

Abstract
Coxsackievirus group B type 3 (CVB3) is an important cause of viral myocarditis. The infiltration of mononuclear cells into the myocardial tissue is one of the key events in viral myocarditis. Immediately after CVB3 infects the heart, the expression of chemokine(s) by infected myocardial cells may be the first trigger for inflammatory infiltration and immune response. However, it is unknown whether CVB3 can induce the chemokine expression in cardiac myocytes. Monocyte chemoattractant protein 1 (MCP-1) is a potent chemokine that stimulates the migration of mononuclear cells. The objective of the present study was to investigate the effect of CVB3 infection on MCP-1 expression in murine cardiac myocytes and the role of MCP-1 in migration of mononuclear cells in viral myocarditis. Our results showed that the expression of MCP-1 was significantly increased in cardiac myocytes after wild-type CVB3 infection in a time- and dose-dependent manner, which resulted in enhanced migration of mononuclear cells in mice with viral myocarditis. The migration of mononuclear cells was partially abolished by antibodies specific for MCP-1 in vivo and in vitro. Administration of anti-MCP-1 antibody prevented infiltration of mononuclear cells bearing the MCP-1 receptor CCR2 in mice with viral myocarditis. Infection by UV-irradiated CVB3 induced rapid and transient expression of MCP-1 in cardiac myocytes. In conclusion, our results indicate that CVB3 infection stimulates the expression of MCP-1 in myocardial cells, which subsequently leads to migration of mononuclear cells in viral myocarditis.
Coxsackievirus group B type 3 (CVB3) is considered the most common cause of viral myocarditis in both humans and animals (35). CVB3 not only causes myocarditis but may also be responsible for dilated cardiomyopathy. Although there has been no clear study about the incidence of viral myocarditis in the population, the incidence studies on dilated cardiomyopathy have reported 2.0 to 8.3 cases per 100,000 per year worldwide (28, 29). In the meantime, the incidence of myocarditis as determined by biopsy has been found in up to 67% of the hearts of dilated cardiomyopathy patients (25).
Despite extensive efforts to date, no specific treatment for viral myocarditis has been demonstrated to be effective in large randomized trials (30). The major reason for the lack of effective treatment is that the pathogenesis of viral myocarditis has not been well clarified. In general, direct virus-induced tissue injury and death of infected cardiac myocytes occur 2 to 4 days after infection. This myocardial injury becomes progressively more noticeable by days 3 and 4 and is significantly advanced by day 5, after which direct injury is complicated by an inflammatory infiltration (6). This inflammatory infiltration includes innate and specific immune participants, and a large number of leukocytes are recruited to the site of infection (22). The process of leukocyte accumulation is dependent upon extravasation or migration of leukocytes across the endothelial barrier into the tissue proper (13). The process of extravasation is thought to occur in a stepwise fashion. Normally, leukocytes maintain close contact with the endothelium by a rolling or tethering motion mediated by weak adhesion molecules, such as selectins. Then, upon appropriate stimulation, the affinities of integrins such as LFA-1, MAC-1, α4β1, and α4β7 expressed on leukocytes change through incompletely understood mechanisms and provide tight adhesion capable of arresting the leukocytes and allowing extravasation. Chemokines have been shown to be particularly effective and important in these processes (14).
Chemokines are an extensive family of related low-molecular-weight proteins grouped into four subfamilies based on the positions of conserved cysteine residues, the CXC, CC, CX3C, and C subfamilies (46). Given their ability to recruit potentially destructive leukocytes into tissues, the regulation of chemokines is very important. The expression of chemokines is under the control of cytokines as well as immunological and inflammatory stimulators (7). Viruses are known to be potent stimulators of chemokine expression in vitro and in vivo (3, 20). It is believed that virus replication is required for virus-induced expression of chemokines (9).
In our previous studies (38, 39), it was found that CVB3 infection in vivo could influence the expression of chemokines in the myocardium of mice. There were significant differences in the species of chemokines in the myocardial tissue of mice with viral myocarditis and normal mice. Three chemokines (MIP-2, MIG, and IP-10) were inducible and 10 chemokines (SDF-1, MIP-1β, MCP-1, MCP-2, MCP-3, MCP-5, MDC, RANTES, FKN and LTN) were constitutive in the viral myocarditis group. The sequence of these chemokines in the myocardial tissue of infected mice from high to low expression level as determined by reverse transcription (RT)-PCR was MCP-3, IP-10, MCP-1, MCP-5, MDC, MCP-2, MIG, MIP-1β, MIP-2, SDF-1, LTN, RANTES, and FKN. The results suggested that, in addition to differences in chemokine species, the expression levels of different chemokines varied significantly in the myocardial tissue of CVB3-infected mice.
Based on our previous results and by comparing the biological features of MCP-3, IP-10, and MCP-1, we speculate that MCP-1 may be one of the key chemokines in a series of reactions induced by CVB3 infection. MCP-1 is a potent chemokine that stimulates migration of mononuclear cells (8). However, it is unknown whether CVB3 can induce the expression of MCP-1 in murine cardiac cells and what role MCP-1 plays in viral myocarditis. In the present study, we found that CVB3 infection stimulated the expression of MCP-1 by murine cardiac myocytes and led to enhanced migration of mononuclear cells in viral myocarditis. The function of MCP-1 in viral myocarditis seemed to be largely mediated by the MCP-1 receptor CCR2. The results of this study will help to understand the molecular mechanism of viral myocarditis and provide a potential target for prevention and therapy of viral myocarditis.
MATERIALS AND METHODS
Cell culture, virus, and mice.
HeLa cells were grown and maintained in RPMI 1640 medium supplemented with 10% heat-inactivated fetal calf serum. CVB3 (Nancy strain) was propagated in HeLa cells and purified by a method previously described by Michael et al. (32). The aliquots were stored at −70°C. Virus titer was routinely determined prior to infection by 50% tissue culture infectious dose (TCID50) assay of HeLa cell monolayers according to previously published procedures (10). UV-irradiated virus was prepared as described elsewhere (1). Briefly, virus preparations were exposed to a UV lamp (125 W) at a distance of 10 cm for 2 h (UV dose = 3.6 × 104 J/cm2). The loss of infectivity was ascertained by the TCID50 assay. Four-week-old male BALB/c mice were purchased from the Experimental Animal Center of Fudan University.
Preparation of neonatal murine cardiac myocytes.
Hearts were removed aseptically from neonatal mice within 72 h of birth. Single-cell suspensions of myocytes were prepared with a method modified from that of Huber et al. (12). Briefly, the hearts were minced finely and subjected to stepwise enzymatic digestion with 0.25% trypsin. The dissociated cells were washed with complete basal Eagle's medium and depleted of endothelial cells and fibroblasts by two sequential 1-h adsorptions to plastic flasks at 37°C. The nonadherent myocytes were removed, washed once, resuspended in complete basal medium, and dispensed into tissue culture wells. After a period of 48 h, required for the myocytes to attach firmly to the plastic, the cells were used as described below. According to observations on the shape and beating activity of the cells obtained, more than 95% cells were identified as cardiac myocytes
RNA extraction and reverse transcription.
Total RNA was extracted from cultured myocytes in vitro by Trizol reagent (Biobasic Inc.) according to the manufacturer's instructions. Briefly, 1 ml of myocytes was pipetted into a sterile microtube and homogenized. The microtube with the homogenate was maintained at room temperature for 5 min, and 0.2 ml of chloroform was added to the microtube. The contents of the microtube was then thoroughly vortexed and left at room temperature for 5 min. The homogenate was centrifuged at 12,000 × g for 10 min at 4°C, and the aqueous layer was pipetted into a new microtube. A volume of 0.5 ml of isopropyl alcohol was added to the homogenate. The homogenate was mixed by vortexing and then kept at room temperature for 10 min. The homogenate mixture was centrifuged at 12,000 × g for 10 min at 4°C, and the RNA pellet was washed in 70% ethanol by centrifugation at 7,500 × g for 5 min. The RNA pellet was briefly air dried and resuspended in diethylpyrocarbonate-treated water. The RNA concentration was estimated by measurement of absorbance at 260 and 280 nm. The integrity of the RNA sample was assessed by resolution on a 1% formaldehyde-agarose gel and ethidium bromide staining. Each RNA sample was aliquoted and stored at −70°C until it was used in an RT-PCR experiment.
To synthesize the first-strand cDNA, approximately 1 μg of total RNA and 1 μl of oligo(dT) (0.5 μg/μl) were added to a microtube, and sterile diethylpyrocarbonate-treated water was added to a total volume of 12 μl. The reaction tube was heated at 70°C for 5 min and then chilled on ice; 1 μl of 10 mM mixed dinucleoside triphosphates, 4 μl of 5× first-strand buffer, 2 μl of 0.5 M dithiothreitol, and 1 μl of Moloney murine leukemia virus reverse transcriptase were added. The reaction mixture was incubated at 37°C for 60 min. The reaction was terminated by incubation at 70°C for 10 min, and the tube was transferred to ice.
External standard for real-time RT-PCR.
To obtain an external standard for the quantitative PCR assay, the MCP-1 cDNA was cloned into plasmid vector and the sequenced was determined. The PCR products of MCP-1 were generated from mouse hearts with 1.5 U of recombinant Taq DNA polymerase (Takara, Kyoto, Japan) and 10 pmol of each of primers 5′-CGGAATTCGCCACCATGCAGGTCCCTGTCAT-3′ (sense) and 5′-GCTCTAGACTAGTTCACTGTCACACT-3′ (antisense). The PCR products were purified and inserted directly into plasmid pVAX1. The recombinant plasmids were then used to transform Escherichia coli DH5α competent cells. The plasmids of individual resistant colonies were isolated, and the inserts were sequenced. The plasmid DNA was quantitated by measurement of absorbance at 260 nm and used as the external standard for the quantitative RT-PCR assay. After quantitation, 5.51 × 10, 5.51 × 102, 5.51 × 103, 5.51 × 104 and 5.51 × 105 copies of the MCP-1 fragment were measured for the standard curve and analysis of the stability of the external standard for real-time RT-PCR. The R2 of the standard curve was 0.994. The coefficients of variation of the interday and intraday experiments were less than 10%.
Quantitative measurement of MCP-1 mRNA by reverse transcription-PCR (RT-PCR).
The quantitative analysis of MCP-1 mRNA was done with a LightCycler (Roche Diagnostic Company, Mannheim, Germany). For direct quantitative analysis, real-time RT-PCR was performed only with the external standard of MCP-1. For relative quantitative analysis, the housekeeping gene for glyceraldehhyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control, and the quantity of MCP-1 was represented relative to the quantity of MCP-1. The reaction mixture consisted of 2 μl of LightCycler DNA Master SYBR Green I. The cycling program had a 10-min initial denaturation at 95°C and then entered a cycle of an instant 95°C denaturation, 10 s of annealing at 60°C, and 12 s of extension at 72°C with a transition rate of 20°C/s between temperature plateaus for a total of 40 cycles. Quantification data is analyzed with the LightCycler analysis software version 3. The standard curve of the MCP-1 plasmid was established, and the coefficient of variation was less than 10%.
Preparation of peripheral blood mononuclear cells and infiltrating mononuclear cells in myocardium.
Under ether anesthesia, blood was aspirated via cardiac puncture, and the hearts were removed. Then peripheral blood mononuclear cells and infiltrating mononuclear cells were isolated by proteolytic enzyme treatment and the density gradient method as described previously (41).
Chemotaxis assay.
Chemotaxis assays were performed as described previously (8). Briefly, myocardial cells were plated dropwise onto 35-mm dishes at a concentration of 105 cells/ml in minimal essential medium (MEM). After 12 h, the myocardial cells were infected with CVB3. After an additional 6 h, the supernatant was harvested and added to the bottom chamber of the chemotaxis chamber. In some cases, 10 ng of MCP-1 (Promega Corporation) per ml was used instead of the supernatant. Anti-MCP-1 antibodies (Santa Cruz Biotechnology Inc., Cambridge, Mass.) or unrelated immunoglobulin G (IgG) antibodies were used where stated in the text. The antibodies were incubated for 30 min at 37°C before addition of the filter to the well. The antibodies were present throughout the chemotaxis assay. Peripheral blood mononuclear cells from mice with viral myocarditis (105 cells in 50 μl) were placed in the upper chamber of the chemotaxis chamber, and the wells were incubated for 8 h in a humidified incubator at 37°C; triplicate wells were used for each data point. At the end of the incubation, the Transwell filters were removed, and the lower surface was swabbed with a pipette tip. The swabbed cells were counted together with the cells in the lower chamber.
Murine model of CVB3 myocarditis.
Four-week-old male BALB/c mice were inoculated intraperitoneally with 105 TCID50 of CVB3 in 100 μl of basal Eagle's medium. Mice inoculated with 100 μl of basal Eagle's medium served as uninfected controls. Hearts were aseptically collected from the mice at the indicated time points postinfection. The collected hearts were washed in sterile basal Eagle's medium, and portions of the hearts were snap-frozen in liquid nitrogen and stored at −70°C for RNA extraction and PCR analysis. The remaining parts of the collected hearts were fixed in formal-buffered saline for histological studies.
Anti-MCP-1 antibody study.
Four-week-old male BALB/c mice were inoculated in the same manner as for the murine model of CVB3 myocarditis. Mice were injected intracardially with 50 μg of anti-MCP-1 antibody diluted in 40 μl of saline on days 3 and 5 and control mice were intracardially injected with goat IgG (50 μg) on days 3 and 5 by the method of Lim et al. (19). Briefly, mice were anesthetized by intraperitoneal injection of 0.75% pentobarbital sodium (50 mg/kg). The skin was dissected, and an incision 2 to 3 cm long was made longitudinally on the left chest. The beat of the heart was observed on the pleura. Then the anti-MCP-1 antibody or goat IgG was injected between the second and third rib with a 1-ml syringe. The depth of the needle was controlled within 2.5 mm. After the injection, the skin was closed. On day 7, the hearts were removed from the injected mice, and the infiltrating mononuclear cells were isolated. Histological analysis was then performed.
Histological studies.
For histological analysis, hearts were fixed in 10% neutral buffered formalin and sectioned into 5-mm slices. Sections were stained with hematoxylin and eosin. Two investigators who were blinded to the experimental treatment analyzed myocardial necrosis and number of infiltrating mononuclear cells in sections. Five fields were analyzed for each sample, and the number of infiltrating mononuclear cells was derived from the mean of the five fields. Alternatively, some portions of the heart were snap-frozen, mounted with ornithine carbamyltransferase compound, and examined immunohistologically. Briefly, sections 5 mm thick were cut from the frozen blocks, and endogenous peroxidase activity was blocked with 0.3% H2O2. Incubation for 30 min followed after addition of 1:100 goat anti-mouse MCP-1 antibody or unrelated antibody, and then the secondary antibody labeled with biotin was incubated with the sections for 30 min. Avidin-biotin complex (ABC) reagent was used, and peroxidase activity was visualized with diaminobenzidine (DAB) as the chromogen. Three hearts were examined in each group.
Flow cytometry.
Unlabeled goat antibody directed against mice CCR2 was purchased from Santa Cruz (Santa Cruz Biotechnology), and fluorescein isothiocyanate-conjugated donkey anti-goat IgG was purchased from Rockland. Single-color flow cytometry was performed by incubating 2 × 105 cells in RPMI 1640 medium plus 2% fetal bovine serum with unlabeled goat antibody against mouse CCR2 and unlabeled goat IgG for the isotype control sample. The incubation (40 min, 4°C) was followed with two washes. The washed cells were then incubated with fluorescein isothiocyanate-conjugated donkey anti-goat IgG. Flow cytometric analysis of 104 cells from each sample was performed on a FACSort flow cytometer (Becton Dickinson) according to standard procedures.
Statistical analysis.
The results were analyzed with the two-tailed independent Student's t test. The level of statistical significance was set at P < 0.05.
RESULTS
CVB3 infection upregulated the expression of MCP-1 in cardiac myocytes in a time- and dose-dependent manner.
We found previously that CVB3 infection upregulates MCP-1 in murine cardiac myocytes (38). To study the time course of MCP-1 expression following CVB3 infection, primary cultures of cardiac myocytes from neonatal BALB/c mice were infected with CVB3 for various time periods, and total cellular RNAs were extracted for RT-PCR analysis. The specificity of the amplified PCR product was confirmed by dot hybridization with a DNA probe specific to MCP-1. The expression of MCP-1 was detected at 0, 2, 4, 6, and 8 h postinfection with 10 TCID50 of CVB3 in cultured cardiac myocytes by real-time RT-PCR in parallel with the external standard. The upregulated expression of MCP-1 was found at the second hour post-CVB3 infection, and the expression of MCP-1 was found to climb to a peak at the sixth hour post-CVB3 infection (Fig. 1A and 1B). By comparison, there was no significant change in the expression of MCP-1 within the 8-h period in uninfected cardiomyocytes. The results indicated that the upexpression of MCP-1 was caused by CVB3 infection and the expression of MCP-1 was upregulated in CVB3-infected myocardial cells in a time-dependent manner in the first 6 h postinfection.
FIG. 1.
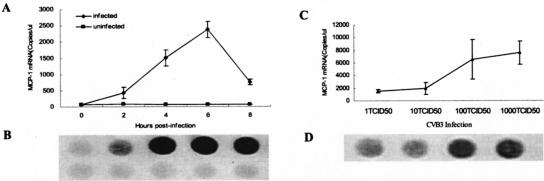
Expression of MCP-1 by cardiac myocytes infected with wild-type CVB3. Cardiac myocytes from neonatal BALB/c mice were incubated with wild-type CVB3 (□) or MEM (▪) for various time periods (A and B) or with different CVB3 doses (C and D). After incubation, total RNA was extracted from the cells at the designated time points, and the expression of MCP-1 was determined by real-time RT-PCR with an external standard (A and C). The specificity of the amplified PCR fragment was confirmed by dot hybridization with a probe labeled with digoxigenin (B and D). The results are presented as the mean ± standard deviation of three separate experiments.
In order to establish the relationship between MCP-1 expression and CVB3 infection dose, primary cultures of cardiac myocytes from neonatal BALB/c mice were infected with different CVB3 doses (1× TCID50, 10× TCID50, 100× TCID50 and 1,000× TCID50). The results showed that the expression of MCP-1 was upregulated as the CVB3 dose increased (Fig. 1C and 1D). The results indicated that CVB3 infection upregulated the expression of MCP-1 in primary cultures of cardiac myocytes in a dose-dependent manner.
Infection by UV-inactivated CVB3 induced transient and low-level expression of MCP-1.
Numerous previous studies have suggested that virus replication is required for the induction and upregulation of chemokines by virus infection (3, 9, 20). To test whether CVB3 replication is required for MCP-1 upregulation, CVB3 was exposed to UV irradiation for 1.5 h, and the replication ability of CVB3 was completely abolished (data not shown). The primary cultures of cardiac myocytes from neonatal BALB/c mice were infected with wild-type CVB3 or UV-irradiated CVB3, and the expression of MCP-1 in the infected cardiac myocytes was detected at 0, 2, 4, 6, and 8 h postinfection. As expected, the expression of MCP-1 started to increase in the wild-type CVB3 group within 2 h postinfection, and the expression level of MCP-1 remained high at 8 h postinfection, with the peak level at around 6 h postinfection (Fig. 2). To our surprise, the expression of MCP-1 also increased in the UV-irradiated CVB3 group, and the trend of MCP-1 upregulation was similar to that in the wild-type CVB3 group in the first 4 h postinfection (Fig. 2). However, the MCP-1 level peaked at around 4 h after infection with UV-irradiated CVB3 and went back almost to its uninduced level 6 h postinfection (P < 0.01 versus UV-irradiated CVB3). The results showed that a virus replication event was not required for the upregulation of MCP-1 by CVB3 infection in cardiac myocytes and that the CVB3 virus itself was responsible for and sufficient to cause the observed upregulation of MCP-1 in the infected cardiac myocytes.
FIG. 2.
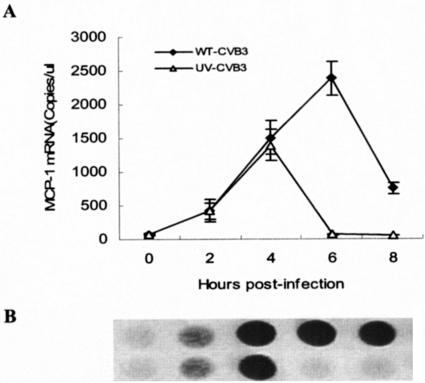
Expression of MCP-1 by myocardial cells infected with wild-type CVB3 or UV-irradiated CVB3. Cardiac myocytes from neonatal BALB/c mice were incubated with UV-irradiated CVB3 (UV-CVB3, ▵) or wild-type CVB3 (WT-CVB3, □) for various periods of time. (A) After incubation, total RNA was extracted from the cells, and the expression of MCP-1 was determined by real-time RT-PCR with an external standard. (B) The specific amplicons were confirmed by dot hybridization with a probe labeled with digoxigenin. The results are represented as the mean ± standard deviation of three separate experiments.
Expression of MCP-1 was correlated with the migration of mononuclear cells in viral myocarditis in vivo.
Because the infiltration of mononuclear cells into the infected site is one of the most important pathological characteristics of viral myocarditis (31), the infiltration of mononuclear cells at indicated time points was studied and compared with the expression of MCP-1 in the myocardium of mice with viral myocarditis. It was found that mononuclear cells started to appear in the myocardium at the fourth day after CVB3 infection (2.9 ± 1.6 cells/field). At the seventh and ninth days, the infiltration of mononuclear cells increased dramatically (17.3 ± 1.8 cells/field and 24 ± 5.2 cells/field, respectively), which was in parallel with the upregulated expression of MCP-1 in myocardium of mice with viral myocarditis(r = 0.873, P < 0.05) (Fig. 3). The results suggested that MCP-1 was correlated with the infiltration of mononuclear cells into the myocardium of mice with viral myocarditis.
FIG. 3.
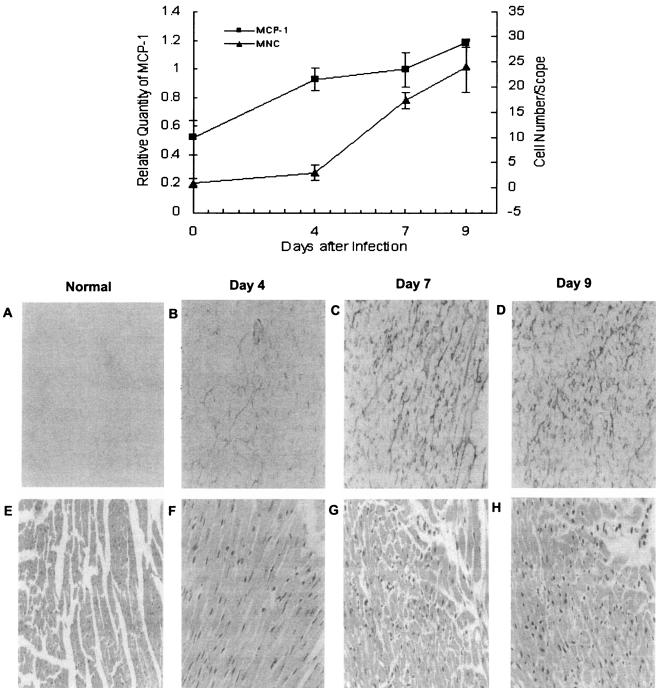
Correlation between expression of MCP-1 and infiltration of mononuclear cells in viral myocarditis. Male BALB/c mice were inoculated intraperitoneally with CVB3 or Eagle's medium. At 0, 4, 7, and 9 days postinfection, mice were exsanguinated under ether anesthesia, and the hearts were collected. Some portions of the heart were snap-frozen and examined immunohistologically by the ABC method. Peroxidase activity was visualized with diaminobenzidine as the chromogen (A, B, C, and D). For analysis of inflammatory infiltration, Sections mounted in paraffin were stained with hematoxylin and eosin. Infiltrating mononuclear cells in interstitial tissue were counted by two investigators who were blinded to the experimental treatment (E, F, G, and H). In the meantime, total RNA was extracted from myocardial tissue. The expression of MCP-1 was detected by RT-PCR. The quantity of MCP-1 is represented relative to that of glyceraldehhyde-3-phosphate dehydrogenase (GAPDH). The number of infiltrating mononuclear cells paralleled the upregulated expression of MCP-1 in the myocardium of mice with viral myocarditis (r = 0.873, P < 0.05).
Expression of MCP-1 in CVB3-infected cardiac myocytes enhanced the migration of mononuclear cells in vitro.
To determine whether MCP-1 plays a role in inducing the migration of peripheral blood mononuclear cells, myocardial cells from neonatal BALB/c mice were infected with CVB3 for 6 h, and aliquots of the culture medium supernatants were then collected for the chemotaxis assay. As shown in Fig. 4, the culture medium supernatant collected from CVB3-infected cardiac myocytes (defined as conditioned medium) significantly stimulated chemotaxis of peripheral blood mononuclear cells (P < 0.01). To determine whether it was MCP-1 that mediated the observed chemotaxis of peripheral blood mononuclear cells, cardiac myocytes were infected with CVB3 and incubated for 6 h, and the conditioned medium supernatants were collected for the chemotaxis assay in the absence and presence of anti-MCP-1 antibodies. Treatment with antibody against MCP-1 (2.5 μg/ml) during the chemotaxis assay abolished the stimulatory effect of conditioned medium supernatant on the chemotaxis of peripheral blood mononuclear cells. These results suggested that MCP-1 was responsible for mediating the observed chemotaxis of peripheral blood mononuclear cells and that CVB3 infection caused enhanced mononuclear cell chemotaxis by inducing MCP-1 expression in myocardial cells (P < 0.01).
FIG. 4.
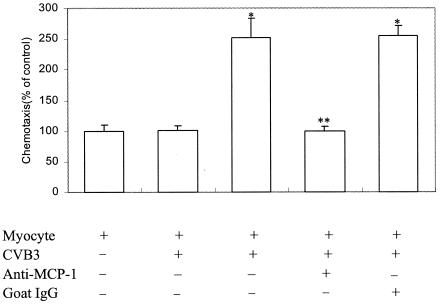
Effects of MCP-1 expression induced by CVB3 infection on chemotaxis of mononuclear cells. Cardiac myocytes were incubated for 6 h in the absence or presence of CVB3. The culture medium were collected (conditioned medium) from cultures of CVB3-infected and uninfected cardiac myocytes at 6 h postinfection. Anti-MCP-1 antibodies (2.5 μg/ml) or an equal amount of goat IgG was added to the conditioned medium 30 min prior to the chemotaxis assay. All samples were tested in duplicate wells. The results are expressed as mean ± standard deviation (error bar) from three separate experiments. *, P < 0.01 versus myocyte control; **, P < 0.01 versus myocytes infected with CVB3.
Blockade of MCP-1 with specific antibodies reduced migration of mononuclear cells in viral myocarditis.
To test whether the migration of mononuclear cells in viral myocarditis was specifically caused by MCP-1, the mice were given intracardiac injections of anti-MCP-1 antibody on days 3 and 5 postinfection with CVB3. On day 7 postinfection, the hearts of the infected mice were removed for histological examination. As shown in Fig. 5, when anti-MCP-1 antibody was used, the infiltration of the mononuclear cells into myocardium decreased by 22 to 56% compared with that in mice infected with CVB3 alone (P < 0.01). The results suggested that MCP-1 was involved in the migration of peripheral blood mononuclear cells in viral myocarditis.
FIG. 5.
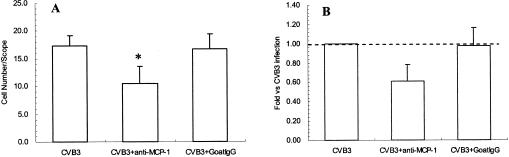
Effect of anti-MCP-1 on the infiltration of mononuclear cells into myocardial tissue. Male BALB/c mice were inoculated intraperitoneally with CVB3. The mice were given intracardiac injections of anti-MCP-1 antibody or goat IgG on days 3 and 5 postinfection with CVB3. On day 7 postinfection, the hearts of the infected mice were removed, and the infiltrating mononuclear cells were counted (A). The results were divided by that of the CVB3 infection group (B). *, P < 0.01 versus CVB3 group only.
MCP-1-induced chemotaxis of CCR2-positive peripheral blood mononuclear cells in viral myocarditis in vitro.
Although CCR2 is known to be the major receptor for MCP-1 (34), little is known about the role of MCP-1 in the chemotaxis of CCR2+ cells in viral myocarditis. In order to determine whether MCP-1 could chemoattract CCR2-positive peripheral blood mononuclear cells in viral myocarditis, the CCR2-positive peripheral blood mononuclear cells were purified from mice 7 days post-CVB3 infection and analyzed in the chemotaxis assay in vitro. As shown in Fig. 6, only 34.46% of the peripheral blood mononuclear cells were CCR2 positive before chemotaxis induced by MCP-1 (Fig. 6B), whereas 87.56% of the peripheral blood mononuclear cells were CCR2 positive after chemotaxis induced by MCP-1 (Fig. 6C). The results suggested that MCP-1 specifically chemoattracted CCR2-positive peripheral blood mononuclear cells purified from CVB3-infected mice.
FIG. 6.
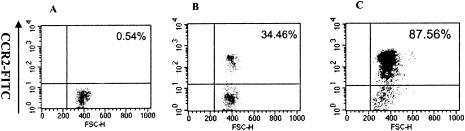
Effect of MCP-1 on chemotaxis of CCR2-positive mononuclear cells in viral myocarditis in vitro. Peripheral blood mononuclear cells were separated from mice 7 days post-CVB3 infection. The purified peripheral blood mononuclear cells were analyzed for the chemotaxis assay in vitro; 105 peripheral blood mononuclear cells were added to the upper chamber of the chemotaxis chamber, and 10 ng of purified MCP-1 protein per ml was included in the lower chamber of the chemotaxis chamber. The details of the chemotaxis assay are described in Materials and Methods. Flow cytometry was employed to detect CCR2-positive cells. The isotype control antibody was used in panel A. The percent CCR2-positive cells detected before (B) and after (C) chemotaxis are shown in the upper sections.
CCR2-positive infiltrating mononuclear cells were the major cells chemoattracted by MCP-1 in viral myocarditis in vivo.
To investigate the effect of MCP-1 on the infiltration of mononuclear cells into the myocardium during viral myocarditis in vivo, mononuclear cells infiltrating the myocardium of CVB3-infected mice were separated and analyzed for changes in CCR2-positive cells by an indirect immunofluorescence technique. As shown in Fig. 7, it was found that 71.75% of the infiltrating mononuclear cells in the myocardium of mice with viral myocarditis were CCR2 positive, whereas only 32.81% of the peripheral blood mononuclear cells from the same mice were CCR2 positive. The results suggested that the CCR2-positive peripheral blood mononuclear cells accumulated in the myocardium during viral myocarditis. When anti-MCP-1 antibody was used to block the function of MCP-1, the percentage of CCR2-positive infiltrating mononuclear cells decreased to 43.05% in the myocardium of CVB3-infected mice. The results demonstrated that CCR2-positive infiltrating mononuclear cells were the major cells chemoattracted by MCP-1 into the myocardium during viral myocarditis.
FIG. 7.
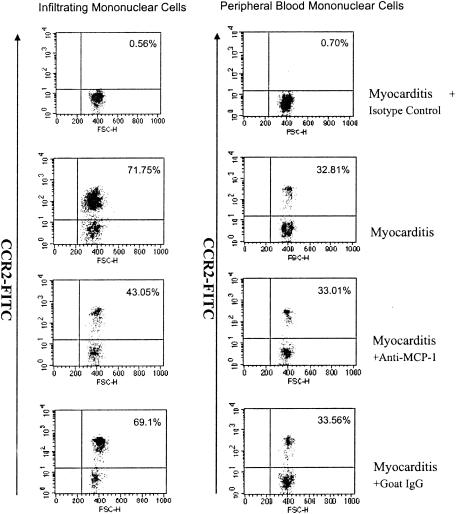
Analysis of CCR2-positive cells in vivo. Infiltrating mononuclear cells in myocardial tissue and peripheral blood mononuclear cells were separated by Ficoll gradient centrifugation from mice with CVB3 infection only (Myocarditis), CVB3 infection combined with anti-MCP-1 goat antibody intervention (Myocarditis + Anti-MCP-1), and CVB3 infection combined with goat IgG intervention (control) (Myocarditis + Goat IgG). Then CCR2 was detected by the indirect immunofluorescence method by flow cytometry. For the control (Myocarditis + Isotype), infiltrating mononuclear cells and peripheral blood mononuclear cells were from mice with CVB3 infection, the first antibody was unlabeled goat IgG, and the secondary antibody was fluorescein isothiocyanate-conjugated donkey anti-goat IgG. For the other three treatment conditions, the first antibody was unlabeled goat antibody against mouse CCR2 and the secondary antibody was fluorescein isothiocyanate-conjugated donkey anti-goat IgG.
DISCUSSION
Our previous studies have shown that MCP-1 may be one of the major chemokines that can be induced by CVB3 infection in mouse myocardium (37, 38). In the present study, it was found that the expression of MCP-1 increased in murine cardiac myocytes infected by CVB3 in vitro in a dose-dependent manner. The time course of MCP-1 expression in cardiac myocytes suggested that induction of MCP-1 was an early event during CVB3 infection. To further investigate the mechanism of MCP-1 induction by CVB3 infection, UV-inactivated CVB3 was used to infect murine cardiac myocytes in vitro. To our surprise, MCP-1 was induced by UV-inactivated CVB3 in cardiac myocytes. The results suggest that the CVB3 virus itself was sufficient to induce MCP-1. Therefore, latent CVB3 virus is enough to induce MCP-1 expression and may be able to cause inflammation and trauma. It is most likely that the CVB3 structural proteins caused the induction of MCP-1 at the early stage of virus infection. In the meantime, compared to that in wild-type CVB3 infection, the transient and low-level expression of MCP-1 induced by UV-inactivated CVB3 suggested that replication is required for the prolonged expression of MCP-1 in cardiac myocytes of CVB3-infected mice.
The infiltration of mononuclear cells into the infected site is one of the most important events in infections caused by viruses like CVB3 (31). Our results indicated that the expression of MCP-1 was correlated with the infiltration of mononuclear cells into the myocardium of CVB3-infected mice. Further studies showed that administration of anti-MCP-1 antibody abolished the effects of MCP-1 on chemotaxis of mononuclear cells in vitro and the infiltration of mononuclear cells into the myocardium of CVB3-infected mice in vivo. These results strongly suggest that MCP-1 plays an important role in enhancing the migration and infiltration of mononuclear cells into the myocardium of CVB3-infected mice.
To study the mechanism of MCP-1-induced migration and infiltration of mononuclear cells in viral myocarditis, we focused on the chemokine receptor CCR2, which is known to be one of the major receptors of MCP-1 (34). The in vitro chemotaxis assay showed that MCP-1 induced chemotaxis of the CCR2-positive peripheral blood mononuclear cells purified from CVB3-infected mice. In addition, the in vivo study of infiltrating mononuclear cells in viral myocarditis showed that CCR2-positive infiltrating mononuclear cells accumulated in the myocardium of CVB3-infected mice (71.75% versus 32.81%), and administration of anti-MCP-1 antibody in vivo reduced the infiltrating mononuclear cells in myocardium to the level comparable to that in control mice (43.05% versus 32.81%). These results jointly suggest that CCR2-positive infiltrating mononuclear cells are the major cells chemoattracted by MCP-1 into the myocardium of CVB3-infected mice, and MCP-1 primarily (if not exclusively) exerts its function through the CCR2 receptor on the surface of peripheral blood mononuclear cells and infiltrating mononuclear cells. From these results, it can also be inferred that CCR2 mediates the chemotactic effect of MCP-1 on mononuclear cells in viral myocarditis. Therefore, MCP-1 is the molecular basis for CVB3-induced inflammation and trauma in viral myocarditis.
Previous studies have suggested that CVB3 influences host gene responses by several mechanisms (21, 42, 45). CVB3 has a short life cycle, which typically culminates in rapid cell death and release of progeny virus. Subsequent to virus attachment to a target cell receptor, CVB3 RNA is released into the cell and acts as a template for translation of the viral polyprotein and replication of the viral genome. Viraus receptors include the coxsackievirus and adenovirus receptor (2, 15, 26, 43) and the decay-accelerating factor coreceptor (27, 36).
The mitogen-activated protein kinases manipulate the signaling machinery to regulate CVB3 replication and host gene responses, which respond to diverse extracellular stimuli and transduce signals from the cell membrane to the nucleus (4, 18). Mitogen-activated protein kinases constitute a superfamily of highly related serine/threonine kinases. At least seven members of the mitogen-activated protein kinase family have been identified in mammals: extracellular signal-regulated kinases 1 and 2 (ERK1/2), c-Jun NH2-terminal kinase/stress-activated protein kinase, p38, big MAPK1, ERK6, and ERK7. ERK 1 and 2 regulate a wide range of cellular functions, including cell proliferation, transformation, differentiation, and notably cell survival and death (5, 24). Small GTP-binding protein Ras has been shown to activate the Raf/MEK/ERK cascade by binding to Raf and anchoring it at the cell membrane, where Raf is phosphorylated and activated by other kinases (23). Recently, the ERK pathway has been implicated in the regulation of viral gene expression and replication for human cytomegalovirus (17), simian virus 40 (40), human immunodeficiency virus type 1 (16, 44), influenza virus (11), and CVB3 (33).
Our finding that upregulation of the expression of MCP-1 can take place in a CVB3 replication-independent manner is worth noticing. Several mechanisms may account for the upregulation of expression of MCP-1 by CVB3 infection in cardiac myocytes. For example, interaction of CVB3 (through its structural protein[s]) with its receptor, replication of CVB3 virus, and signal transduction by the ERK1 and ERK2 pathways may all contribute to the upregulation of MCP-1 in CVB3-infected cardiac myocytes.
In summary, the present study revealed the specific effect and target of MCP-1 in the pathogenesis of viral myocarditis. The results from this study will contribute to the understanding of the mechanism of viral myocarditis and may be extended to other inflammation-associated infectious diseases. The predominant role of MCP-1 in the pathogenesis of viral myocarditis provides a potential target for prevention and therapy of viral myocarditis in the future.
Acknowledgments
This work was supported by the Major State Basic Research Development Program of the People's Republic of China (2001CB510005), the Key Scientific and Technological Program of the Ministry of Education, and partially by a National Scientific Grant for Excellent Youth (39925031).
REFERENCES
- 1.Beck, M. A., N. M. Chapman, B. M. McManus, J. C. Mullican, and S. Tracy. 1990. Secondary enterovirus infection in the murine model of myocarditis. Pathologic and immunologic aspects. Am. J. Pathol. 136:669-681. [PMC free article] [PubMed] [Google Scholar]
- 2.Bergelson, J. M., J. A. Cunningham, G. Droguett, E. A. Kurt-Jones, A. Krithivas, J. S. Hong, M. S. Horwitz, R. L. Crowell, and R. W. Finberg. 1997. Isolation of a common receptor for coxsackie B viruses and adenoviruses 2 and 5. Science 275:1320-1323. [DOI] [PubMed] [Google Scholar]
- 3.Billstrom, S. M., and G. S. Worthen. 2002. Viral regulation of RANTES expression during human cytomegalovirus infection of endothelial cells. J. Virol. 75:3383-3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blenis, J. 1993. Signal transduction via the MAP kinases: proceed at your own RSK. Proc. Natl. Acad. Sci. USA 90:5889-5892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davis, R. J. 1993. The mitogen-activated protein kinase signal transduction pathway. J. Biol. Chem. 268:14553-14556. [PubMed] [Google Scholar]
- 6.Gebhard, J. R., C. M. Perry, S. Harkins, T. Lane, I. Mena, V. C. Asensio, I. L. Campbell, and J. L. Whitton. 1998. Coxsackievirus B3-induced myocarditis:perforin exacerbates disease, but plays no detectable role. Am. J. Pathol. 153:417-428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gerard, C., and B. J. Rollins. 2001. Chemokines and disease. Nat. Immunol. 2:108-115. [DOI] [PubMed] [Google Scholar]
- 8.Gunn, M. D., N. A. Nelken, X. Liao, and L. T. Williams. 1997. Monocyte chemoattractant protein-1 is sufficient for the chemotaxis of monocytes and lymphocytes in transgenic mice but requires an additional stimulus for inflammatory activation. J. Immunol. 158:376-383. [PubMed] [Google Scholar]
- 9.Heim, A., S. Zeuke, S. Weiss, W. Ruschewski, and I. M. Grumbach. 2000. Transient induction of cytokine production in human myocardial fibroblasts by coxsackievirus B3. Circ. Res. 86:753-759. [DOI] [PubMed] [Google Scholar]
- 10.Henke, A., S. A. Huber, A. Stelzner, and J. L. Whitton. 1995. The role of CD8+ T lymphocytes in coxsackievirus B3-induced myocarditis. J. Virol. 69:6720-6728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hirono, Y., K. Okura, and Y. Kato. 2001. Influenza virus propagation is impaired by inhibition of the Raf/MEK/ERK signalling cascade. Nat. Cell Biol. 3:301-305. [DOI] [PubMed] [Google Scholar]
- 12.Huber, S. A., D. Sartini, and M. Exley. 2002. Vγ4+ T cells promote autoimmune CD8+ cytolytic T-lymphocyte activation in coxsackievirus B3-induced myocarditis in mice: the role for CD4+ Th1 cells. J. Virol. 76:10785-10790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Imhof, B. A., and D. Dunon. 1997. Basic mechanism of leukocyte migration. Horm. Metab. Res. 29:614-621. [DOI] [PubMed] [Google Scholar]
- 14.Imhof, B. A., B. Engelhardt, and M. A. Vadas. 2001. Novel mechanisms of the transendothelial migration of leukocytes. Immunol. Today 22:411-414. [DOI] [PubMed] [Google Scholar]
- 15.Ito, M., M. Kodama, M. Masuko, M. Yamaura, K. Fuse, Y. Uesugi, S. Hirono, Y. Okura, K. Kato, Y. Hotta, T. Honda, R. Kuwano, and Y. Aizawa. 2000. Expression of coxsackievirus and adenovirus receptor in hearts of rats with experimental autoimmune myocarditis. Circ. Res. 86:275-280. [DOI] [PubMed] [Google Scholar]
- 16.Jacque, J. M., A. Mann, H. Enslen, N. Sharova, B. Brichacek, R. J. Davis, and M. Stevenson. 1998. Modulation of HIV-1 infectivity by MAPK, a virion-associated kinase. EMBO J. 17:2607-2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnson, R. A., X. L. Ma, A. D. Yurochko, and E. S. Huang. 2001. The role of MKK1/2 kinase activity in human cytomegalovirus infection. J. Gen. Virol. 82:493-497. [DOI] [PubMed] [Google Scholar]
- 18.Lange-Carter, C. A., C. M. Pleiman, A. M. Gardner, K. J. Blumer, and G. L. Johnson. 1993. A divergence in the MAP kinase regulatory network defined by MEK kinase and Raf. Science 260:315-319. [DOI] [PubMed] [Google Scholar]
- 19.Lim, B. K., S. C. Choe, J. O. Shin, S. H. Ho, J. M. Kim, S. S. Yu, S. Y. Kim, E. S. Jeon. 2002. Local expression of interleukin-1 receptor antagonist by plasmid DNA improves mortality and decreases myocardial inflammation in experimental coxsackieviral myocarditis. Circulation 105:1278-1281. [PubMed] [Google Scholar]
- 20.Lim, S. P., and A. Garzino-Demo. 2000. The human immunodeficiency virus type 1 Tat protein upregulates the promoter activity of the beta-chemokine monocyte chemoattractant protein 1 in the human astrocytoma cell line U-87 MG: role of Sp1, Ap1, and NF-κb consensus sites. J. Virol. 74:1632-1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu, P. K., Y. Y. Kong, and M. A. Opavsky. 2000. The tyrosine kinase p56lck is essential in coxsackievirus B3-mediated heart disease. Nat. Med. 6:429-434. [DOI] [PubMed] [Google Scholar]
- 22.Luster, A. D. 2002. The role of chemokines in linking innate and adaptive immunity. Curr. Opin. Immunol. 14:129-135. [DOI] [PubMed] [Google Scholar]
- 23.Marais, R. Y., H. F. Light, Paterson, and C. J. Marshall. 1995. Ras recruits Raf-1 to the plasma membrane for activation by tyrosine phosphorylation. EMBO J. 14:3136-3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marshall, C. J. 1995. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell 80:179-185. [DOI] [PubMed] [Google Scholar]
- 25.Martin, A. B., S. Webber, F. J. Fricker, R. Jaffe, G. Demmler, D. Kearney, Y. H. Zhang, J. Bodurtha, B. Gelb, and J. Ni. 1994. Acute myocarditis. Rapid diagnosis by PCR in children. Circulation 90:330-339. [DOI] [PubMed] [Google Scholar]
- 26.Martino, T. A., M. Petric, H. Weingartl, J. M. Bergelson, M. A. Opavsky, C. D. Richardson, J. F. Modlin, R. W. Finberg, K. C. Kain, N. Willis, C. J. Gauntt, and P. P. Liu. 2000. The coxsackie-adenovirus receptor (CAR) is used by reference strains and clinical isolates representing all six serotypes of coxsackievirus group B and by swine vesicular disease virus. Virology 271:99-108. [DOI] [PubMed] [Google Scholar]
- 27.Martino, T. A., M. Petric, M. Brown, K. Aitken, C. J. Gauntt, C. D. Richardson, L. H. Chow, and P. P. Liu. 1998. Cardiovirulent coxsackieviruses and the decay-accelerating factor (CD55) receptor. Virology 244:302-314. [DOI] [PubMed] [Google Scholar]
- 28.Mason, J. W., J. B. O'Connell, A. Herskowitz, N. R. Rose, B. M. McManus, M. E. Billingham, and T. E. Moon. 1995. A clinical trial of immunosuppressive therapy for myocarditis. The myocarditis treatment trial investigators. N. Engl. J. Med. 333:269-275. [DOI] [PubMed] [Google Scholar]
- 29.McCarthy, R. E., J. P. Boehmer, R. H. Hruban, G. M. Hutchins, E. K. Kasper, J. M. Hare, and K. L. Baughman. 2000. Longterm outcome of fulminant myocarditis as compared with acute (nonfulminant) myocarditis. N. Engl. J. Med. 342:690-695. [DOI] [PubMed] [Google Scholar]
- 30.Mendes, L., M. Picard, G. Dec, V. Hartz, I. Palacios, and R. Davidoff. 1999. Ventricular remodeling in active myocarditis. Myocarditis treatment trial. Am. Heart J. 138:303-308. [DOI] [PubMed] [Google Scholar]
- 31.Merkle, I., M. Tonew, B. Gluck, M. Schmidtke, R. Egerer, and A. Stelzner. 1999. Coxsackievirus B3-induced chronic myocarditis in outbred NMRI mice. J. Hum. Virol. 2:369-379. [PubMed] [Google Scholar]
- 32.Michael, H., K. A. Watson, H. C. Selinka, C. M. Carthy, K. Klingel, B. M. Mcmanus, and R. Kandolf. 1999. Cleavage of RasGAP and phosphorylation of mitogen-activated protein kinase in the course of coxsackievirus B3 replication. J. Virol. 73:3587-3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Opavsky, M. A., T. Martino, M. Rabinovitch, J. Penninger, C. Richardson, M. Petric, C. Trinidad, L. Butcher, J. Chan, and P. P. Liu. 2002. Enhanced ERK-1/2 activation in mice susceptible to coxsackievirus-induced myocarditis. J. Clin. Investig. 109:1561-1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peters, W., and I. F. Charo. 2001. Involvement of chemokine receptor 2 and its ligand, monocyte chemoattractant protein-1, in the development of atherosclerosis: lessons from knockout mice. Curr. Opin. Lipidol. 12:175-180. [DOI] [PubMed] [Google Scholar]
- 35.Satoh, M., G. Tamura, I. Segawa, K. Hiramori, and R. Satodate. 1994. Enteroviral RNA in dilated cardiomyopathy. Eur. Heart J. 15:934-939. [DOI] [PubMed] [Google Scholar]
- 36.Shafren, D. R., R. C. Bates, M. V. Agrez, R. L. Herd, G. F. Burns, and R. D. Barry. 1995. Coxsackieviruses B1, B3, and B5 use decay accelerating factor as a receptor for cell attachment. J. Virol. 69:3873-3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shen, Y., R. Z. Chen, W. Xu, and S. D. Xiong. 2003. Detection of MIP-1β and MCP-1 in myocardium of viral myocarditis and its implications. J. Shanghai Immunol. 23:95-98. [Google Scholar]
- 38.Shen, Y., S. D. Xiong, X. A. Shao, X. J. Zheng, R. Z. Chen, and Y. Z. Yang. 2003. Chemokine expression profile and its implications in viral myocarditis. Chin. J. Microbiol. Immunol. 23:178-182. [Google Scholar]
- 39.Shen, Y., W. Xu, R. Z. Chen, Y. Z. Yang, and S. D. Xiong. 2003. Infection of coxsackievirus group B type 3 regulates the expression profile of chemokines in myocardial tissue/cells. Nat. Med. J. China 83:981-985. [PubMed] [Google Scholar]
- 40.Sontag, E., S. Fedorov, C. Kamibayashi, D. Robbins, M. Cobb, and M. Mumby. 1993. The interaction of SV40 small tumor antigen with protein phosphatase 2A stimulates the map kinase pathway and induces cell proliferation. Cell 75:887-897. [DOI] [PubMed] [Google Scholar]
- 41.Tanuma, N., T. Kojima, T. Shin, Y. Aikawa, T. Kohji, Y. Ishihara, and Y. Matsumoto. 1997. Competitive PCR quantification of pro- and anti-inflammatory cytokine mRNA in the central nervous system during autoimmune encephalomyelitis. J. Neuroimmunol. 73:197-206. [DOI] [PubMed] [Google Scholar]
- 42.Taylor, L. A., C. M. Carthy, D. Yang, K. Saad, D. Wong, G. Schreiner, L. W. Stanton, and B. M. McManus. 2000. Host gene regulation during coxsackievirus B3 infection in mice: assessment by microarrays. Circ. Res. 87:328-334. [DOI] [PubMed] [Google Scholar]
- 43.Wang, X., and J. M. Bergelson. 1999. Coxsackievirus and adenovirus receptor cytoplasmic and transmembrane domains are not essential for coxsackievirus and adenovirus infection. J. Virol. 73:2559-2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang, D., J. Yu, and Z. Luo. 1999. Regulation of human immunodeficiency virus type 1 infectivity by the ERK mitogen-activated protein kinase signaling pathway. J. Virol. 73:3460-3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang, D. J., Z. Yu, and C. M. Luo. 1999. Viral myocarditis: identification of five differentially expressed genes in coxsackievirus B3-infected mouse heart. Circ. Res. 84:704-712. [DOI] [PubMed] [Google Scholar]
- 46.Zlotnic, A., and O. Yoshie. 2000. Chemokines: a new classification system and their role in immunity. Immunity 12:121-125. [DOI] [PubMed] [Google Scholar]