:317-322. [DOI] [PubMed] [Google Scholar]
- 6.Burke, T. W., J. G. Cook, M. Asano, and J. R. Nevins. 2001. Replication factors MCM2 and ORC1 interact with the histone acetyltransferase HBO1. J. Biol. Chem. 276:15397-15408. [DOI] [PubMed] [Google Scholar]
- 7.Burysek, L., and P. M. Pitha. 2001. Latently expressed human herpesvirus 8-encoded interferon regulatory factor 2 inhibits double-stranded RNA-activated protein kinase. J. Virol. 75:2345-2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cesarman, E. 2002. The role of Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8) in lymphoproliferative diseases. Recent Results Cancer Res. 159:27-37. [DOI] [PubMed] [Google Scholar]
- 9.Cesarman, E., Y. Chang, P. S. Moore, J. W. Said, and D. M. Knowles. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 332:1186-1191. [DOI] [PubMed] [Google Scholar]
- 10.Chang, Y., E. Cesarman, M. S. Pessin, F. Lee, J. Culpepper, D. M. Knowles, and A. P. S. Moore. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266:1865-1869. [DOI] [PubMed] [Google Scholar]
- 11.Chaudhuri, B., H. Xu, I. Todorov, A. Dutta, and J. L. Yates. 2001. Human DNA replication initiation factors, ORC and MCM, associate with oriP of Epstein-Barr virus. Proc. Natl. Acad. Sci. USA 98:10085-10089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chittenden, T., S. Lupton, and A. J. Levine. 1989. Functional limits of oriP, the Epstein-Barr virus plasmid origin of replication. J. Virol. 63:3016-3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Collins, C. M., M. M. Medveczky, T. Lund, and P. G. Medveczky. 2002. The terminal repeats and latency-associated nuclear antigen of herpesvirus saimiri are essential for episomal persistence of the viral genome. J. Gen. Virol. 83:2269-2278. [DOI] [PubMed] [Google Scholar]
- 14.Collins, C. M., and P. G. Medveczky. 2002. Genetic requirements for the episomal maintenance of oncogenic herpesvirus genomes. Adv. Cancer Res. 84:155-174. [DOI] [PubMed] [Google Scholar]
- 15.Cotter, M. A., II, and E. S. Robertson. 1999. The latency-associated nuclear antigen tethers the Kaposi's sarcoma-associated herpesvirus genome to host chromosomes in body cavity-based lymphoma cells. Virology 264:254-264. [DOI] [PubMed] [Google Scholar]
- 16.Cotter, M. A., II, and E. S. Robertson. 2002. Molecular biology of Kaposi's sarcoma-associated herpesvirus. Front. Biosci. 7:d358-d375. [DOI] [PubMed] [Google Scholar]
- 17.Cotter, M. A., II, C. Subramanian, and E. S. Robertson. 2001. The Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen binds to specific sequences at the left end of the viral genome through its carboxy-terminus. Virology 291:241-259. [DOI] [PubMed] [Google Scholar]
- 18.Deng, Z., L. Lezina, C. J. Chen, S. Shtivelband, W. So, and P. M. Lieberman. 2002. Telomeric proteins regulate episomal maintenance of Epstein-Barr virus origin of plasmid replication. Mol. Cell 9:493-503. [DOI] [PubMed] [Google Scholar]
- 19.DePamphilis, M. L. 2003. The ′ORC cycle': a novel pathway for regulating eukaryotic DNA replication. Gene 310:1-15. [DOI] [PubMed] [Google Scholar]
- 20.Dhar, S. K., L. Delmolino, and A. Dutta. 2001. Architecture of the human origin recognition complex. J. Biol. Chem. 276:29067-29071. [DOI] [PubMed] [Google Scholar]
- 21.Dhar, S. K., K. Yoshida, Y. Machida, P. Khaira, B. Chaudhuri, J. A. Wohlschlegel, M. Leffak, J. Yates, and A. Dutta. 2001. Replication from oriP of Epstein-Barr virus requires human ORC and is inhibited by geminin. Cell 106:287-296. [DOI] [PubMed] [Google Scholar]
- 22.Dignam, J. D., R. M. Lebovitz, and R. G. Roeder. 1983. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 11:1475-1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dittmer, D., M. Lagunoff, R. Renne, K. Staskus, A. Haase, and D. Ganem. 1998. A cluster of latently expressed genes in Kaposi's sarcoma-associated herpesvirus. J. Virol. 72:8309-8315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fejer, G., M. M. Medveczky, E. Horvath, B. Lane, Y. Chang, and P. G. Medveczky. 2003. The latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus interacts preferentially with the terminal repeats of the genome in vivo and this complex is sufficient for episomal DNA replication. J. Gen. Virol. 84:1451-1462. [DOI] [PubMed] [Google Scholar]
- 25.Garber, A. C., J. Hu, and R. Renne. 2002. Latency-associated nuclear antigen (LANA) cooperatively binds to two sites within the terminal repeat, and both sites contribute to the ability of LANA to suppress transcription and to facilitate DNA replication. J. Biol. Chem. 277:27401-27411. [DOI] [PubMed] [Google Scholar]
- 26.Garber, A. C., M. A. Shu, J. Hu, and R. Renne. 2001. DNA binding and modulation of gene expression by the latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus. J. Virol. 75:7882-7892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gilbert, D. M. 2001. Making sense of eukaryotic DNA replication origins. Science 294:96-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guo, H. G., M. Sadowska, W. Reid, E. Tschachler, G. Hayward, and M. Reitz. 2003. Kaposi's sarcoma-like tumors in a human herpesvirus 8 ORF74 transgenic mouse. J. Virol. 77:2631-2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hager, G. L., and G. Fragoso. 1999. Analysis of nucleosome positioning in mammalian cells. Methods Enzymol. 304:626-638. [DOI] [PubMed] [Google Scholar]
- 30.Higuchi, R., G. Dollinger, P. S. Walsh, and R. Griffith. 1992. Simultaneous amplification and detection of specific DNA sequences. Biotechnology (New York) 10:413-417. [DOI] [PubMed] [Google Scholar]
- 31.Higuchi, R., C. Fockler, G. Dollinger, and R. Watson. 1993. Kinetic PCR analysis: real-time monitoring of DNA amplification reactions. Biotechnology (New York) 11:1026-1030. [DOI] [PubMed] [Google Scholar]
- 32.Hsieh, D. J., S. M. Camiolo, and J. L. Yates. 1993. Constitutive binding of EBNA1 protein to the Epstein-Barr virus replication origin, oriP, with distortion of DNA structure during latent infection. EMBO J. 12:4933-4944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hu, J., Garber, A. C., and R. Renne. 2002. The latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus supports latent DNA replication in dividing cells. J. Virol. 76:11677-11687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iizuka, M., and B. Stillman. 1999. Histone acetyltransferase HBO1 interacts with the ORC1 subunit of the human initiator protein. J. Biol. Chem. 274:23027-23034. [DOI] [PubMed] [Google Scholar]
- 35.Jenuwein, T., and C. D. Allis. 2001. Translating the histone code. Science 293:1074-1080. [DOI] [PubMed] [Google Scholar]
- 36.Kedes, D. H., M. Lagunoff, R. Renne, and D. Ganem. 1997. Identification of the gene encoding the major latency-associated nuclear antigen of the Kaposi's sarcoma-associated herpesvirus. J. Clin. Investig. 100:2606-2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kirchmaier, A. L., and B. Sugden. 1995. Plasmid maintenance of derivatives of oriP of Epstein-Barr virus. J. Virol. 69:1280-1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Komatsu, T., M. E. Ballestas, A. J. Barbera, and K. M. Kaye. 2002. The KSHV latency-associated nuclear antigen: a multifunctional protein. Front. Biosci. 7:d726-d730. [DOI] [PubMed] [Google Scholar]
- 39.Krithivas, A., D. B. Young, G. Liao, D. Greene, and S. D. Hayward. 2000. Human herpesvirus 8 LANA interacts with proteins of the mSin3 corepressor complex and negatively regulates Epstein-Barr virus gene expression in dually infected PEL cells. J. Virol. 74:9637-9645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lim, C., Y. Gwack, S. Hwang, S. Kim, and J. Choe. 2001. The transcriptional activity of cAMP response element-binding protein-binding protein is modulated by the latency associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus. J. Biol. Chem. 276:31016-31022. [DOI] [PubMed] [Google Scholar]
- 41.Lim, C., H. Sohn, D. Lee, Y. Gwack, and J. Choe. 2002. Functional dissection of latency-associated nuclear antigen 1 of Kaposi's sarcoma-associated herpesvirus involved in latent DNA replication and transcription of terminal repeats of the viral genome. J. Virol. 76:10320-10331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Linskens, M. H., and J. A. Huberman. 1988. Organization of replication of ribosomal DNA in Saccharomyces cerevisiae. Mol. Cell. Biol. 8:4927-4935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lomvardas, S., and D. Thanos. 2002. Modifying gene expression programs by altering core promoter chromatin architecture. Cell 110:261-271. [DOI] [PubMed] [Google Scholar]
- 44.Lu, F., J. Zhou, A. Wiedmer, K. Madden, Y. Yuan, and P. M. Lieberman. 2003. Chromatin remodeling of the Kaposi's sarcoma-associated herpesvirus ORF50 promoter correlates with reactivation from latency. J. Virol. 77:11425-11435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Martienssen, R. A. 2003. Maintenance of heterochromatin by RNA interference of tandem repeats. Nat. Genet. 35:213-214. [DOI] [PubMed] [Google Scholar]
- 46.Mattsson, K., C. Kiss, G. M. Platt, G. R. Simpson, E. Kashuba, G. Klein, T. F. Schulz, and L. Szekely. 2002. Latent nuclear antigen of Kaposi's sarcoma herpesvirus/human herpesvirus-8 induces and relocates RING3 to nuclear heterochromatin regions. J. Gen. Virol. 83:179-188. [DOI] [PubMed] [Google Scholar]
- 47.McNairn, A. J., and D. M. Gilbert. 2003. Epigenomic replication: linking epigenetics to DNA replication. Bioessays 25:647-656. [DOI] [PubMed] [Google Scholar]
- 48.Muralidhar, S., A. M. Pumfery, M. Hassani, M. R. Sadaie, M. Kishishita, J. N. Brady, J. Doniger, P. Medveczky, and L. J. Rosenthal. 1998. Identification of kaposin (open reading frame K12) as a human herpesvirus 8 (Kaposi's sarcoma-associated herpesvirus) transforming gene. J. Virol. 72:4980-4988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pasero, P., A. Bensimon, and E. Schwob. 2002. Single-molecule analysis reveals clustering and epigenetic regulation of replication origins at the yeast rDNA locus. Genes Dev. 16:2479-2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Platt, G. M., G. R. Simpson, S. Mittnacht, and T. F. Schulz. 1999. Latent nuclear antigen of Kaposi's sarcoma-associated herpesvirus interacts with RING3, a homolog of the drosophila female sterile homeotic (fsh) gene. J. Virol. 73:9789-9795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rainbow, L., G. M. Platt, G. R. Simpson, R. Sarid, S.-J. Gao, H. Stoiber, C. S. Herrington, P. S. Moore, and T. F. Schulz. 1997. The 222- to 234-kilodalton latent nuclear protein (LNA) of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) is encoded by orf73 and is a component of the latency-associated nuclear antigen. J. Virol. 71:5915-5921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Richards, E. J., and S. C. Elgin. 2002. Epigenetic codes for heterochromatin formation and silencing: rounding up the usual suspects. Cell 108:489-500. [DOI] [PubMed] [Google Scholar]
- 53.Russo, J. J., R. A. Bohenzky, M. C. Chien, J. Chen, M. Yan, D. Maddalena, J. P. Parry, D. Peruzzi, I. S. Edelman, Y. Chang, and P. S. Moore. 1996. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc. Natl. Acad. Sci. USA 93:14862-14867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ryan, M. P., G. A. Stafford, L. Yu, K. B. Cummings, and R. H. Morse. 1999. Assays for nucleosome positioning in yeast. Methods Enzymol. 304:376-399. [DOI] [PubMed] [Google Scholar]
- 55.Sadler, R., L. Wu, B. Forghani, R. Renne, W. Zhong, B. Herndier, and D. Ganem. 1999. A complex translational program generates multiple novel proteins from the latently expressed kaposin (K12) locus of Kaposi's sarcoma-associated herpesvirus. J. Virol. 73:5722-5730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sakakibara, S., K. Ueda, K. Nishimura, E. Do, E. Ohsaki, T. Okuno, and K. Yamanishi. 2004. Accumulation of heterochromatin components on the terminal repeat sequence of Kaposi's sarcoma-associated herpesvirus mediated by the latency-associated nuclear antigen. J. Virol. 78:7299-7310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 58.Schepers, A., M. Ritzi, K. Bousset, E. Kremmer, J. L. Yates, J. Harwood, J. F. Diffley, and W. Hammerschmidt. 2001. Human origin recognition complex binds to the region of the latent origin of DNA replication of Epstein-Barr virus. EMBO J. 20:4588-4602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Soulier, J., L. Grollet, E. Oksenhendler, P. Cacoub, D. Cazals-Hatem, P. Babinet, M. F. d'Agay, J. P. Clauvel, M. Raphael, L. Degos, et al. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman's disease. Blood 86:1276-1280. [PubMed] [Google Scholar]
- 60.Strahl, B. D., and C. D. Allis. 2000. The language of covalent histone modifications. Nature 403:41-45. [DOI] [PubMed] [Google Scholar]
- 61.Tye, B. K. 1999. MCM proteins in DNA replication. Annu. Rev. Biochem. 68:649-686. [DOI] [PubMed] [Google Scholar]
- 62.Vashee, S., C. Cvetic, W. Lu, P. Simancek, T. J. Kelly, and J. C. Walter. 2003. Sequence-independent DNA binding and replication initiation by the human origin recognition complex. Genes Dev. 17:1894-1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wittwer, C. T., M. G. Herrmann, A. A. Moss, and R. P. Rasmussen. 1997. Continuous fluorescence monitoring of rapid cycle DNA amplification. BioTechniques 22:130-131, 134-138. [DOI] [PubMed] [Google Scholar]
- 64.Wysokenski, D. A., and J. L. Yates. 1989. Multiple EBNA1-binding sites are required to form an EBNA1-dependent enhancer and to activate a minimal replicative origin within oriP of Epstein-Barr virus. J. Virol. 63:2657-2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yates, J. L., S. M. Camiolo, and J. M. Bashaw. 2000. The minimal replicator of Epstein-Barr virus oriP. J. Virol. 74:4512-4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.