

Abstract
Members of the ArsR/SmtB family of transcriptional repressors, such as CadC, regulate the intracellular levels of heavy metals like Cd(II), Hg(II), and Pb(II). These metal sensing proteins bind their target metals with high specificity and affinity, however, a lack of structural information about these proteins makes defining the coordination sphere of the target metal difficult. Lingering questions as to the identity of Cd(II) coordination in CadC are addressed via protein design techniques. Two designed peptides with tetrathiolate metal binding sites were prepared and characterized, revealing fast exchange between CdS3O and CdS4 coordination spheres. Correlation of 111mCd PAC spectroscopy and 113Cd NMR spectroscopy suggests that Cd(II) coordinated to CadC is in fast exchange between CdS3O and CdS4 forms, which may provide a mechanism for rapid sensing of heavy metal contaminants by this regulatory protein.
Introduction
Metal ions are required by all forms of life to sustain cellular processes, but cadmium, lead, and other thiophilic heavy metals can disrupt intracellular processes by binding to protein thiols. DNA-binding metalloregulatory proteins, such as CadC, which is a member of the ArsR/SmtB family of transcriptional repressors, are responsible for sensing the presence of these metals.1 Highly specific metal–protein interactions govern proper metal homeostasis by the specific uptake, efflux, storage, and trafficking of the metal of interest. While some metals are toxic at nearly all concentrations, others that are often required for life only become toxic at high concentrations. Understanding how these metal–protein interactions can result in detrimental pathologies is important to understanding processes ranging from bioremediation to metal toxicity to neurodegenerative conditions in which an imbalance of metals has been implicated, such as Alzheimer’s disease.2 In particular, the levels of metals are controlled by a complex system of metalloregulatory proteins and chaperones that form specific metal–protein interactions.1,3 Metalloregulatory proteins often bind only a small subset of metals with high selectivity but how this selectivity is achieved is still not well understood. In general, distorted or unusual coordination spheres are a common feature of metal sensors.1,4,5 In particular, CadC from S. aureus pI258 confers resistance to Cd(II), Pb(II), Zn(II), and Bi(III). CadC binds Cd(II) tetrahedrally and Pb(II) trigonally at the α3N (or type 1) site that consists of four cysteines donated from α-helices in the core of the protein and from the N terminal loop.6–8 Mutational analysis studies have revealed that the cysteines that coordinate Cd(II) and Pb(II) in CadC are nonequivalent; removal of a particular cysteine residue can still result in metal binding, but the presence of the bound metal is not “sensed” and the repressor does not dissociate from DNA.6 UV-visible spectroscopy and X-ray absorption spectroscopy suggest that Cd(II) coordinates tetrahedrally with a CdS4 primary coordination; however, 113Cd NMR revealed a chemical shift of 622 ppm,4 which is significantly lower than that of bonafide Cd-substituted S4 proteins such zinc fingers, the structural site metal site in horse liver alcohol dehydrogenase,9 and rubredoxins.10 In light of the functional asymmetry of the site due to the non-equivalence of coordinating cysteines and the fact that no high resolution structures of Cd(II)-bound CadC yet exist, it has been suggested that Cd(II) bound to CadC assumes a highly distorted tetrahedral geometry with one very long Cd–S bond,4,6 but alternatively, the spectroscopic characteristics could imply that Cd(II) experiences exchange between CdS3O and CdS4 coordination modes.
Since few members of the ArsR/SmtB family have been extensively characterized structurally, questions about unusual, intermediate, or distorted coordination spheres are unresolved. Therefore, our group has turned to protein design methods and specialized spectroscopic techniques to understand and define the relevant coordination chemistry for toxic heavy metals. Previous studies have focused on generating thiolate-rich metal binding sites in three-stranded a-helical coiled-coils (3SCCs). Amphipathic α-helices are generated using the heptad repeat strategy with sequences of seven amino acids (with residues a–g) in which the a and d positions are occupied by hydrophobic residues that face towards the interior of the peptide bundle and drive the folding and association of the peptides. Replacement of these interior-facing hydrophobic residues with metal chelating residues creates the metal binding site. As(III) coordinated to triscysteine sites,11 as well as trigonal Hg(II) sites12,13 have been characterized with these models. Our group has also been able to control the coordination sphere—both geometry and coordination number—for Cd(II) and Hg(II) within the hydrophobic interior of peptides, as well as bind Cd(II) in a site-selective manner based on ion recognition.14–16
In particular, the work on Cd(II) has resulted in detailed correlations between Cd(II) spectroscopic characteristics and the relative proportion of CdS3 and CdS3O coordination present in a protein,17 as well as studies on Cd(II) exchange in 3SCCs.18 More recently, we have sought to move this chemistry into a single-stranded scaffold, which adopts more of a globular fold like that of a native protein. A single-stranded construct is capable of enforcing asymmetry, which is essential to understanding how most native proteins function. Many of the suspected Cd(II)-binding sensors utilize 3–4 cysteines to generate CdS3, CdS3O, or distorted CdS sites.4–6 To this end, α3DIV, a three-helix bundle peptide, was created that can bind Cd(II), Pb(II), and Hg(II).19 This construct incorporates three cysteine residues without greatly perturbing the folding of the peptide, which then are available for metal binding.19,20 Furthermore, the heptad repeat structure used in designed α-helices places potential substitutions either three or four residues away from the next hydrophobic residue, creating the motifs, CXXL or CXXXL. Substitution in the α3D scaffold, which is inherently asymmetric, could then easily lead to a controlled number of CXXC (or potentially CXXXC) motifs as are found in most native Cd(II)-binding proteins.1
Herein, we report the introduction of a fourth cysteine residue in two different locations in α3DIV (Fig. 1) to create sites that can mimic native Cd(II)-binding proteins, and in particular, CadC. Constructing such a site in α3DIV presents a significant challenge because native sites that feature tetrathiolate coordination typically utilize at least one loop or hairpin secondary structural element to coordinate the metal. Two different peptides with tetrathiolate metal binding sites were designed and characterized and reveal different proportions of CdS4 and CdS3O coordination environments. Correlation of UV-visible, 113Cd NMR, and 111mCd perturbed angular correlation (PAC) spectroscopy suggests that Cd(II) coordinated to CadC is in fast exchange between CdS4 and CdS3O forms. These observations are used to develop a model for dynamic exchange that is necessary for proper metalloregulatory protein function.
Fig. 1.
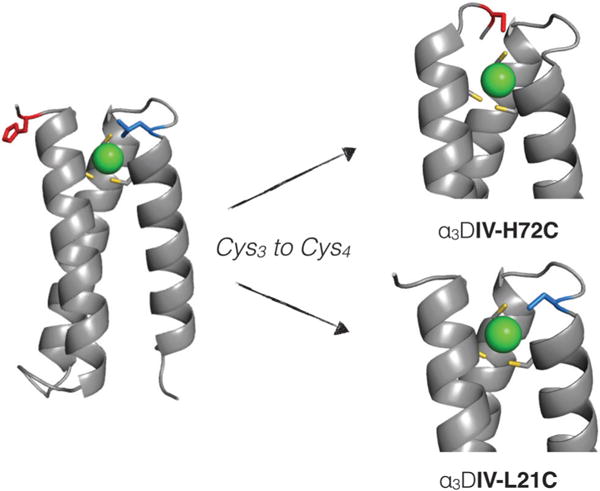
Design strategy for the generation of a tetrathiolate metal-binding motif in the α3DIV scaffold. Here, the substitution of His72 (red residue), which coordinates Cd(II) in α3DIV could allow for tetrathiolate coordination of Cd(II). An alternative approach substitutes Leu21 (pictured in blue) to create a CXXC binding motif. Models are based on PDB: 2MTQ.
Materials and methods
Protein production and purification
The gene for α3DIV-H72C was ordered from Celtek genes (Franklin, TN) subcloned into vector pET15b with ApR as a selective marker. The construct α3DIV-L21C was generated by sequential site directed mutagenesis of α3DIV-H72C by QuikChange kit (Stratagene) to replace the histidine in the 72nd position (C72H) and incorporating the L21C change. The plasmids were transformed into E. coli BL21 (DE3) and plated on LB Amp plates. A single colony was grown overnight in a starter colony and used to inoculate auto-induction media.21 The cells were grown for 15–20 hours at 25 °C. The cells were resuspended in 1 × PBS with lysozyme and 5 mM DTT then lysed by microfluidizer. After 30 min of heat denaturation at 55 °C, the cell lysate was acidified and the soluble fraction collected after ultracentrifugation at 15 K rpm at 4 °C. The pure proteins were isolated by HPLC on a C18 reverse phase column and lyophilized to yield pure, white powder yielding approximately 8 mg L−1 of α3DIV-H72C and 20 mg L−1 of α3DIV-L21C. Identity of the proteins was confirmed by ESI-MS.
UV-visible spectroscopy
All experiments were carried out using a Cary 100 spectrophotometer. Purified, lyophilized proteins were resuspended and the concentration determined using the calculated molar extinction coefficient based on the aromatic residue content. Cadmium titrations were done in 50 mM CHES buffer at pH 8.6 at a peptide concentration of 20 μM. A stock solution of 0.0146 M CdCl2, which was standardized by ICP, was titrated into the peptide solution anaerobically.
113Cd nuclear magnetic resonance spectroscopy
Samples were prepared anaerobically in 10% D2O with 0.8 equivalents of enriched 113Cd with peptide concentrations of 2–3 mM, and the pH was adjusted to 8.6. Samples were added to a Shigemi solvent-matched NMR tube and sealed with parafilm. The spectra were collected on a 500 MHz Varian spectrometer using a 90° pulse (5 μs) and 1 s acquisition with no delay. Spectra were processed on Mestre-C software with 100 Hz line broadening.
111mCd perturbed angular correlation spectroscopy
All perturbed angular correlation (PAC) experiments were performed using a six detector instrument. The sample was kept at a temperature of 1 °C controlled by a Peltier element. The radioactive cadmium was produced on the day of the experiment at the University Hospital cyclotron in Copenhagen and extracted as described previously,17 except that the HPLC separation of zinc and cadmium was omitted in order to avoid chloride contamination of the sample. This procedure may lead to zinc contamination of the sample, but the level of contamination (a few micromolar) should not interfere with the experiment. The 111mCd dissolved in ion exchanged water (10–40 μL) was mixed with nonradioactive cadmium acetate and TRIS buffer in appropriate amounts to achieve the desired final concentrations. The α3DIV-H72C or α3DIV-L21C peptide was then added (dissolved in ion-exchanged water), and the sample was left to equilibrate for 10 min to allow for metal binding. Finally, sucrose was added to produce a 55% w/w solution, and the pH of the solution was adjusted with H2SO4 or KOH. To measure the pH, a small volume of sample was removed from the solution to avoid chloride contamination of the sample. The pH was measured at room temperature the following day in the actual sample. Because of the pH dependence on the temperature of TRIS solutions, the pH of the solution at 1 °C was calculated using pH(1 °C) = 0.964[pH(25 °C)] + 0.86. The samples were either used immediately after preparation or left on ice for a few hours until the measurement was started. All buffers were purged with Ar and treated so as to lower metal contamination. Time resolution of the measurement was 0.860 ns, and the time per channel was 0.562 ns. All fits were carried out with 300 data points, disregarding the 5 first points due to systematic errors in these. The actual data-analysis is performed on the time dependence of the gamma–gamma angular correlation function.22 Two NQIs were included in the analysis. For the minor species the linewidth (Δω0/ω0) and the asymmetry parameter (η) were fixed at the value obtained from the other spectrum (where it is the major species).
Results
There were two likely options for designing a tetrathiolate metal site based on the three metal-binding cysteine residues from α3DIV as shown in Fig. 1.20 One involved replacing a histidine in a loop region that previously had been implicated in binding Cd(II),19 and the other incorporated a cysteine two residues away from one of α3DIV cysteine residues to generate a single CXXC binding motif (Table 1). Both peptides fold into a three-helix bundle in the absence of metal (Fig. S1, ESI†). Cd(II) binds extremely tightly to both α3DIV-H72C and α3DIV-L21C to form 1:1 complexes (Fig. S2, ESI†). The UV-visible spectra are characterized by ligand-to-metal charge transfer (LMCT) transitions in the UV region. The complex formed by Cd(II) binding to α3DIV-H72C is characterized by a single broad peak at 232 nm with a molar extinction coefficient of 25 000 M−1 cm−1 (Fig. 2). That of α3DIV-L21C shows a strong transition at 224 nm with a shoulder at 243 nm and molar extinction coefficients of 33 000 M−1 cm−1 and 18 500 M−1 cm−1, respectively (Fig. 2 and Table 2).
Table 1.
Sequences of peptides
| Peptide | Sequence |
|---|---|
| α3DIV | MGS WAEFKQR LAAIKTR CQAL GG SEAECAAFEKE IAAFESE LQAY KGKG NPE VEALRKE AAAIRDE CQAY RHN |
| α3DIV-H72C | MGS WAEFKQR LAAIKTR LQAC GG SEAECAAFEKE IAAFESE LQAY KGKG NPE VEALRKE AAAIRDE CQAY RCN GSGC |
| α3DIV-L21C | MGS WAEFKQR LAAIKTR CQAC GG SEAECAAFEKE IAAFESE LQAY KGKG NPE VEALRKE AAAIRDE CQAY RHN GSGC |
Fig. 2.
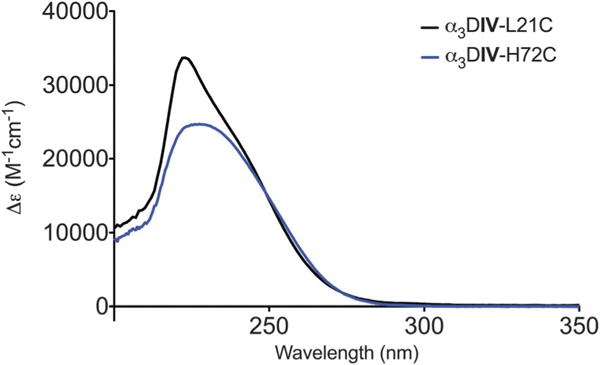
UV-visible spectroscopy of Cd(II) bound to α3DIV-H72C and α3DIV-L21C in 50 mM CHES buffer at pH 8.6.
Table 2.
Tabulated values for UV-visible spectroscopy and 113Cd NMR
The coordination of Cd(II) in α3DIV-H72C and α3DIV-L21C was assessed using 113Cd NMR. Spectra from α3DIV-H72C with 0.8 eq. 113Cd(II) in 10% D2O show a single peak at 595 ppm, which can also be seen in Cd(II)-bound α3DIV (Fig. 3 and Table 2). While very few native proteins exhibit chemical shifts in this region, previous studies of designed proteins have shown that this chemical shift range is often associated with CdS3O coordination.13,17,23 On the other hand, the chemical shift of α3DIV-L21C is 685 ppm (Fig. 2 and Table 2), a chemical shift which has been more typical of CdS3 sites in designed proteins, although native proteins with pure S3 coordination of Cd(II) are not known. The chemical shift of 113Cd-substituted rubredoxin has been reported at 730 ppm.24 Thus, this NMR signal is most consistent with a high proportion of CdS4 coordination that may be undergoing exchange that is faster than the millisecond timescale of NMR.
Fig. 3.
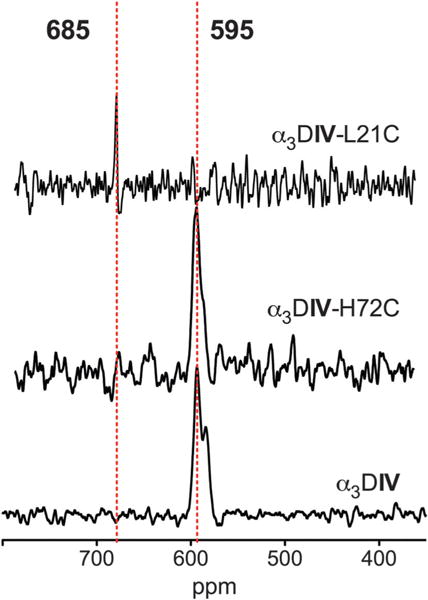
113Cd NMR reveals different coordination environments for α3DIV-H72C and α3DIV-L21C, while the coordination environments in α3DIV and α3DIV-H72C are similar. All experiments were conducted with 2–3 mM peptide and 0.8 eq. of enriched 113Cd in 10% D2O.
Since 111mCd PAC can resolve multiple species on a faster timescale than NMR, we turned to this technique to understand the Cd coordination in these two peptides. The data sets for α3DIV-H72C and α3DIV-L21C were initially analyzed with one NQI, capturing the major species, but this did not give satisfactory fits, and thus two NQIs (ω0 ~ 0.075 rad ns−1 and ~0.35 rad ns−1) were included for each sample (Fig. 4 and Table 3). The ω0 ~ 0.075 rad ns−1 NQI was the major species for α3DIV-H72C, while ω0 ~ 0.35 rad ns−1 was the major species recorded for α3DIV-L21C. This resulted in acceptable fits, and agreed with visual inspection of the spectra (Fig. 4). In other words, for α3DIV-H72C the major species (ω0 = 0.356 rad ns−1, 79%) is accompanied by a minor species (ω0 = 0.082 rad ns−1, 21%), which is highly similar to the major species recorded (72.5%) for α3DIV-L21C. Conversely, the minor species (27.5%) recorded for α3DIV-L21C resembles the major species recorded for α3DIV-H72C. The relative populations for each site were derived from the amplitudes (A) in Table 3. The NQI with ω0 around 0.350 rad ns−1 is comparable to that reported previously in the literature reflecting a CdS3O coordination geometry, although with slightly higher frequency.13,14 The NQI with ω0 of about 0.080 rad ns−1 is highly similar to that reported in the literature for the structural (Cys4) site of horse liver alcohol dehydrogenase9 and for a peptide that reproduces this site.25 Thus, the dominating species for α3DIV-H72C is most likely a CdS3O coordination geometry, accompanied by a minor fraction, which is most likely a CdS4 coordination geometry. Conversely, the dominating species for α3DIV-L21C is most likely a CdS4 site accompanied by a minor fraction, which is a CdS3O site.
Fig. 4.
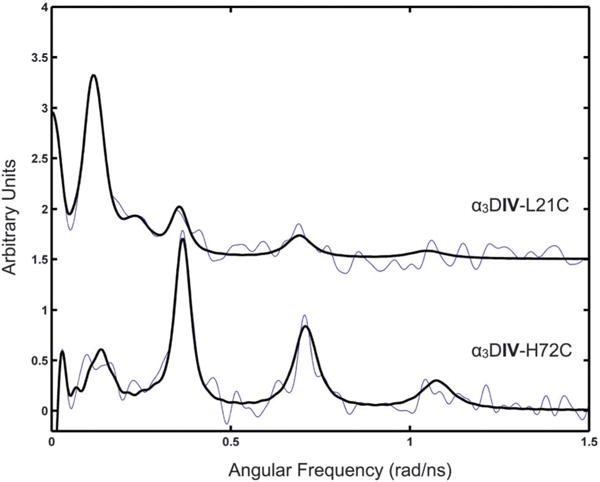
111mCd PAC spectra for α3DIV-L21C (top) and α3DIV-H72C (bottom) with the data (fine line) and fit (thick line). Experiment was performed at 300 μM peptide, 20 mM pH 8.6 TRIS and a peptide/Cd(II) ratio of 12:1.
Table 3.
Parameters fitted to PAC-data
| Peptide | pH | ω0 (rad ns−1) | η | Δω0/ω0 × 100 | 1/τc (μs−1) | A × 100 | χr2 |
|---|---|---|---|---|---|---|---|
| α3DIV-H72Ca | 8.6 | 0.082 ± 0.003 | 0.79f | 15f | 4.3 ± 0.7 | 1.7 ± 0.2 | 1.26 |
| 0.356 ± 0.001 | 0.16 ± 0.01 | 4.2 ± 0.5 | 4.3 ± 0.7 | 6.3 ± 0.4 | |||
| α3DIV-L21Ca | 8.6 | 0.071 ± 0.002 | 0.79 ± 0.06 | 15 ± 3 | 5.2 ± 0.8 | 5.0 ± 0.2 | 1.28 |
| 0.347 ± 0.003 | 0.16f | 4f | 5.2 ± 0.8 | 1.9 ± 0.2 | |||
| TRIL16Cb,c | 8.7 | 0.337 ± 0.002 | 0.23 ± 0.02 | 5.1 ± 0.7 | 8 ± 5 | 5.1 ± 0.6 | 1.10 |
| 0.438 ± 0.004 | 0.20 ± 0.03 | 5.4 ± 0.7 | 3 ± 3 | 3.6 ± 0.4 | |||
| TRIL12AL16Cd,e | 8.8 | 0.3405 ± 0.0003 | 0.141 ± 0.004 | 1.7 ± 0.1 | 5.4 ± 0.5 | 8.3 ± 0.2 | 1.06 |
| C4 peptidef | 0.075 ± 0.001 | 0.60 ± 0.09 | 16 ± 5 | 38 ± 5 | |||
| LADH C4 siteg | 0.065 ± 0.001 | 0.81 ± 0.06 | 10 ± 3 | 8.7 ± 0.1 |
The amplitudes of each species from the fitting of the PAC data, can be used to analyze the NMR data in order to generate a correlation between the proportion of each species to extract the “pure” chemical shift for each species. By converting the amplitudes to the fraction represented by each species, a simple system of equations can be written to calculate the chemical shift of the pure species,
where x and y are the chemical shift for CdS3O and CdS4, respectively. Solving this system of equations predicts a chemical shift of 730 ppm for pure CdS4 and 560 ppm for pure CdS3O, both of which are reasonable values that fall in line with what has previously been observed for these CdS424 and CdS3O17 species, respectively. The observed chemical shift for the CdS3O species is slightly lower (~20 ppm) than has been observed for both our previous designed peptides, as well as for native proteins with this center, but compared over the ~900 ppm range accessible by 113Cd NMR, this is likely due to systematic error in the calculation of the PAC amplitudes. Indeed, the value of 560 ppm is within error for that previously determined in our lab based on a comparison of S3 and S3O sites.17 These calculations suggest that the observed behavior is due to coalescence of the NMR signals caused by rapid water/thiolate exchange from the cadmium ion.
Discussion
A tris-thiolate metal binding site was previously incorporated into a single-stranded three-helix bundle, α3D,26,27 to generate the construct, α3DIV,19 which is capable of binding Pb(II), Hg(II), and Cd(II). A minor species with possible CdS3N coordination sphere was measured by 111mCd PAC spectroscopy, suggesting that a nearby histidine residue was capable of interacting with bound Cd(II). In seeking to incorporate a fourth cysteine residue to create a tetrathiolate metal binding site, a logical first step would be the replacement of this nearby histidine residue, with a cysteine generating α3DIV-H72C. While histidine has a significantly longer extension than cysteine, the location of this residue on a flexible loop region could allow for insertion of the thiolate sulfur into the Cd(II) first coordination sphere. As an alternate approach, we also identified another residue, Leu21, which is located two residues away from Cys18, one of the cysteines incorporated into α3DIV. This site would incorporate a CXXC motif, much as is found in zinc fingers, rubredoxin, and Cd(II)-sensing proteins. These two constructs represent two reasonable approaches to generating S4 sites, and characterizing them could also provide some insights into the basic requirements for tetrahedral metal binding sites.
The UV-visible spectroscopy of both α3DIV-H72C and α3DIV-L21C are consistent with Cd(II) coordinated to cysteine thiolates. Both of these constructs have much higher molar extinction coefficients than previously designed Cd(II)-binding peptides from our laboratory,13,19,23,28 but are consistent with native Cd(II)-binding proteins.6 Previous studies have elucidated that molar extinction coefficients increase linearly with respect to the increasing number of thiolates coordinated to Cd(II),29 so the increase in molar extinction coefficient observed for these peptides compared to other designed peptides (e.g., Cd(TRIL12AL16C)3−)23 is consistent with increased cysteine coordination. CadC shows a single, broad peak at 238 nm with a molar extinction coefficient of 25 000 M−1 cm−1.4 This differs greatly from the absorption of Cd-substituted rubredoxin, which is centered at 232 nm with three resolvable transitions at 245 nm, 229 nm, and 213 nm.29 The molar extinction coefficient of the lowest energy transition can be used to estimate the number of thiolates bonded to the Cd(II) based on ~6000 M−1 cm−1 per thiolate.29 Other studies, including those on CadC, have used this rule of thumb even when the spectrum only has a single broad peak and the lowest energy transition was not resolvable.6 The extinction coefficient of the shoulder at 243 nm of α3DIV-L21C is lower than the 24 000 M−1 cm−1 benchmark for tetrathiolate coordination, which could signal that less than 100% of the protein in the sample binds to Cd(II) with four thiolates. The lack of multiple resolvable transitions in CadC and α3DIV-H72C is interesting and suggests that α3DIV-H72C is in some ways a good spectroscopic model for CadC. The band structure found in rubredoxins and zinc fingers likely arises from the tetrahedral geometry of the site, and the lack of this structure in CadC indicates that the Cd(II) coordination could be highly distorted with one very long Cd–S bond, or in equilibrium between CdS3O and CdS4, creating the broader transition at an intermediate energy (238 nm vs. 229 nm and 245 nm as found in rubredoxin). This appears to also be the case in α3DIV-H72C, which features a similar broad band at an intermediate energy (232 nm vs. 224 nm and 243 nm as found in α3DIV-L21C). In comparison to α3DIV, α3DIV-H72C has a higher molar extinction coefficient (18 200 vs. 25 000), which supports the concept that this may represent a mixture of two coordination environments and that some greater amount of thiolate coordination is present.
113Cd NMR was used to investigate Cd(II) coordination in α3DIV-H72C and α3DIV-L21C. The chemical shifts of α3DIV-H72C and α3DIV-L21C were 595 ppm and 685 ppm, respectively. As a result of this data, the original assignments for α3DIV 113Cd NMR have been revisited. Initially it was thought that the signal at 595 ppm represented a CdS3N species while the signal at 583 ppm reflected a CdS3O species. Yet comparison of 113Cd NMR data from α3DIV-H72C and α3DIV reveals that the 595 ppm resonance persists in the two proteins indicating that this signal represents the CdS3O species. While very few native proteins exhibit chemical shifts in this region, studies in our lab have shown that this chemical shift range is often associated with CdS3O. A series of studies on rubredoxin mutants with cysteine to serine mutations measured the chemical shifts of these S3O sites to be 605–645 ppm, while the chemical shift of 113Cd-substituted rubredoxin has been reported at 730 ppm.24 However, there are Cd-substituted S4 sites in native proteins that have shown chemical shifts lower than 700 ppm, although many of these tend to be S4 bimetallic bridged structures.10 The origin of the chemical shift of 685 ppm for α3DIV-L21C could be due to a distorted site or to an equilibrium between CdS3O and CdS4 forms that exchange faster than the NMR timescale, leading to coalescence of the signals from the two species to a single, intermediate chemical shift.
The 111mCd PAC data indicate that both CdS3O and CdS4 species are found in both peptides in inverse proportions. Taken together, the NMR and PAC spectroscopic data suggest that α3DIV-H72C adopts a primarily CdS3O coordination with a small proportion of CdS4. The UV-visible spectroscopy supports this model as the spectrum is characterized by a single broad transition. The molar extinction coefficient is higher than those previously reported for these sites by our group, but this could be due to the small proportion of CdS4, which can contribute up to 6000 M−1 cm−1 to the molar extinction coefficient.29,30 The PAC and UV-visible spectroscopy indicate that the major species for α3DIV-L21C is CdS4. The chemical shift for this peptide is smaller than that for a Cd-substituted classically tetrahedral protein, such as rubredoxin. Given the two species in the PAC data and the good agreement in energy for the LMCT, it seems most likely that coalescence of the signals causes the smaller chemical shift in the 113Cd NMR. Assuming the molar extinction coefficient for the resolved low energy transition in α3DIV-L21C is only due to CdS4, a correction can be derived to calculate the real molar extinction coefficient for the CdS4 species. This calculation yields ε243nm of 25500 M−1 cm−1, which reflects the expected molar extinction coefficient for a CdS4 site.
Based on the correlation between the PAC amplitudes and the NMR data, the chemical shift of the pure CdS3O and CdS4 species was calculated to be 560 ppm and 730 ppm, respectively. These values agree very well with what has previously been determined for these species. The value for CdS3O coordination is within 20 ppm of what has previously been determined in our lab for a correlation between CdS3 and CdS3O coordinations,17 which is within the margin of error for the determination of the PAC amplitudes. This rationale and calculated values for the pure species can be applied to CadC. The NMR signal for CadC, 622 ppm, is then made up of two signals at differing proportions. The equation for this system can be written as,
where x is the proportion of S4 species. This analysis suggests that the NMR signal for CadC is produced by coalescence between the two forms, where 40% exists as S4 and 60% exists as S3O. This NMR-derived speciation model also explains the λmax and molar extinction coefficient from the UV spectra of CadC. Finally, the EXAFS data recorded for CadC are fitted almost equally well with a CdS4 and a CdS3O coordination sphere, but with a very large Debye–Waller factor for the oxygen.4 Thus, we conclude that the results from the α3DIV-H72C and α3DIV-L21C provide compelling evidence supporting a rapid exchange between S3O and S4 species in CadC, and that this model provides a coherent interpretation of UV-absorption, 113Cd NMR, and EXAFS spectroscopic data recorded for CadC.
The function of CadC is to serve as a sensitive metal ion sensor that can respond rapidly to variations of Cd(II) concentrations in cells. When Cd(II) levels are extremely low, it must repress synthesis of the heavy metal detoxification pathway by binding to DNA, while falling off the repressor region when concentrations of Cd(II) increase. As this sensor works at picomolar Cd(II) concentrations, this requires CadC to have a very high affinity for its target metal. However, this switch must be reversible as when Cd(II) levels are diminished, the protein must be reused to repress gene expression. Rapid exchange between two coordination spheres can serve as a functional feature of CadC such that this fast exchange can facilitate transfer of Cd(II) from small molecule ligands, such as glutathione, to the metal binding site. Furthermore, the dynamic nature of the site can allow it to sense small changes in the intracellular concentration of Cd(II) resulting in a highly sensitive and responsive sensor. Similarly, PAC data clearly demonstrate the presence of two co-existing coordination spheres for Ag(I) and possibly for Cd(II) bound to the ArsR/SmtB family member BxmR.31 Thus, it seems plausible that an intrinsic feature of such metalloregulating proteins is the ability to have a dynamic coordination site that can facilitate exchange so that the protein does not irreversibly lock a metal into the regulatory domain.
In summary, we have reported the design and characterization of two three-helix bundles with four cysteine residues. Both peptides bind Cd(II) tightly in a 1:1 complex that is characterized by high energy LMCT. The two peptides, α3DIV-H72C and α3DIV-L21C, were then characterized with 113Cd NMR, suggesting that each assumes a different coordination. 111mCd PAC revealed that both peptides coordinate Cd(II) in a mixture of CdS3O and CdS4 with opposite major species such that the major species of α3DIV-H72C was CdS3O and α3DIV-L21C was CdS4. Correlating the NMR chemical shifts and the proportion of each species derived from the PAC fits results in the pure species being represented by chemical shifts of 730 ppm and 560 ppm for CdS4 and CdS3O, respectively. Applying these values to CadC suggests that CadC also coordinated Cd(II) as a mixture of CdS3O and CdS4 under rapid exchange with 60% CdS3O and 40% CdS4, which agrees well with reported spectroscopy for this protein. This data suggests a model for metalloregulatory proteins in which apparently distorted coordination spheres are actually in fast exchange between two different forms, which may function to facility metal binding from low molecular weight compounds in the cytosol and sense small changes in the concentration of metals.
Acknowledgments
A.G.T. acknowledges training grant support from the University of Michigan Chemistry-Biology Interface (CBI) training program (NIH grant 5T32GM008597) and V.L.P. would like to thank the National Institutes of Health (NIH) for financial support for this research (ES012236).
Footnotes
Electronic supplementary information (ESI) available. See DOI: 10.1039/c5mt00228a
References
- 1.Ma Z, Jacobsen FE, Giedroc DP. Chem Rev. 2009;109:4644–4681. doi: 10.1021/cr900077w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gaggelli E, Kozlowski H, Valensin D, Valensin G. Chem Rev. 2006;106:1995–2044. doi: 10.1021/cr040410w. [DOI] [PubMed] [Google Scholar]
- 3.Argüello JM, Raimunda D, González-Guerrero M. J Biol Chem. 2012;287:13510–13517. doi: 10.1074/jbc.R111.319343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Busenlehner LS, Cosper NJ, Scott RA, Rosen BP, Wong MD, Giedroc DP. Biochemistry. 2001;40:4426–4436. doi: 10.1021/bi010006g. [DOI] [PubMed] [Google Scholar]
- 5.Wang Y, Kendall J, Cavet JS, Giedroc DP. Biochemistry. 2010;49:6617–6626. doi: 10.1021/bi100490u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Busenlehner LS, Weng TC, Penner-Hahn JE, Giedroc DP. J Mol Biol. 2002;319:685–701. doi: 10.1016/S0022-2836(02)00299-1. [DOI] [PubMed] [Google Scholar]
- 7.Ye J, Kandegedara A, Martin P, Rosen BP. J Bacteriol. 2005;187:4214–4221. doi: 10.1128/JB.187.12.4214-4221.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Busenlehner LS, Giedroc DP. J Inorg Biochem. 2006;100:1024–1034. doi: 10.1016/j.jinorgbio.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 9.Hemmingsen L, Bauer R, Bjerrum MJ, Zeppezauer M, Adolph HW, Formicka G, Cedergren-Zeppezauer E. Biochemistry. 1995;34:7145–7153. doi: 10.1021/bi00021a028. [DOI] [PubMed] [Google Scholar]
- 10.Armitage IM, Drakenberg T, Reilly B. In: Cadmium: From Toxicity to Essentiality. Sigel A, Sigel H, Sigel RK, editors. Vol. 11. Springer Netherlands; Dordrecht: 2012. pp. 117–144. [Google Scholar]
- 11.Touw DS, Nordman CE, Stuckey JA, Pecoraro VL. Proc Natl Acad Sci U S A. 2007;104:11969–11974. doi: 10.1073/pnas.0701979104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dieckmann GR, McRorie DK, Tierney DL, Utschig LM, Singer CP, O’Halloran TV, Penner-Hahn JE, DeGrado WF, Pecoraro VL. J Am Chem Soc. 1997:6195–6196. [Google Scholar]
- 13.Matzapetakis M, Farrer BT, Weng TC, Hemmingsen L, Penner-Hahn JE, Pecoraro VL. J Am Chem Soc. 2002;124:8042–8054. doi: 10.1021/ja017520u. [DOI] [PubMed] [Google Scholar]
- 14.Peacock AFA, Hemmingsen L, Pecoraro VL. Proc Natl Acad Sci U S A. 2008;105:16566–16571. doi: 10.1073/pnas.0806792105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iranzo O, Cabello C, Pecoraro VL. Angew Chem, Int Ed. 2007;46:6688–6691. doi: 10.1002/anie.200701729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iranzo O, Chakraborty S, Hemmingsen L, Pecoraro VL. J Am Chem Soc. 2011;133:239–251. doi: 10.1021/ja104433n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iranzo O, Jakusch T, Lee KH, Hemmingsen L, Pecoraro VL. Chem – Eur J. 2009;15:3761–3772. doi: 10.1002/chem.200802105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chakraborty S, Iranzo O, Zuiderweg ERP, Pecoraro VL. J Am Chem Soc. 2012;134:6191–6203. doi: 10.1021/ja210510g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chakraborty S, Kravitz JY, Thulstrup PW, Hemmingsen L, DeGrado WF, Pecoraro VL. Angew Chem, Int Ed. 2011;50:2049–2053. doi: 10.1002/anie.201006413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Plegaria JS, Dzul S, Zuiderweg ERP, Stemmler TL, Pecoraro VL. Biochemistry. 2015;54:2858–2873. doi: 10.1021/acs.biochem.5b00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Studier FW. Protein Expression Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 22.Hemmingsen L, Sas KN, Danielsen E. Chem Rev. 2004;104:4027–4062. doi: 10.1021/cr030030v. [DOI] [PubMed] [Google Scholar]
- 23.Iranzo O, Ghosh D, Pecoraro VL. Inorg Chem. 2006;45:9959–9973. doi: 10.1021/ic061183e. [DOI] [PubMed] [Google Scholar]
- 24.Xiao Z, Lavery MJ, Ayhan M, Scrofani SDB, Wilce MCJ, Guss JM, Tregloan PA, George GN, Wedd AG. J Am Chem Soc. 1998;120:4135–4150. [Google Scholar]
- 25.Heinz U, Hemmingsen L, Kiefer M, Adolph HW. Chem – Eur J. 2009;15:7350–7358. doi: 10.1002/chem.200900870. [DOI] [PubMed] [Google Scholar]
- 26.Walsh ST, Cheng H, Bryson JW, Roder H, DeGrado WF. Proc Natl Acad Sci U S A. 1999;96:5486–5491. doi: 10.1073/pnas.96.10.5486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bryson JW, Desjarlais JR, Handel TM, DeGrado WF. Protein Sci. 1998;7:1404–1414. doi: 10.1002/pro.5560070617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matzapetakis M, Ghosh D, Weng TC, Penner-Hahn JE, Pecoraro VL. JBIC, J Biol Inorg Chem. 2006;11:876–890. doi: 10.1007/s00775-006-0140-7. [DOI] [PubMed] [Google Scholar]
- 29.Henehan CJ, Pountney DL, Vasák M, Zerbe O. Protein Sci. 1993;2:1756–1764. doi: 10.1002/pro.5560021019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pountney DL, Tiwari RP, Egan JB. Protein Sci. 1997;6:892–902. doi: 10.1002/pro.5560060416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu T, Chen X, Ma Z, Shokes J, Hemmingsen L, Scott RA, Giedroc DP. Biochemistry. 2008;47:10564–10575. doi: 10.1021/bi801313y. [DOI] [PMC free article] [PubMed] [Google Scholar]