

Abstract
The fusion inhibitor T20 belongs to a new class of anti-human immunodeficiency virus type 1 (HIV-1) drugs designed to block entry of the virus into the host cell. However, the success of T20 has met with the inevitable emergence of drug-resistant HIV-1 variants. We describe an evolutionary pathway taken by HIV-1 to escape from the selective pressure of T20 in a treated patient. Besides the appearance of T20-resistant variants, we report for the first time the emergence of drug-dependent viruses with mutations in both the HR1 and HR2 domains of envelope glycoprotein 41. We propose a mechanistic model for the dependence of HIV-1 entry on the T20 peptide. The T20-dependent mutant is more prone to undergo the conformational switch that results in the formation of the fusogenic six-helix bundle structure in gp41. A premature switch will generate nonfunctional envelope glycoproteins (dead spikes) on the surface of the virion, and T20 prevents this abortive event by acting as a safety pin that preserves an earlier prefusion conformation.
Despite the success of highly active antiretroviral therapy, the emergence of drug-resistant human immunodeficiency virus type 1 (HIV-1) strains has become a widespread and growing problem (25). The search for new antiviral drugs therefore has to continue, and of special interest are inhibitors that target previously unexploited steps in the HIV-1 life cycle (30). One interesting avenue that is currently an area of intense investigation is the targeting of the viral entry process (1, 29). The entry process is a critical step in the HIV-1 replication cycle that is mediated by the envelope (Env) glycoproteins, gp120 and gp41. Env is arranged on the virus particle as trimeric spikes, comprising three gp120 and three gp41 molecules, anchored within the viral membrane via the gp41 transmembrane (TM) domain. Binding of the surface subunit gp120 to CD4 and a coreceptor on the T-cell surface triggers conformational changes in the Env complex, leading to the insertion of the hydrophobic N-terminal fusion peptide of gp41 into the target cell membrane (reviewed in reference 11).
Subsequent changes within the gp41 ectodomain (gp41e) involve two leucine zipper-like motifs, heptad repeat 1 (HR1) and heptad repeat 2 (HR2). Ultimately, HR1 and HR2 from three gp41 molecules assemble into a highly stable six-helix bundle structure, which juxtaposes the viral and cellular membranes for the fusion event (5, 38, 41). The change in free energy associated with this structural transition within gp41e is predicted to be sufficient to cause lipid mixing and membrane fusion (14, 28). Peptide fusion inhibitors that bind to one of the HR motifs can block this conformational switch and thus inhibit viral entry (1, 6, 26, 43).
The fusion inhibitor T20 (also called enfuvirtide and fuzeon) is the most clinically advanced drug of a new class of antivirals designed to inhibit viral entry (23). T20 is a synthetic 36-amino-acid peptide derived from the C-terminal region of HR2 (42, 43). By competitively binding to HR1, T20 blocks the formation of the six-helix bundle, a prerequisite for membrane fusion and viral entry (20, 43). HIV-1 variants that are resistant to this compound have already emerged (19, 33, 40). Continuous in vitro passaging of HIV-1 in the presence of increasing T20 concentrations resulted in the selection of resistant virus variants within 6 weeks (34). Sequence analysis of the resistant viral population revealed the acquisition of mutations within a stretch of three HR1 amino acids, glycine-isoleucine-valine (further referred to as the GIV sequence; HXB2 amino acid positions 36 to 38 of gp41) that is highly conserved within HIV-1 (10, 13, 35, 44, 45). Resistance mutations have also been identified within the viral quasispecies of patients on T20 therapy (33, 40). Specifically, the change of valine to alanine (GIV to GIA; mutated amino acid underlined) provided 45-fold resistance to T20.
In this study, we performed a genetic analysis on the entire HIV-1 gp41 ectodomain from a patient who failed on T20 therapy. Sequence analysis revealed the acquisition of novel T20 resistance mutations in the HR1 domain of gp41. We also document, for the first time, changes in the three-amino-acid SNY sequence of the HR2 domain. Surprisingly, we demonstrate that an HR1-HR2 double mutant (GIA-SKY), which dominates the viral population after 32 weeks of therapy, is not only highly resistant to T20 but also critically dependent on the T20 peptide for its replication. We propose a mechanistic model that explains this novel feature of drug-dependent viral entry.
MATERIALS AND METHODS
HIV-1 RNA isolation, PCR amplification, and sequencing.
The patient in this study was part of the TORO-2 study trial (23). The plasma viral RNA load was measured with the Versant HIV-1 RNA 3.0 assay (bDNA) kit (Bayer Corporation, Tarrytown, N.Y.). Total RNA was isolated from 200 μl of EDTA-treated plasma with the Boom method (3) and eluted in a total volume of 40 μl of water. Primer and template RNAs (20 μl) were allowed to anneal for 2 min at room temperature in a mixture containing 4 ng of antisense primer (gp41-3′outer, 5′-GTGAGTATCCCTGCCTAACTCTAT-3′) in CMB buffer (0.01 M Tris-HCl, pH 8.3, 0.05 M KCl, 0.1% Triton), 0.8 mM deoxynucleoside triphosphates, and 20 U of Rnasin (Promega, Madison, Wis.). Upon annealing, we added 100 U of Moloney murine leukemia virus reverse transcriptase (Life Technologies, Paisley, Scotland) and MgCl2 to a final concentration of 2.4 mM, followed by incubation for 2 h at 37°C in a total volume of 40 μl. The reaction mixture was subsequently inactivated at 95°C for 5 min.
The complete reverse transcription reaction mixture (40 μl) was added to 60 μl of a PCR mixture with 100 ng of a sense primer (gp41-5′outer, 5′-GAGGGACAATTGGAGAAGTGAATT-3′) and the antisense primer (gp41-3′outer), CMB buffer, 0.8 mM deoxynucleoside triphosphates, 1.65 mM MgCl2 and 2 U of Taq polymerase (Perkin-Elmer Cetus). After incubation for 5 min at 95°C, the reaction mixture was subjected to 35 PCR cycles in a type 9700 DNA thermal cycler (Perkin Elmer Cetus), with each cycle including a denaturation step for 1 min at 95°C, an annealing step for 1 min at 55°C, and an extension step for 2 min at 72°C. This was followed by a final extension step of 10 min at 72°C. The nested PCR was performed with 5 μl of the first PCR and 50 ng of the sense and antisense primers (gp41-5′inner, 5′-GGAGAAGTGAATTATATAAATATAAAG-3′, and gp41-3′inner, 5′-CACTATAGAAAGTACAGCAAAAACTAT-3′) in a 50-μl PCR as above but with 1.2 mM MgCl2. DNA products were analyzed on a 2% agarose gel that was prestained with ethidium bromide.
PCR products were sequenced directly with the nested set of primers (gp41-5′inner and gp41-3′inner) with the DNA Big Dye terminator sequencing kit (ABI, Foster City, Calif.) and an ABI 377 automated sequencer. PCR products were also cloned with the TA cloning kit (Invitrogen, San Diego, Calif.). White colonies were screened by PCR with primers M13 reverse and M13 (−21) provided in the TA cloning kit, and the products were analyzed on a 2% agarose gel for the correct size. Positive clones were sequenced with the M13 reverse and M13 (−21) primers as described above.
Construction of LAI molecular clones.
The full-length molecular HIV-1 clone pLAI was used to produce wild-type and mutant viruses (31). We first constructed a wild-type virus with the GIV-SNY sequence as observed in the patient isolate in place of the GIV-NNY sequence that is present in the LAI molecular clone. Plasmid pRS1, designed to subclone mutant env genes, was generated as follows. First, the SalI-BamHI fragment from pLAI was cloned into pUC18 (Roche, Indianapolis, Ind.). Second, a PstI-StuI fragment from the resulting plasmid was cloned into plasmid pBS-SK(+)-gp160 containing the SalI-XhoI sequences of pLAI. Mutations were introduced in pRS1 with the Quickchange mutagenesis kit (Stratagene, La Jolla, Calif.) and verified by DNA sequencing. Mutant env genes in pRS1 were cloned back into pLAI as SalI-BamHI fragments.
Transfections and CA-p24 determination.
The SupT1 T-cell line was maintained in RPMI 1640 supplemented with 10% fetal calf serum and penicillin and streptomycin (both at 100 U/ml) and incubated at 37°C with 5% CO2. SupT1 cells were transfected with HIV-1 molecular clones by electroporation. Briefly, 5 × 106 cells were washed in RPMI 1640 with 20% fetal calf serum, mixed with 1 μg of DNA in 0.4-cm cuvettes, and electroporated at 250 V and 975 μF, followed by resuspension of cells in RPMI 1640 with 10% fetal calf serum. The transfected cells were split at day 1 posttransfection and cultured with and without T20 (Trimeris, Durham, N.C.). CA-p24 production was determined from culture supernatant taken at various days posttransfection with an in-house CA-p24 antigen capture enzyme-linked immunosorbent assay (Abbott).
Cell-cell fusion assay.
SupT1 cells (5 × 106) were transfected with 5 μg of the indicated pLAI variants and/or the pcDNA3Tat expression plasmid as described above. The cells were cultured for 24 h, spun at 1,200 rpm for 10 min, and resuspended in fresh medium containing saquinavir (1 μM final concentration), with or without T20 at various concentrations. Cells were subsequently mixed with a SupT1 culture that had been transfected the previous day with 5 μg of the long terminal repeat-luciferase reporter plasmid. Cells were cultured for 24 h and scored for syncytium formation, and the cell lysate was assayed for luciferase production (Promega, Madison, Wis.), which was measured with a luminometer. The output was expressed as relative light units.
Single-cycle virus infection assay.
SupT1 cells were transfected with 5 μg of the long terminal repeat-luciferase reporter vector. These cells (5 × 106) were infected 24 h posttransfection with an equal amount of different virus variants (20 ng of CA-p24) in the presence or absence of T20. The cell lysate was assayed 24 h later for luciferase production as described above. To eliminate the possibility of reinfection, saquinavir (1 μM final concentration) was included. The GIA-SKY virus was produced from a 5-μg DNA transfection into SupT1 cells in the presence (100 ng/ml) and absence of T20 as described above.
Peptide production.
The pN36/C34 plasmid, encoding the patient isolate N36(L6)C34 model peptide, was derived from pN36/C34JR-FL (37). Amino acid substitutions were introduced into pN36/34 by the method of Kunkel et al. (21) and then verified by DNA sequencing. All recombinant peptides were expressed in Escherichia coli strain BL21(DE3)/pLysS (Novagen, Madison, Wis.). The bacteria were grown at 37°C in LB medium to an optical density of 0.8 at 600 nm and induced with isopropylthio-β-d-galactoside for 3 to 4 h. Cells were lysed at 0°C with glacial acetic acid. The bacterial lysate was centrifuged (35,000 × g for 30 min) to separate the soluble fraction from inclusion bodies. The soluble fraction, containing denatured peptide, was dialyzed into 5% acetic acid overnight at room temperature. Peptides were purified from the soluble fraction to homogeneity by reverse-phase high-performance liquid chromatography (Waters, Milford, Mass.) with a Vydac C-18 preparative column (Vydac, Hesperia, Calif.) and a water-acetonitrile gradient in the presence of 0.1% trifluoroacetic acid and then lyophilized. The molecular weight of each peptide was confirmed by matrix-assisted laser desorption ionization-time-of-flight mass spectrometry (PerSeptive Biosystems, Framingham, Mass.). The concentration of each peptide was determined at 280 nm after solubilization in 6 M guanidinium chloride (12).
Circular dichroism spectroscopy.
High-pressure liquid chromatography-purified peptides were solubilized in 6 M guanidinium chloride and 10 mM Tris-HCl (pH 7.0) and refolded by dilution into phosphate-buffered saline at neutral pH. The single-point-substituted variant peptides were named according to the position of the substitution. Circular dichroism experiments were performed with an Aviv 62A DS circular dichroism spectrometer. The wavelength dependence of molar ellipticity, [θ], was monitored at 4°C, with a 10 μM peptide solution in 100 mM NaCl-50 mM sodium phosphate, pH 7.0 (phosphate-buffered saline). Helix content was calculated by the method of Chen et al. (7). Thermal stability was determined by monitoring the change in the circular dichroism signal at 222 nm ([θ]222) as a function of temperature. Thermal melts were performed in 2°C increments with an equilibration time of 2 min at the desired temperature and an integration time of 30 s. All melts were reversible. Superimposable folding and unfolding curves were observed, and >90% of the signal was regained upon cooling. The melting temperatures or midpoints of the cooperative thermal unfolding transitions (Tm) were determined from the maximum of the first derivative with respect to the reciprocal of the temperature of the [θ]222 values (4). The error in estimation of Tm is ±0.5°C.
Sedimentation equilibrium analysis.
A Beckman XL-A (Beckman Coulter, Fullerton, Calif.) analytical ultracentrifuge equipped with an An-60 Ti rotor (Beckman Coulter) was used. Peptide solutions were dialyzed overnight against phosphate-buffered saline (pH 7.0), loaded at initial concentrations of 10, 30, and 100 μM, and analyzed at rotor speeds at 20,000 and 23,000 rpm at 20°C. Data sets were fitted simultaneously to a single-species model of ln(absorbance) versus radial distance squared with the program Nonlin (15). Protein partial specific volume and solvent density were calculated as described by Laue et al. (22).
RESULTS
HIV-1 escapes from T20 therapy.
The patient involved in this study received T20 as 90-mg subcutaneous injections twice daily for 50 weeks in combination with reverse transcriptase and protease inhibitors (Fig. 1A). The start of T20 therapy resulted in a significant decline in HIV-1 viral RNA load from 84,377 to 356 copies/ml, followed by a rebound to 5,352 copies/ml at week 16, suggesting the emergence of a T20-resistant HIV-1 population. We analyzed the virus population present in this individual at multiple time points before, during, and after T20 therapy.
FIG. 1.
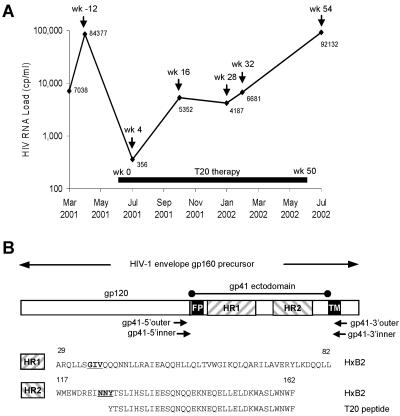
(A) Plasma HIV-1 RNA load during T20 therapy. Plasma samples that were analyzed by sequencing are marked in the graph as week -12 (pretherapy), week 16, week 28, week 32, and week 54. T20 therapy was started at week 0 and stopped at week 50. At the initiation of T20 therapy, the patient also received 300 mg of ziduvidine, 150 mg of lamivudine, and three caps of lopinavir twice daily. The patient's treatment regimen was modified at week 16 to 150 mg of lamivudine, 600 mg of amprenavir, 100 mg of ritonavir, 90 mg of T20 twice daily, and 300 mg of tenofovir disoproxil fumarate once daily. (B) PCR amplification of the gp41 ectodomain. The HIV-1 env gene is shown (not to scale) with the location of primers used to amplify the gp41e region. The HXB2 amino acid sequences for HR1 and HR2 are indicated. The GIV sequence in HR1 and SNY sequence in HR2 (NNY for strain HXB2) are underlined and bolded. The T20 peptide sequence within HR2 is indicated.
To determine whether the patient's HIV-1 population had acquired T20 resistance mutations, we sequenced the complete gp41e region (Fig. 1B). The viral RNA was isolated from plasma and amplified by nested RT-PCR. Sequencing of the HIV-1 quasispecies revealed the acquisition of mutations in the GIV and SNY sequences of HR1 and HR2 in the course of T20 therapy, but no other changes became fixed in the gp41e sequence (Fig. 2). Changes in the GIV sequence of HR1 have previously been correlated with T20 resistance (34, 40), but the description of the HR2 change is new.
FIG. 2.

Amino acid sequence of gp41e in individual HIV-1 clones from a patient under T20 therapy. The PCR fragments that were sequenced directly were ligated into the TA cloning vector, and 20 to 38 individual clones per time point were sequenced. Only the sequences that passed the quality control are shown (full-length sequence of correct product). All clones were aligned to a consensus sequence based on the 15 sequences of the pretreatment sample (week -12). Dots mark amino acids that are identical to this consensus sequence. Stop codons are indicated with *. The GIV and SNY motifs are underlined and in bold. The HR2 region corresponding to the T20 peptide sequence (amino acids 127 to 162) is underlined.
We further performed clonal sequencing of 153 individual clones to obtain a more accurate picture of the relative frequency of the mutational events (Fig. 2), which allowed us to build a detailed evolutionary scheme (Fig. 3). Analysis of the pretreatment sample confirmed the presence of the well-conserved GIV sequence in HR1 and the SNY sequence in HR2. At week 16 of T20 therapy, 97% of the viral population contained the GIG sequence in HR1 (30 of 31 clones). In the week 28 sample, we observed the complete disappearance of the GIG sequence at the expense of GIA (16 clones), GIW (13 clones), and the GKA double mutant (1 clone).
FIG. 3.
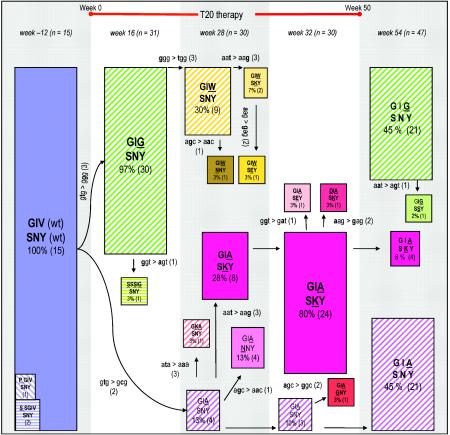
Evolution of T20 resistance in the ectodomain of HIV-1 gp41. This schematic of the viral quasispecies in the course of T20 therapy focuses exclusively on the GIV sequence in HR1 and the SNY sequence in HR2. The situation is depicted at week -12 (pretherapy), weeks 16, 28, and 32 (during T20 therapy), and week 54 (posttherapy). The number of clones that were sequenced per time point is indicated by n. The initial virus population present at week -12 consists of the fully wild-type sequence (GIV-SNY). The starting virus population is shown as a blue box, all single mutants are shown as hatched and colored boxes (e.g., GIG-SNY at week 16) and the double mutants in full color boxes (e.g., GIA-SKY at weeks 28 and 32). The sizes of the boxes reflect the abundance of that particular variant within the viral population. The actual percentage of that variant in the viral quasispecies and the number of clones with that sequence are indicated within the boxes. The evolution scheme was derived in part from inspection of the actual codon changes that are indicated alongside the arrows. The mutations are ranked according to their likelihood (2, 17, 18): the easy G-to-A and C-to-T transitions (1), the more difficult A-to-G and T-to-C transitions (2), and all transversions (3).
Inspection of the underlying codon changes indicates that GIW is made from the initial GIG variant, whereas GIA represents an independent evolutionary path that originates from the wild-type GIV sequence (see discussion). The week 28 sample shows the first changes in the SNY sequence of HR2. The wild-type SNY sequence is still dominant (14 clones), but we also observed the variants SKY (10 clones), NNY (5 clones), and SEY (1 clone). Inspection of the sequences indicates that the SKY mutation in HR2 is coupled to the GIA mutation in HR1. Specifically, of the 10 SKY sequences, eight are linked to GIA and only two to GIW, and the latter combines preferentially with the wild-type SNY sequence (9 of 13 clones). This GIA-SKY double mutant prevails at week 32, being present in 80% of the HIV-1 sequences (24 of 30 clones). Once the patient stopped T20 therapy after 50 weeks, the GIA-SKY double mutant disappears almost completely within 4 weeks (8%, 4 of 47 clones), which suggests that this variant is not very fit in the absence of T20. In this sample of week 54, we see the reappearance of the GIG-SNY (21 of 47 clones) and GIA-SNY (21 of 47 clones) single mutants.
T20-resistant and T20-dependent HIV-1 variants.
The major gp41e mutations observed under T20 therapy (GIG, GIW, and GIA in HR1 and SKY in HR2) were introduced in the LAI molecular clone, a CXCR4-with primary HIV-1 isolate from a French AIDS patient, and tested for their impact on in vitro virus replication and resistance to T20. Viral DNA constructs were transfected into the SupT1 T-cell line and cultured in the presence or absence of T20 (Fig. 4a). Replication of the wild-type virus was strongly inhibited by T20. In contrast, all HR1 variants (GIG, GIW and GIA) were highly resistant to T20. Surprisingly, the GIA-SKY double mutant was not only highly resistant to T20, but also critically dependent on T20 for its replication. In the absence of the T20 peptide, the GIA-SKY double mutant is unable to initiate a spreading infection. At 0.5 μg/ml of T20, the GIA-SKY mutant was able to replicate at levels comparable to the wild-type virus in the absence of the drug. At higher T20 concentrations, replication of the GIA-SKY mutant was inhibited, indicating that there is a certain T20 concentration window to elicit the drug-dependence phenotype.
FIG. 4.
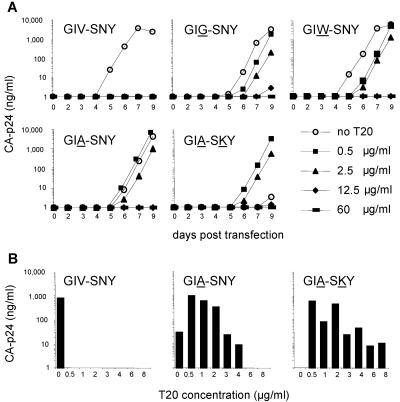
Replication of wild-type and mutant HIV-1 LAI molecular clones. (A) Viral replication curves over a 9-day period. The open-circle curve represents replication fitness in the absence of T20, and the other curves show the replication fitness of the mutants in the presence of different T20 concentrations. Similar results were obtained from six separate transfection experiments. (B) We used T20 concentrations that are comparable to that of patient plasma after twice daily subcutaneous 100-mg T20 injections (19). We only show CA-p24 values for day 6 posttransfection, which best represent the relative differences in replication capacity in the absence of T20 and the resistance-dependence phenotypes observed for the GIA-SNY and GIA-SKY mutants.
We also tested the replication capacity of the GIA and GIA-SKY mutants with concentrations of T20 comparable to levels in patient plasma (Fig. 4b) (19). The GIA mutant was resistant to relatively high levels of T20 (4 μg/ml). Interestingly, low amounts of T20 seem to have a stimulatory effect on this mutant. However, the GIA-SKY double mutant is critically dependent on T20 for its replication and more resistant to high T20 concentrations (6 to 8 μg/ml) than the GIA single mutant, which may explain why the GIA-SKY double mutant is selected in vivo.
The T20-dependent Env variant is hyperfusogenic.
We next tested the hypothesis that the addition of the K mutation in the GIA-SKY double mutant represented a compensatory mutation to enhance the rate of the structural transition in Env, but in a way that was now partially controlled by the presence of T20. To test this mechanistic interpretation, we performed additional experiments. First, such a mechanism predicts that fusogenic activity is reduced for the GIA resistant variant, but increased for the GIA-SKY double mutant. This was tested in a cell-cell fusion assay, which measures Env activity before a potential premature switch can occur, because the Env molecules can be engaged in the fusion process as soon as they appear at the cell surface. In this assay, one cell expresses the wild-type or mutant Env protein and the other cell the appropriate receptors (CD4 and CXCR4). Fusion was scored by the formation of syncytia. In addition, we introduced a long terminal repeat-luciferase reporter in the acceptor cell that is activated upon cell fusion by Tat protein that is expressed in the donor cell.
We consistently measured reduced fusion activity (syncytia and luciferase counts) for the GIA mutant and increased activity for the GIA-SKY mutant. The representative experiment in Fig. 5A shows 126% fusion activity for the GIA-SKY mutant compared to the wild-type control (set at 100%), and similar results were obtained in independent experiments (134%, 136%, and 150%, results not shown). This finding confirms the hyperactivity of the GIA-SKY Env mutant, which is an important aspect of our model.
FIG. 5.
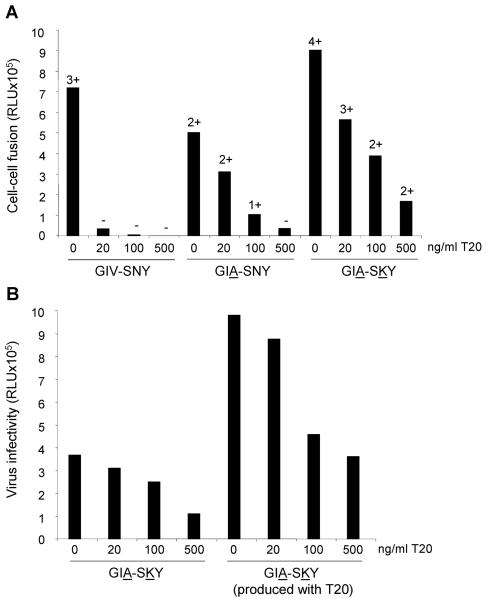
(A) T20-dependent Env has increased fusogenic activity. SupT1 cells were transfected with the mutants indicated below the x axis. One day later, transfected cells were mixed with SupT1 cells containing a Tat-responsive long terminal repeat-luciferase reporter gene construct. After 24 h, formation of syncytia was analyzed by light microscopy (−, no syncytia; ++++, all cells involved in syncytia) and quantitated by measurement of luciferase activity in cell extracts. We consistently measured reduced fusion activity (syncytia and luciferase counts) for the GIA mutant and increased activity for the GIA-SKY mutant. This is a representative experiment, which shows 126% fusion activity for the GIA-SKY mutant compared to the wild-type control (set at 100%). Similar results were obtained in independent experiments (134%, 136%, and 150%; results not shown). (B) T20 prevents the loss of infectivity of GIA-SKY virions. SupT1 cells containing a Tat-responsive long terminal repeat-luciferase reporter gene construct were infected with equal amounts of the GIA-SKY double mutant virus, which was produced in the presence (100 ng/ml) or absence of T20 and scored for infectivity in a single-cycle infection assay. Virus infectivity is expressed in relative light units (RLU) from a luciferase assay performed on the cell lysate.
We also tested cell-cell fusion activity in the presence of T20. Not surprisingly, wild-type Env is inhibited by T20 and GIA is relatively resistant. Interestingly, the GIA-SKY mutant displays a very similar resistance phenotype. These results indicate that the GIA mutation in HR1 is responsible for reduced T20 affinity (resistance) and reduced HR2 affinity (fusion activity) and that the SKY mutation in HR2 creates a hyperfusogenic Env. The latter property may seem to contradict the observation of impaired virus replication, but a premature switch will result in dead Env spikes on the surface of virus particles. T20 may prevent such premature inactivation and thus rescue virion infectivity.
T20 prevents inactivation of the peptide-dependent virus.
We produced the GIA-SKY mutant virus particles in both the presence and absence of T20 and scored their infectivity in a single-cycle infection assay in which a long terminal repeat-luciferase reporter is activated upon successful infection by newly synthesized viral Tat protein. Even though an equivalent amount of HIV-1 virions (based on CA-p24) was used in this assay, we measured a two- to threefold increase in infectivity when virions were produced with T20 (Fig. 5B). When (additional) T20 was added during the infection experiment, we measured a dose-dependent decrease in the infectivity of both samples, confirming the resistance phenotype. Thus, T20 needs to be present early during virion production in order to prevent premature switching and the consequent loss of infectivity. When added later during the infection process, T20 does not activate but rather inhibits the GIA-SKY mutant. This temporal separation of the positive and negative effects of T20 is consistent with the proposed mechanism.
Biophysical properties of T20-resistant and T20-dependent six-helix bundles.
The proposed mechanism suggests that the T20-dependent variant undergoes an accelerated transition from the native state to the postfusion six-helix bundle conformation of gp41. This should be reflected in the relative stabilities of the various conformations that gp41 has to go through during fusion. However, an adequate model system is only available for the postfusion six-helix bundle conformation of gp41.
To determine the effects of the T20-resistant and T20-dependent mutations on the overall structure and stability of the six-helix bundle, we constructed a soluble recombinant model of the gp41e core. This model polypeptide, designated N36(L6)C34, consists of the N36 and C34 peptides connected via a short peptide linker that replaces the disulfide-bonded loop region of gp41 (Fig. 6A). The N36 peptide consists of residues 35 to 70 and the C34 peptide consists of residues 117 to 150 of the patient gp41 sequence. The GIA and GIA-SKY substitutions were introduced into N36(L6)C34. On the basis of circular dichroism measurements, the wild-type protein and the two variants are well folded, with greater than 95% α-helical content (results not shown). Sedimentation equilibrium experiments indicate that the GIA-SKY peptide exists in a discretely trimeric state over a 10-fold range of protein concentrations (10 to 100 μM) (Fig. 6C). We conclude that, to a good approximation, the introduction of T20 resistance and T20 dependence substitutions into the HR1 and HR2 regions of gp41e does not affect the overall protein folding of the six-helix bundle. In addition, we obtained crystal structures for both the wild-type and T20-dependent GIA-SKY variant six-helix bundles, showing that the structures are indeed very similar (results not shown).
FIG. 6.
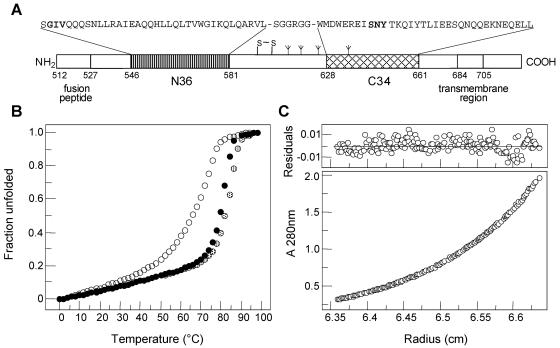
Biophysical characterization of the wild-type and mutant gp41 ectodomain cores. (A) Schematic representation of HIV-1 gp41. The important functional features of gp41 and the sequences of the N36 and C34 segments are shown. Recombinant N36(L6)C34 protein consists of the N36 and C34 peptides plus a six-residue linker. The disulfide bond and four potential N-glycosylation sites are depicted. The residues are numbered according to their position in gp160 of the HIV-1 HXB2 strain. (B) Thermal melts of the wild-type peptide N36(L6)C34 (solid fill) and the GIA (open) and GIA-SKY (dotted fill) mutants monitored by circular dichroism at 222 nm at 10 μM protein concentration in phosphate-buffered saline. We performed thermal unfolding experiments four times for the wild-type and mutant peptides. The error of Tm determination is ±0.5°C. It should be emphasized that melting temperature is useful only as a qualitative guide to stability. (C) Representative sedimentation equilibrium data for the GIA-SKY mutant (≈30 μM) were collected at 20°C and 18,000 rpm in phosphate-buffered saline. The data fit closely to a trimer model. The deviation in the data from the linear fit for a trimer model is plotted (upper).
The thermal stability of the wild-type and mutant N36(L6)C34 six-helix bundles was assessed by circular dichroism by monitoring ellipticity at 222 nm as a function of temperature. The circular dichroism spectra of each of the proteins showed a typical loss of α-helical content when the temperature was raised (Fig. 6B). The pre- and posttransitional slopes and the shape of the main transition were very similar for the wild-type and variant peptides, but there was a profound difference in thermal stability. At a concentration of 10 μM, the midpoint (Tm) of thermal denaturation of the wild-type protein is 80°C, compared with Tm values of 69 and 83°C for the GIA and GIA-SKY mutants, respectively. Taken together, these results indicate that the T20-resistant mutation GIA leads to an appreciable destabilization of the six-helix bundle structure. In contrast, the T20-dependent SKY mutation stabilizes the six-helix bundle conformation. This is striking because the destabilizing GIA mutation is also present in this variant. Thus, the decreased six-helix bundle stability may explain why the T20-resistant virus is less fusogenic, while the T20-dependent double mutant, with increased six-helix bundle stability, is hyperfusogenic.
DISCUSSION
The HIV-1 evolution scheme depicted in Fig. 3 summarizes the multistep development of T20 resistance within the ectodomain of the envelope glycoprotein gp41 in a patient who failed on T20 therapy. This evolution scheme was derived from inspection of the actual codon changes that are indicated alongside the arrows. We initially did not observe the GIA variant, which has previously been implicated in T20 resistance (19, 40). Instead, we initially detected other GIV variants, such as GIG, by a single T-to-G substitution (week 16) and subsequently GIW by an additional G-to-T substitution within the same codon (week 28). Both GIG and GIW are novel variants, and the latter is much more difficult to generate than GIA (double transversion versus a single transition) (2, 17, 18).
We propose that the GIA variant is made directly from the GIV starting population by a simple T-to-C substitution. The GIW variant is eventually taken over by a virus variant that combines the GIA mutation in HR1 with an SKY mutation in HR2 to give rise to a GIA-SKY double mutant, which dominates the viral population after 32 weeks of therapy, presumably due to its increased resistance to T20 (Fig. 4B and results not shown). Interestingly, once the patient stops T20 therapy, the GIA-SKY double mutant disappears almost completely within 4 weeks, suggesting that this variant is not very fit in the absence of the drug. In vitro, this GIA-SKY mutant replicates only in the presence of T20, making it a drug-dependent virus (Fig. 4). This T20 dependence obviously explains the rapid disappearance of this variant once therapy is stopped.
Inspection of the HR1 and HR2 sequences in natural subtype B HIV-1 isolates (http://www.hiv.lanl.gov/) (1, 36) indicates that the GIV sequence in HR1 is highly conserved, with only four isolates among 338 sequences that deviate from this sequence (of which two are GIA variants). The SNY sequence, in particular the first residue, is somewhat more variable (DNY, NNY, SNY, and ENY represent 305 of the 332 sequences). The SKY sequence that is selected under T20 therapy is never observed in natural isolates, although a single DKY variant was found. The loss of the glycosylation site is extremely rare (only 8 of 332 isolates). Strikingly, one of the two natural GIA variants in HR1 is combined with an exceptional DSY sequence in HR2. This finding may relate to our observation that changes in HR2 are coupled to changes in HR1. A more extensive screening of the HIV-1 sequence database further strengthens this idea. Among 417 sequences of HIV-1 non-B subtypes, we found one additional isolate with the GIA sequence, and this sequence also encodes an unusual HR2 sequence RNI.
Resistance is caused by mutation of the valine (V) residue within the GIV sequence of HR1 into a number of alternative amino acids: methionine (34); alanine (40; this paper); and glycine and tryptophan (this paper). The wild-type valine normally interacts with residues in the critical center of the HR2-derived T20 peptide, and the mutation could thus weaken the interaction with both T20 and HR2. The smaller glycine and alanine residues could leave an unfavorable cavity at the HR1-HR2/HR1-T20 interface, whereas the bulky tryptophan side chain may sterically interfere with the interaction. Direct HR1-T20 binding experiments are consistent with this proposal (34). Reduced HR1-T20 affinity may explain the resistance phenotype, but reduced HR1-HR2 interaction may also negatively impact Env-mediated fusion and HIV-1 fitness. Indeed, we measured reduced fitness for the T20 resistance variants GIA, GIG, and GIW compared to the wild-type virus (Fig. 4 and results not shown). Nevertheless, the slight reduction in viral fitness indicates that the HR1-HR2 interaction is not affected in a major way. The impact on the HR1-HR2 interaction may be less severe than on the HR1-T20 interaction because the former is an intramolecular interaction (34).
Several mechanistic models can be proposed for the unique T20-dependent replication phenotype. First, it can be envisaged that the T20 peptide is actively involved in formation of the six-helix bundle structure of the GIA-SKY double mutant. Thus, the wild-type T20 peptide could replace one or multiple mutant HR2 domains to enable the formation of this fusion-competent structure. This scenario is unlikely because it would require the formation of a six-helix bundle with a much shorter T20-based HR2 domain and with dangling free HR2 domains that may interfere with the fusion process. Second, T20 dependence may mimic the process of enhancement of virus infectivity that is often observed in antibody neutralization experiments. However, such enhancement does not explain the nearly complete inactivity of the GIA-SKY virus in the absence of T20. Our results suggest a third model for T20-dependent replication of the GIA-SKY double mutant that builds on the resistance phenotype of the GIA single mutant (Fig. 7).
FIG. 7.
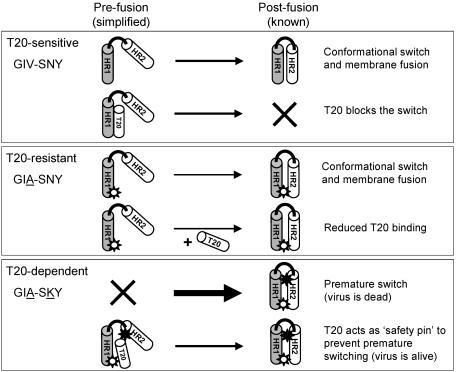
Proposed model for T20-dependent viral entry. Each box depicts one of three scenarios: T20-sensitive (GIV-SNY), T20-resistant (GIA-SNY), and T20-dependent (GIA-SKY). A simplified gp41 ectodomain comprising only one subunit of HR1 (shaded cylinder) and HR2 (white cylinder) joined by a loop region (black line) is used to depict the prefusion and postfusion states of the peptide. The thickness of the arrows represents the speed of the conformational switch between pre- and postfusion conformations. A white star represents the GIA mutation in HR1, and a black star represents the SKY mutation in HR2. Explanations for each reaction are provided on the right hand side.
We now propose that part of the GIA phenotype (weakened HR1-HR2 interaction and reduced fusion) is reversed by the SKY mutation in HR2. This mutation creates an Env variant that is more prone to undergo the conformational switch leading to six-helix bundle formation. Premature switching of GIA-SKY will make a dead Env spike and thus effectively kill virus infectivity, which is what we measured (Fig. 4). More importantly, this scenario easily explains the observed T20 dependency, as HR1-T20 binding will block the premature switch and Env inactivation. This T20 blockade should obviously be transient, and release of the T20 peptide may be facilitated by the reduced HR1-T20 affinity due to the GIA resistance mutation. This mechanism also explains why the GIA-SKY double mutant is inhibited at high T20 levels, which freezes Env in a prefusion conformation.
The key mutation for the T20-dependent phenotype is the SNY to SKY change within HR2. This change creates a hyperfusogenic Env approximately 1.5-fold more active than the wild-type control, but around twofold more active than the GIA precursor form (Fig. 5A). We propose that the GIA-SKY double mutant hardly replicates because this Env variant is too aggressive, leading to a premature switch of the spring-loaded Env, which results in nonfunctional six-helix bundles (dead spikes) on the virus particle. The SKY mutation is solely responsible for this enhanced fusogenicity and lack of replication capacity because these properties were not observed for the GIA single mutant (in fact, reduced fusogenicity was measured; see Fig. 5A). Furthermore, the SKY single mutant is replication impaired (results not shown). This also means that evolution of the GIA-SKY mutant must follow the precise two-step scenario; first GIA-mediated resistance and then SKY-mediated dependence.
The GIA mutation is also important for the ability of the double mutant to replicate in the presence of T20. The GIA-SKY double mutant exhibits a T20 resistance phenotype that is very similar to that of the GIA single mutant, the difference being that T20 is required for GIA-SKY to maintain a prefusion Env conformation, but the presence of T20 during the infection process partially inhibits virus entry (Fig. 5B). In other words, the T20 peptide acts as a safety pin that locks or stabilizes a prefusion conformation of the gp41 protein, but it presumably has to be removed at the right time to ensure correct Env function (Fig. 7).
Further biophysical studies were performed to reveal how the SKY mutation in HR2 triggers the conformational switch within Env. There are several possibilities; the HR2 mutation may either destabilize the initial Env conformation or an intermediate structure, or it may stabilize the final six-helix bundle structure. A direct stabilizing effect on the latter structure was indeed measured for the GIA-SKY double mutant. This effect is likely caused by the lysine residue, which strongly favors an α-helix conformation. However, this amino acid substitution also leads to the removal of N-linked glycosylation at the asparagine (N) of the SNY sequence (8, 9, 16, 24, 32). This loss may also have gross structural repercussions, although it cannot be measured in our studies that were performed with E. coli expressed gp41 peptides.
Our model suggests that the T20-dependent variant is prone to undergo premature conformational changes resulting in the formation of six-helix bundle dead spikes. This might imply that the T20-dependent variant is also more prone to spontaneous shedding of gp120. We have evidence that this is indeed the case (results not shown).
We anticipate that our T20-dependent virus will provide a useful tool to dissect molecular details of the Env-mediated entry process and such studies are ongoing. Further research could yield novel information on structural intermediates that may facilitate Env-based vaccine research and the construction of improved immunogens. The T20-dependent phenotype may be a useful tool to control or to synchronize the entry of viral vectors. Conditional viral entry may also add to the safety of a doxycycline-dependent HIV-1 variant that was constructed as a live attenuated virus vaccine (27, 39). In fact, it seems possible to make a virus that can be controlled from the outside both at the level of cell entry (T20) and viral gene expression (doxycycline). The patient data in Fig. 2 and 3 indicate that such a T20 switch can operate in vivo.
Finally, the evolution of drug-dependent HIV-1 variants has an obvious clinical relevance. The appearance of such variants during antiviral therapy may be an indication to modify the drug regimen. It is therefore important to test if the next generation of T20-like compounds are able to trigger the replication of the GIA-SKY mutant. It is already known that patients harboring T20-resistant viruses do not show cross resistance to the related T1249 compound, and we have preliminary evidence that cross-dependence of the GIA-SKY double mutant does not occur either (results not shown). An interesting difference between drug-resistant and drug-dependent viruses is at the level of the human population and virus transmission. Drug-resistant viruses are known to spread within the current epidemic, but this would seem impossible for T20-dependent viruses because the antiviral inducer T20 is not available in the newly infected individual.
Acknowledgments
We thank Daniel Blanckenberg and Nicole Back for clinical samples. We also thank Marion Cornelissen, Els Busser, Stephen Heynen, and Remco van den Burg for help and advice on protocols and Hendrik Huthoff and Bill Paxton for critical reading of the manuscript. We thank Trimeris (Durham, N.C.) for providing us with the T20 peptide used in our in vitro assays.
This research was supported by the International Antiretroviral Therapy Evaluation Centre; the Netherlands AIDS Fund (grant 5003); the NWO-VICI program; and the National Institutes of Health (AI42382).
REFERENCES
- 1.Baldwin, C. E., R. W. Sanders, and B. Berkhout. 2003. Inhibiting HIV-1 entry with fusion inhibitors. Curr. Med. Chem. 10:1633-1642. [DOI] [PubMed] [Google Scholar]
- 2.Berkhout, B., A. T. Das, and N. Beerens. 2001. HIV-1 RNA editing, hypermutation and error-prone reverse transcription. Science 292:7. [DOI] [PubMed] [Google Scholar]
- 3.Boom, R., C. J. A. Sol, M. M. M. Salimans, C. L. Jansen, P. M. E. Wertheim-van Dillen, and J. Van der Noordaa. 1990. A rapid and simple method for purification of nucleic acids. J. Clin. Microbiol. 28:495-503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cantor, C., and P. Schimmel. 1980. Biophysical chemistry, part III. W. H. Freeman and Company, New York, N.Y.
- 5.Chan, D. C., D. Fass, J. M. Berger, and P. S. Kim. 1997. Core structure of gp41 from the HIV envelope glycoprotein. Cell 89:263-273. [DOI] [PubMed] [Google Scholar]
- 6.Chen, S. S. 1994. Functional role of the zipper motif region of human immunodeficiency virus type 1 transmembrane protein gp41. J. Virol. 68:2002-2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen, Y. H., J. T. Yang, and K. H. Chau. 1974. Determination of the helix and beta form of proteins in aqueous solution by circular dichroism. Biochemistry 13:3350-3359. [DOI] [PubMed] [Google Scholar]
- 8.Dash, B., A. McIntosh, W. Barrett, and R. Daniels. 1994. Deletion of a single N-linked glycosylation site from the transmembrane envelope protein of human immunodeficiency virus type 1 stops cleavage and transport of gp160 preventing env-mediated fusion. J. Gen. Virol. 75:1389-1397. [DOI] [PubMed] [Google Scholar]
- 9.Dedera, D. A., R. L. Gu, and L. Ratner. 1992. Role of asparagine-linked glycosylation in human immunodeficiency virus type 1 transmembrane envelope function. Virology 187:377-382. [DOI] [PubMed] [Google Scholar]
- 10.Dong, X. N., Y. Xiao, M. P. Dierich, and Y. H. Chen. 2001. N- and C-domains of HIV-1 gp41: mutation, structure and functions. Immunol. Lett. 75:215-220. [DOI] [PubMed] [Google Scholar]
- 11.Eckert, D. M., and P. S. Kim. 2001. Mechanisms of viral membrane fusion and its inhibition. Annu. Rev. Biochem. 70:777-810. [DOI] [PubMed] [Google Scholar]
- 12.Edelhoch, H. 1967. Spectroscopic determination of tryptophan and tyrosine in proteins. Biochemistry 6:1948-1954. [DOI] [PubMed] [Google Scholar]
- 13.Hanna, S. L., C. Yang, S. M. Owen, and R. B. Lal. 2002. Resistance mutation in HIV entry inhibitors. AIDS 16:1603-1608. [DOI] [PubMed] [Google Scholar]
- 14.Jelesarov, I., and M. Lu. 2001. Thermodynamics of trimer-of-hairpins formation by the SIV gp41 envelope protein. J. Mol. Biol. 307:637-656. [DOI] [PubMed] [Google Scholar]
- 15.Johnson, M. L., J. J. Correia, D. A. Yphantis, and H. R. Halvorson. 1981. Analysis of data from the analytical ultracentrifuge by nonlinear least- squares techniques. Biophys. J. 36:575-588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johnson, W. E., J. M. Sauvron, and R. C. Desrosiers. 2001. Conserved, N-linked carbohydrates of human immunodeficiency virus type 1 gp41 are largely dispensable for viral replication. J. Virol. 75:11426-11436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Keulen, W., N. K. T. Back, A. van Wijk, C. A. B. Boucher, and B. Berkhout. 1997. Initial appearance of the 184Ile variant in lamivudine-treated patients is caused by the mutational bias of the human immunodeficiency virus type 1 reverse transcriptase. J. Virol. 71:3346-3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Keulen, W., C. Boucher, and B. Berkhout. 1996. Nucleotide substitution patterns can predict the requirements for drug resistance of HIV-1 proteins. Antiviral Res. 31:45-57. [DOI] [PubMed] [Google Scholar]
- 19.Kilby, J. M., J. P. Lalezari, J. J. Eron, M. Carlson, C. Cohen, R. C. Arduino, J. C. Goodgame, J. E. Gallant, P. Volberding, R. L. Murphy, F. Valentine, M. S. Saag, E. L. Nelson, P. R. Sista, and A. Dusek. 2002. The safety, plasma pharmacokinetics, and antiviral activity of subcutaneous enfuvirtide (T-20), a peptide inhibitor of gp41-mediated virus fusion, in HIV-infected adults. AIDS Res. Hum. Retroviruses 18:685-693. [DOI] [PubMed] [Google Scholar]
- 20.Kilgore, N. R., K. Salzwedel, M. Reddick, G. P. Allaway, and C. T. Wild. 2003. Direct evidence that C-peptide inhibitors of human immunodeficiency virus type 1 entry bind to the gp41 N-helical domain in receptor-activated viral envelope. J. Virol. 77:7669-7672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kunkel, T. A. 1985. Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc. Natl. Acad. Sci. USA 82:488-492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Laue, T. M., B. D. Shah, T. M. Ridgeway, and S. L. Pelletier. 1992. Computer-aided interpretation of analytical sedimentation data for proteins, p. 90-125. In S. E. Harding, A. J. Rowe, and J. C. Horton (ed.), Analytical ultracentrifugation in biochemistry and polymer science. Royal Society of Chemistry, Cambridge, England.
- 23.Lazzarin, A., B. Clotet, D. Cooper, J. Reynes, K. Arasteh, M. Nelson, C. Katlama, H. J. Stellbrink, J. F. Delfraissy, J. Lange, L. Huson, R. DeMasi, C. Wat, J. Delehanty, C. Drobnes, and M. Salgo. 2003. Efficacy of enfuvirtide in patients infected with drug-resistant HIV-1 in Europe and Australia. N. Engl. J. Med. 348:2186-2195. [DOI] [PubMed] [Google Scholar]
- 24.Lee, W. R., X. F. Yu, W. J. Syu, M. Essex, and T. H. Lee. 1992. Mutational analysis of conserved N-linked glycosylation sites of human immunodeficiency virus type 1 gp41. J. Virol. 66:1799-1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Little, S. J., S. Holte, J. P. Routy, E. S. Daar, M. Markowitz, A. C. Collier, R. A. Koup, J. W. Mellors, E. Connick, B. Conway, M. Kilby, L. Wang, J. M. Whitcomb, N. S. Hellmann, and D. D. Richman. 2002. Antiretroviral-drug resistance among patients recently infected with HIV. N. Engl. J. Med. 347:385-394. [DOI] [PubMed] [Google Scholar]
- 26.Lu, M., S. C. Blacklow, and P. S. Kim. 1995. A trimeric structural domain of the HIV-1 transmembrane glycoprotein. Nat. Struct. Biol. 2:1075-1082. [DOI] [PubMed] [Google Scholar]
- 27.Marzio, G., K. Verhoef, M. Vink, and B. Berkhout. 2001. In vitro evolution of a highly replicating, doxycycline-dependent HIV for applications in vaccine studies. Proc. Natl. Acad. Sci. USA 98:6342-6347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Melikyan, G. B., R. M. Markosyan, H. Hemmati, M. K. Delmedico, D. M. Lambert, and F. S. Cohen. 2000. Evidence that the transition of HIV-1 gp41 into a six-helix bundle, not the bundle configuration, induces membrane fusion. J. Cell Biol. 151:413-423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moore, J. P., and R. W. Doms. 2003. The entry of entry inhibitors: A fusion of science and medicine. Proc. Natl. Acad. Sci. USA 100:10598-10606. [DOI] [PMC free article] [PubMed]
- 30.Moore, J. P., and M. Stevenson. 2000. New targets for inhibitors of HIV-1 replication. Nat. Rev. Mol. Cell. Biol. 1:40-49. [DOI] [PubMed] [Google Scholar]
- 31.Peden, K., M. Emerman, and L. Montagnier. 1991. Changes in growth properties on passage in tissue culture of viruses derived from infectious molecular clones of HIV-1LAI, HIV-1MAL, and HIV-1ELI. Virol. 185:661-672. [DOI] [PubMed] [Google Scholar]
- 32.Perrin, C., E. Fenouillet, and I. M. Jones. 1998. Role of gp41 glycosylation sites in the biological activity of human immunodeficiency virus type 1 envelope glycoprotein. Virology 242:338-345. [DOI] [PubMed] [Google Scholar]
- 33.Poveda, E., B. Rodes, C. Toro, L. Martin-Carbonero, J. Gonzalez-Lahoz, and V. Soriano. 2002. Evolution of the gp41 env region in HIV-infected patients receiving T-20, a fusion inhibitor. AIDS 16:1959-1961. [DOI] [PubMed] [Google Scholar]
- 34.Rimsky, L. T., D. C. Shugars, and T. J. Matthews. 1998. Determinants of human immunodeficiency virus type 1 resistance to gp41-derived inhibitory peptides. J. Virol. 72:986-993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roman, F., D. Gonzalez, C. Lambert, S. Deroo, A. Fischer, T. Baurith, T. Staub, R. Boulme, V. Arendt, F. Schneider, R. Hemmer, and J. C. Schmit. 2003. Uncommon mutations at residue positions critical for enfuvirtide (T-20) resistance in enfuvirtide-naive patients infected with subtype B and non-B HIV-1 strains. J. Acquir. Immune Defic. Syndr. 33:134-139. [DOI] [PubMed] [Google Scholar]
- 36.Sanders, R. W., B. Korber, M. Lu, B. Berkhout, and J. P. Moore. 2002. Mutational analyses and natural variability of the gp41 ectodomain, p. 43-68. In C. Kuiken, B. Foley, E. Freed, B. Hahn, P. Marx, F. McCutchan, J. Mellors, S. Wolinsky, and B. Korber (ed.), HIV sequence compendium 2002. Los Alamos National Laboratory Theoretical Biology and Biophysics Group, Los Alamos, N.Mex.
- 37.Sanders, R. W., M. Vesanen, N. Schuelke, A. Master, L. Schiffner, R. Kalyanaraman, M. Paluch, B. Berkhout, P. J. Maddon, W. C. Olson, M. Lu, and J. P. Moore. 2002. Stabilization of the soluble, cleaved, trimeric form of the envelope glycoprotein complex of human immunodeficiency virus type 1. J. Virol. 76:8875-8889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tan, K., J. Liu, J. Wang, S. Shen, and M. Lu. 1997. Atomic structure of a thermostable subdomain of HIV-1 gp41. Proc. Natl. Acad. Sci. USA 94:12303-12308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Verhoef, K., G. Marzio, W. Hillen, H. Bujard, and B. Berkhout. 2001. Strict control of human immunodeficiency virus type 1 replication by a genetic switch: Tet for Tat. J. Virol. 75:979-987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wei, X., J. M. Decker, H. Liu, Z. Zhang, R. B. Arani, J. M. Kilby, M. S. Saag, X. Wu, G. M. Shaw, and J. C. Kappes. 2002. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob. Agents Chemother. 46:1896-1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weissenhorn, W., A. Dessen, S. C. Harrison, J. J. Skehel, and D. C. Wiley. 1997. Atomic structure of the ectodomain from HIV-1 gp41. Nature 387:426-430. [DOI] [PubMed] [Google Scholar]
- 42.Wild, C., J. W. Dubay, T. Greenwell, T. Baird, Jr., T. G. Oas, C. McDanal, E. Hunter, and T. Matthews. 1994. Propensity for a leucine zipper-like domain of human immunodeficiency virus type 1 gp41 to form oligomers correlates with a role in virus-induced fusion rather than assembly of the glycoprotein complex. Proc. Natl. Acad. Sci. USA 91:12676-12680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wild, C. T., D. C. Shugars, T. K. Greenwell, C. B. McDanal, and T. J. Matthews. 1994. Peptides corresponding to a predictive alpha-helical domain of human immunodeficiency virus type 1 gp41 are potent inhibitors of virus infection. Proc. Natl. Acad. Sci. USA 91:9770-9774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu, L., S. Hue, S. Taylor, D. Ratcliffe, J. A. Workman, S. Jackson, P. A. Cane, and D. Pillay. 2002. Minimal variation in T-20 binding domain of different HIV-1 subtypes from antiretroviral-naive and -experienced patients. AIDS 16:1684-1686. [DOI] [PubMed] [Google Scholar]
- 45.Zollner, B., H. H. Feucht, M. Schroter, P. Schafer, A. Plettenberg, A. Stoehr, and R. Laufs. 2001. Primary genotypic resistance of HIV-1 to the fusion inhibitor T-20 in long-term infected patients. AIDS 15:935-936. [DOI] [PubMed] [Google Scholar]