

Abstract
Autophagy is a highly controlled lysosome-mediated function in eukaryotic cells to eliminate damaged or aged long-lived proteins and organelles. It is required for restoring cellular homeostasis in cell survival under multiple stresses. Autophagy is known to be a double-edged sword because too much activation or inhibition of autophagy can disrupt homeostatic degradation of protein and organelles within the brain and play a role in neuronal cell death. Many factors affect autophagy flux function in the brain, including endoplasmic reticulum (ER) stress, oxidative stress, and aging. Newly emerged research indicates that altered autophagy flux functionality is involved in neurodegeneration of the aged brain, chronic neurological diseases, and after traumatic and ischemic brain injuries. In search to identify neuroprotective agents that may reduce oxidative stress and stimulate autophagy, one particular neuroprotective agent docosahexaenoic acid (DHA) presents unique functions in reducing ER and oxidative stress and modulating autophagy. This review will summarize the recent findings on changes of autophagy in aging, neurodegenerative diseases, and brain injury after trauma or ischemic strokes. Discussion of DHA functions is focused on modulating ER stress and autophagy in regard to its neuroprotection and anti-tumor functions.
Keywords: autophagy, brain aging, docosahexaenoic acid, lysosome, stroke, traumatic brain injury
Graphical Abstract
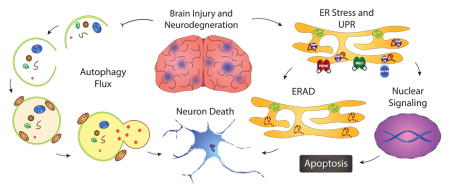
1. Introduction
Degradation of unfolded/misfolded proteins via ER associated degradation (ERAD) and autophagy is an evolutionally conserved cellular function and critical for cell survival and tissue repair. Sustained ER stress and dysfunction of autophagy are associated with many neurological diseases. ER stress stimulates the unfolded protein response (UPR) (Bravo et al., 2013). The primary goal of the UPR is to maintain ER homeostasis by halting protein translation, promoting refolding of unfolded proteins, and degrading misfolded proteins via ERAD. In situations where the cell experiences chronic and prolonged ER stress, the UPR directs it towards apoptosis (Lenna et al., 2014). The UPR is involved in a number of neurological disorders, such as amyotrophic lateral sclerosis (ALS), Alzheimer’s disease (AD), Parkinson’s disease (PD), or ischemic stroke and traumatic brain injury (TBI) (Perri et al., 2016, Truettner et al., 2009, Begum et al., 2014). Thus, ER stress is now wildly recognized as a new therapeutic target for neurological diseases.
Impaired autophagy flux participates in many human diseases, including cancer, cardiovascular diseases, immune-mediated disorders, myopathy, and neurodegenerative diseases (Zhang et al., 2013). In particular, the dysfunction of autophagy flux plays an important role in neurological disorders. In a study using Huntington’s disease (HD) knock-in mice, polyglutamine-expanded huntingtin (polyQ-htt) was found to disrupt autophagosome transport, leading to defected degradation of cargo (Wong and Holzbaur, 2014). Furthermore, either delayed autophagosome maturation, damage of autophagic-vesicular structures, impaired lysosomal hydrolysis, or impaired autophagosome transport has been detected in experimental studies of AD or PD (Tanik et al., 2013; Lee et al., 2011). On the other hand, neuronal cell death after TBI is in part attributed to an impaired autophagic clearance due to lysosomal dysfunction (Sarkar et al., 2014). In cerebral ischemia, autophagy is required for neuronal survival against ischemia-induced death (Yan et al., 2013). For readers who are specifically interested in review of ER stress and autophagy in chronic degenerative diseases such as AD, PD, and HD, please read other recent reviews (Morimoto et al., 2014, Mollereau et al., 2016). In this review, we will discuss the cellular mechanisms of ER stress and autophagy flux, their changes in brain injury after ischemic stroke and TBI, as well as in the aged brain. In addition, effects of DHA on these cellular changes will also be discussed.
2. ER stress
2.1 ER stress
The endoplasmic reticulum (ER) is an important organelle in eukaryotic cells, responsible for protein synthesis, lipid formation, Ca2+ storage and signaling (Díaz-Villanueva et al., 2015, Weber-Boyvat et al., 2013, van der Kant et al., 2014). Various intracellular and extracellular factors that disturb any of these three homeostatic functions in the ER will induce ER stress (Schröder, 2008). In the ER, secretory and membrane proteins are folded, modified and assembled to yield the final, functional protein structures (Ellgaard and Helenius, 2001). However, the export of such proteins from the ER is precisely regulated and only properly folded proteins can exit the ER. Proteins that are not properly folded are degraded through the ubiquitin-proteasome system, ERAD (Duncan et al., 2013). Accumulation of unfolded or misfolded proteins in the ER, which results from the imbalance of ER homeostasis, can cause ER stress and trigger UPR (Schönthal, 2012).
The UPR is essentially initiated by three ER-transmembrane sensors (Figure 1), PERK (PKR-like ER kinase), IRE1 (inosital requiring kinase 1), and ATF6 (activating transcription factor 6) (Kim et al., 2008). All these three effector proteins are regulated by the ER chaperone GRP78/BIP (immunoglobulin heavy chain binding protein). Normally, GRP78 is bound to all of them by its peptide-binding domains, suppressing their activity and keeping them in an inactive state (Rao and Bredesen, 2004). In addition, the peptide-binding domain of GRP78 acts as the binding region for misfolded proteins. Upon ER stress and accompanying UPR (Figure 1), GRP78 binds to the misfolded proteins to dissociate itself from these sensors (PERK, IRE, and ATF6), which subsequently activates the sensors (Rao and Bredesen, 2004). As a consequence, first, PERK becomes activated by autophosphorylation to dimerize and subsequently triggers global protein translation inhibition by phosphorylation of eukaryotic translation initiation factor 2α (eIF2α), which decreases mRNA translation in an attempt to halt the accumulation of protein in the ER (Kim et al., 2008, Larner et al., 2006). Also, such activation allows translation of ATF4, a UPR-dependent signaling protein that upregulates the expression of ER chaperones and the key transcription factor CHOP, which translocates to the nucleus, increases transcription of those genes, and is associated with ER stress-mediated apoptosis (Rzymski et al., 2010). Similarly, inositol-requiring enzyme 1 (IRE1) is initially bound by GRP78 and activated by the separation of GRP78 to allow dimerization (Figure 1). Activated IRE1 (an mRNA endoribonuclease) mediates unconventional splicing of X-box binding protein 1 (XBP1) coding mRNA to activate another transcriptional factor, xbp-1, that crosses into the nucleus to generate more ER-resident chaperone proteins (Flamment et al., 2012). Additionally, although the pathway is unclear, activated IRE1 is associated with JUN N-terminal Kinase (JNK) that can lead to autophagy or apoptosis (Todd et al., 2008). Finally, the third arm of ER stress involves ATF6 (Figure 1). The dissociation of ATF6 and GRP78 allows ATF6 to translocate to the Golgi where it gets cleaved, and the modified protein, now a transcription factor, can induce the production of ER chaperones and ERAD components like IRE1 (Flamment et al., 2012). Taken together, UPR activation involves protein synthesis inhibition and selective activation of stress gene expression for cells to regulate signaling pathways for damage repair or their own death.
Figure 1. ER stress and the subsequent unfolded protein response (UPR).

Upon the accumulation of unfolded and misfolded proteins, GRP78 is dissociated from PERK, IRE1, and ATF6 to bind with the aberrant proteins, which triggers the activation of 3-arm UPR. The autophosphorylation of PERK leads to the phosphorylation of eIF2α, which inhibits the translation of proteins and activates ATF4. ATF4 will enter the nucleus and transcribe ER stress response elements (ERSEs) and CHOP to lead apoptosis. Activation of IRE1 splices XBP1 mRNA so that it can translate the protein XBP1, which enters the nucleus to transcribe proteins leading to ER associated degradation (ERAD). Activation of ATF6 involves its release from the ER membrane to undergo cleavage in the Golgi. The modified ATF6 enters the nucleus to transcribe ERSEs as well as CHOP and leads to apoptosis.
2.2 ER-associated degradation (ERAD)
In eukaryotic cells, both ERAD and autophagy pathways can be triggered by UPR to restore cellular homeostasis (Milisav et al., 2015). ERAD is also known as a ubiquitin–proteasome pathway because it targets misfolded proteins of the ER for ubiquitination and delivers them to the proteasome for degradation and clearance (Schönthal et al., 2012). ERAD is a degradation mechanism integral to ER protein quality control (Tsai et al., 2010). When ERAD is impaired or unable to clear proteins, autophagy induced by ER stress can also degrade the misfolded proteins (Fujita et al., 2007, Jia et al., 2015). However, if those misfolded proteins are not sufficiently degraded via autophagy, cells may activate cell death programs (Fujita et al., 2007).
3. Autophagy and autophagy flux
Autophagy, a Greek word meaning self-eating, is a highly controlled lysosome-mediated function in eukaryotes for cells to eliminate damaged or aged, long-lived proteins and organelles like ER, mitochondria or ribosomes (Ravikumar et al., 2010, Feng et al., 2014). These contents following lysosomal degradation can be recycled for making new proteins and organelles (Yin et al., 2008). Autophagy plays a crucial role in maintaining cell survival under multiple stresses to restore cellular homeostasis (Glick et al., 2010). However, autophagy is a double-edged sword where under certain conditions, autophagy results in cell death (Button et al., 2015). Based on the way that prodegradation products (cargo) are delivered into the lysosomes, autophagy can be divided into three forms, chaperone-mediated autophagy (CMA), microautophagy, and macroautophagy (Pereira et al., 2012). CMA is an extremely selective pathway, and only soluble proteins containing the recognition site for chaperones like heat shock protein 70 (Hsc70) complex and others can be its substrates (Duraes et al., 2015). Once the Hsc70 complex combines with lysosome associated membrane protein 2A (LAMP2A), a transmembrane protein, cargo is delivered to the lysosome to be degraded. In microautophagy, cargo is directly engulfed into the lysosome by inward folding lysosomal membrane or cellular protrusion (Česen et al., 2012). Microautophagy mediates both non-selective and selective elimination of cytoplasmic cargo and is very similar to macroautophagy, except that cargo is directly engulfed by the lysosome and does not involve the formation of the autophagosome (Li et al., 2012). Of the three types of autophagy, macroautophagy has been most extensively studied and is the best characterized form of autophagy. Usually, the term autophagy refers to macroautophagy and involves non-selective or bulk degradation of cargo (Mizushima et al., 2008).
3.1 Autophagy steps and function
As shown Figure 2, autophagy process consists of 4 sequential steps, including 1) induction and phagophore formation; 2) autophagosome formation; 3) autophagosome-lysosome fusion (autolysosome); and 4) degradation and recycling. To start, nutrient deficiency, rapamycin, or various other stimuli inhibit the mechanistic target of rapamycin (mTOR), which itself is an inhibitor of unc-51-like kinase complex (ULK, the mammalian equivalent of autophagy-related gene Atg 1) in the induction and phagophore formation step (Dunlop and Tee, 2014). ULK complex is composed of multiple protein subunits, ULK 1 and 2 as well as Atg13 and FIP200. The mTOR pathway is made of two complexes, mTORC1 and mTORC2, which directly and indirectly inhibit autophagy, respectively (Spilman et al., 2010). mTORC1 phosphorylates and inhibits ULK1 and mAtg13, which are involved in preautophagosomal structure formation (Mizushima, 2010). mTORC2, on the other hand, phosphorylates and activates Akt, which not only stimulates mTORC1, but also inhibits FOXO3, which promotes the expression of autophagy-related genes (Mammucari et al., 2007). Indeed, transcriptional control of autophagy and lysosomal function has emerged as a subject of intensive research in the last several years due to identification of transcription factors EB and E3 (TFEB and TFE3) as drivers of lysosomal biogenesis and autophagy. Under normal conditions, TFEB/TFE3 are phosphorylated by mTORC1, which promotes its 14-3-3 binding and retention in the cytoplasm, but energy deficits and lysosomal stress inactivate mTORC1 (Raben and Puertollano, 2016). In parallel, activation of the lysosomal ion channel TRPML1 leads to calcineurin activation, which dephosphorylates TFEB/TFE3 and promotes its nuclear translocation (Sardiello, 2016). For more details of this process, please refer to the series of excellent recent reviews (Raben and Puertollano, 2016; Sardiello, 2016).
Figure 2. Autophagy and its regulation.

Through macroautophagy, cargo such as malfunctioning organelles and proteins can be recycled. Step 1: Induction and phagophore formation. An isolation membrane is gathered by the ULK and PI3K-III complexes. Step 2: Formation of autophagosome. The membrane is elongated by the Atg16-Atg12-Atg5 complex while LC3-II is embedded on both the outer and inner membranes to form the phagophore. Step3: Autophagosome-lysosome fusion. The autophagosome fuses with the lysosome to form the autolysosome. P62 will begin to degrade under normal autophagy flux. Step 4: Degradation and recycling. The lysosomal acidic pH and enzymes can degrade the cargo for release back into the cell, and the membrane is disintegrated for a new cycle.
In addition, class 3 phosphoinositide 3-kinase (PIK3C3, or VPS34) together with beclin 1 (Atg 6) and other proteins, such as VPS15, AMBRA1, ATG14, form the PI3K-III complex, which works together with the ULK complex to recruit the isolation membrane and form the beginning of the phagophore (Flemming et al., 2011). During the autophagosome formation step, the elongation of the phagophore leads to the formation of a typical double-membrane vesicle autophagosome. To do so requires the implementation of two different pathways simultaneously. The first involves Atg7 activating Atg12 followed by Atg10 mediating the conjugation of Atg12 and Atg5 (Glick et al., 2010). This Atg12-Atg5 complex is then linked with Atg16 to yield a Atg12-Atg5-Atg16 complex, that is bound to the developing membrane and is responsible for the recruitment of Microtubule-associated protein light chain 3 (LC3) (Fujita, N., Itoh, T. et al, 2008). As shown in Figure 2, LC3 (or Atg8) is cleaved by ATG4 protease to become LC3-I and afterward is activated by the same Atg7, allowing the subsequent conjugation to phosphatidylethanolamine (PE) via Atg3 to yield LC3-II (Kirkin et al., 2009, Glick et al., 2010).
LC3 is the most commonly used marker to monitor autophagic activation because it is tightly conjugated to the outer autophagosome membrane and can be detected by fluorescence and electron microscopy (Mizushima et al., 2010). Because LC3 II runs faster than LC3 I in SDS-PAGE gels, the ratio of LC3-II/LC3-I is commonly utilized in Western blot analysis to measure autophagy function (Aparicio et al., 2016). LC3-II, can bind to the inner autophagosome membrane and is conveyed and degraded in the lysosomes, making it also a good marker to rate autophagy flux (Nakatogawa et al., 2009).
However, LC3 cannot measure cargo digestion. Autophagosomes are initiated by the elongation of the phagophore and are completely formed by the enclosed phagophore. After formation, autophagosomes are then able to deliver cargo, including long-lived proteins, organelles, protein aggregates, and invasive pathogens (Weidberg et al., 2011), to the lysosome for degradation and recycling. Autophagosomes undergo a maturation process through fusion with lysosomes. The autophagosome forms quickly, taking 4–5 min in yeast (Geng et al., 2008) and 5–10 min in mammals (Fujita, N., Hayashi-Nishino, M. et al., 2008). Next, during autophagosome-lysosome fusion (autolysosome), the autophagosome fuses with the lysosomes to form autolysosomes, and LC3-II is completely dissociated from the membrane of the autolysosome. Subsequently, p62 binds to ubiquitinated substrates and LC3 on the inner membrane of the completed autophagosome, which is degraded in the autolysosome (Bitto et al., 2014). In the last degradation and recycling step, the autophagy cargo is digested in the autolysosome by lysosomal hydrolases. The resulting small molecules are recycled and reused. 3-methyladenine (3-MA) and wortmannin are both autophagy inhibitors for their suppression of PIK3C3 (Jaber et al., 2012) while Bafilomycin A1 and chloroquine inhibit the autophagy by neutralizing the lysosomal pH and blocking autophagosome-lysosome fusion (Li and He, 2014).
Autophagosome function
Increasing evidence suggests multiple organelles, including the ER, mitochondria, the plasma membrane, the Golgi, and recycling endosomes, as the possible source of the autophagosomal membranes (Mari et al., 2011). Autophagy flux is defined as the whole process of cargo moving through the autophagy system, from phagophore formation to autophagosome formation, autophagosome-lysosome fusion, and eventually cargo degradation plus recycling. Autophagy is a dynamic, not static process, and TEM is the gold standard method for assessing the presence of autophagic vesicles, like autophagosomes, lysosomes, autolysosomes (Dutta et al., 2016). Autophagosomes are confirmed by transmission electron microscopy (TEM) as double-membrane organelles with diameters of about 0.3–0.9 μm in yeast (Baba et al., 1997) and 0.5–1.5 μm in mammals (Mizushima et al., 2002). Additionally, the adaptor protein p62/SQSTM1 is also used to measure autophagy flux (Pankiv et al., 2007). p62 binds to ubiqutinated substrates and LC3 to be degraded along with its cargo. Therefore, the decreased p62 levels indicate the enhancement of autophagy flux, and when autophagy flux is inhibited, the p62 levels increase (Sharifi et al., 2015). LC3 binds both the outer and inner membrane of the autophagosome, and thus, it can be detected by fluorescence, electron microscopy, and Western blot. In neurons, new studies have discovered the unique dynamics of autophagosomes, the latter are distally generated (at the neurite tip), and envelop both organelle and cytosolic cargo in primary neurons. Once formed, mature autophagosomes travel towards the cell soma along the axon driven by the kinesin and dynein motor (Maday et al., 2012). Both the autophagy inhibitors like 3-MA, Bafilomycin A1, and chloroquine, as well as activators like Rapamycin, are used to reveal autophagic mechanisms by affecting on autophagy flux (Yang et al., 2013).
3.2 Autophagy in aging brain and in neurodegenerative diseases
Autophagy plays a vital role in maintaining a healthy brain resistant to senescence. As the brain ages, oxidative stress induces damage/modification of DNA, lipids, and proteins, which is an indicator of aging and neurodegeneration (Giordano et al., 2014). The efficiency of the macroautophagic system decreases with age and determines the cell lifespan (Bergamini, 2006). Consequently, a healthy brain that resists aging requires the maintenance of high levels of autophagy throughout the lifespan (Triplett et al., 2015).
Autophagy is regulated by many factors under several proposed mechanisms. One classic example is the regulation of autophagy via caloric restriction (CR). The mechanism by which CR promotes autophagy likely involves mTORC1 inhibition and TFEB/TFE3 activation, it has been shown that high caloric intake suppresses autophagy and CR results in significant learning and memory improvements in mice (Dong et al., 2015). Recent studies have shown that the senescence-associated secretory phenotype (SASP), a pro-inflammatory response linked to aging, is regulated by GATA4. Under normal conditions, GATA4 binds the p62 autophagy adaptor to be degraded by autophagy, but when there is decreased interaction between GATA4 and p62, autophagy is suppressed, leading to the overexpression of GATA4 that leads to senescence (Kang et al., 2015). Further evidence for autophagy’s role in preventing senescence has been found in a recent study of MTMR14, a myotubularin-related phosphatase, in antagonizing the formation of autophagic membrane structures. Blocking MTMR14 by autophagy enhancer-67 (AUTEN-67) stimulates autophagic flux and increases the lifespan of model organisms (Papp et al., 2016).
mTOR is a protein kinase involved in the regulation of cell growth and synthesis of protein in response to growth factors, nutrients, and stress, and is an important inhibitor of autophagy (Mizushima et al., 2010). Thus, mTOR may be involved in the age-related decline of autophagy and accumulation of subcellular detritus. Inhibition of the mTOR pathway by rapamycin has been found to extend life span when both male and female specific pathogen free mice were fed with rapamycin (beginning at 270 and 600 days of age) (Harrison et al., 2009). The lifespan of males and females increased by 9% and 13%, respectively, which further support that mTOR regulates and inhibits autophagy.
Many neurodegenerative diseases such as Alzheimer’s disease are linked with aberrant modification of proteins in neurons (Butterfield and Lauderback, 2002, Carvalho et al., 2015). A classic example of these aberrant proteins is amyloid-beta peptide (Aβ) accumulation, one of the hallmarks of AD. Aβ is a product of the abnormal cleavage of a precursor protein and aggregates to form toxic plaques in brains (Masters et al., 1985, Yankner et al., 1990). The aggregation of such damaged proteins in the brain will disrupt the regulation of apoptosis and autophagy. The failure to remove these aberrant proteins in the brain will cause cellular damage and ultimately cell death (Caballero and Coto-Montes, 2012). Aβ has been found to disrupt autophagy by interrupting the formation of lysosomes (Yu et al., 2005). Indeed, Aβ tends to gather within autophagic vacuoles (AVs), a precursor to the formation of lysosomes. The abundance of AVs within dystrophic neurites of AD brains implies that the lysosome maturation pathway is arrested before full lysosomal maturation, causing dysfunction of autophagy (Yu et al., 2005).
Likewise, the accumulation of dysfunctional mitochondria will also lead to neurodegenerative disease. In PD, mutations in protein deglycase DJ-1 (PARK7) cause a change of protein structure and loss of its function (McCoy and Cookson, 2011). The accumulation of inactive PARK7 therefore results in the subsequent accumulation of dysfunctional mitochondria by making mitochondria more vulnerable to oxidative stress. Additionally, mitochondria collected from the brains of triple transgenic AD mice have been observed to have decreased biogenesis and autophagy, which prevent the recycling of damaged mitochondria (Carvalho et al., 2015). CMA, however, can target and reduce the amount of aberrant PARK7 to maintain healthy mitochondria (Wang, B et al., 2016).
Another protein related to autophagy in the aged brain is progranulin (PGRN), a larger protein precursor to peptide granulin, both of which are linked to cancer, inflammation, and mediation of the cell cycle (Cenik et al. 2012). Progranulin is involved in neuronal ceroid lipofuscinosis (NCL) and frontotemporal lobar degeneration (FTLD) (Baker et al., 2006). It has been shown that the complete loss of the PGRN-coding gene GRN results in adult-onset neuronal ceroid lipofuscinosis, which is a lysosomal storage disease (LSD), and the heterogeneous loss of GRN results in a subtype of FTLD, FTLD-TDP (Benussi et al., 2008, Smith et al., 2012). Although much more investigation is required, it appears that PGRN is the common link between FTLD-TDP and LSD, suggesting that PGRN has an effect on the lysosomal pathway, which in turn has an effect on the development of neurodegenerative diseases (Götzl et al., 2014).
Although the link between ER stress and altered autophagy in the aging brain remains unknown, there have been some recent reports on the topic. XBP1 is involved in the regulation of key ER-associated degradation (ERAD) genes that were activated during ER stress (Lee et al., 2003). Reduced ERAD activity results in increased levels of misfolded proteins and stimulates autophagy to compensate, which can be observed in XBP1 deficient mice (Hetz et al., 2009). In fact, XBP1 may be an inhibitor of autophagy by negatively regulation of FOXO1, which controls the transcription of autophagy genes in neurons (Garcia-Huerta et al., 2016). Therefore, it can be speculated that proper response to ER stress through ERAD pathways can prevent the necessity of the XBP1 activation and causes impairment of autophagy. Obviously, further research is needed to investigate the relationship between ER stress and altered autophagy in many neurodegenerative diseases.
4. Changes of ER stress and autophagy after traumatic brain injury and ischemic stroke
Traumatic brain injury (TBI) is composed of an initial, physical damage to the brain followed by secondary, prolonged deterioration of brain tissue (Stoica and Faden, 2010). This secondary injury can be present long after the initial blow and involves many complications such as Ca2+ concentration disruption, free radical generation, ischemia, edema, excitotoxicity, and intracranial hypertension, and Ca2+ concentration disruption in particular can lead to increased protein unfolding and misfolding to result in ER stress (Greve and Zink, 2009, Begum et al., 2014). On the other hand, ischemic strokes are the leading cause of death, cognitive and motor dysfunction, and neurodegenerative diseases (Wei et al., 2012). Similarly, ER stress has been found in neurons after ischemic stroke (Yu et al., 2016).
Additionally, many studies have found autophagy to be affected by TBI and ischemia, and it may have positive and/or negative effects on the damaged brain tissues. Although the activation of autophagy may either protect or destroy the neuronal cells, the levels of autophagy determine which role autophagy plays (Chen et al., 2014, Wei et al., 2012). Because there is both ER stress and autophagy observed after TBI and ischemic stroke, investigation between any possible links between the two are of great interest.
4.1 Changes of ER stress after TBI and ischemic stroke
TBI is often associated with Ca2+ disruptions and perturbations that could result in the aggregation of unfolded and misfolded proteins within cells of the central nervous system (CNS). In the spinal cord, after traumatic injury spinal cord white matter can die after release of ER Ca2+ mediated by the ryanodine (RyR) and inositol (1,4,5)-triphosphate (IP3R) receptors (Thorell et al., 2002). In the brain, elevated Ca2+ levels have been found to be a hallmark of neuronal injury and death after traumatic injury (McIntosh et al., 1997; Raghupathi, 2004). It has even been suggested that neurological disorders such as epilepsy are caused from the injury, not death, of neurons caused by elevated Ca2+ levels (Delorenzo et al., 2005).
Ca2+ homeostasis is important to TBI and ischemia because a disruption of ER Ca2+ homeostasis may result in ER stress and subsequent UPR activation. One study of astrocytes exposed to oxygen/glucose deprivation and reoxygenation (OGD/REOX) simulating ischemia caused an increase in ER Ca2+ concentrations that coincided with increased ER stress (Begum et al., 2012). In rats, TBI can lead to disruption of Ca2+ homeostasis for as long as 30 days post injury (Sun et al., 2008). Moreover, in cardio myocytes depletion of Ca2+ within the ER leads to activation of the ATF6 arm of the UPR (Thuerauf et al., 2001). Thus, Ca2+ dysregulation after TBI or ischemia plays a central role in triggering UPR.
Well established evidence shows that ER stress followed by UPR activation is involved in TBI and ischemic stroke. For example, one study has found that ER stress increases via the PERK arm after blast-induced traumatic brain injury (bTBI) in rats (Logsdon et al., 2014). The ATF6 arm is activated after the concussive mild traumatic injury (mTBI) in the mouse model (Rubovitch et al., 2011). Furthermore, low oxygen and low nutrient environments similar to that of ischemia have been found to induce ER stress through IRE1 activation (Yoneda et al., 2001). ER stress after ischemic stroke has been observed to act through all three arms as well, but interactions between all three arms have yet to be fully elucidated (Hetz et al., 2011, Zhang et al., 2014).
The determination of all these pathways after TBI and ischemia could not have been possible without the use of ER stress marker proteins. One of the most commonly used ER stress marker proteins is C/EBP homologous protein (CHOP), which is also known as growth arrest- and DNA damage-inducible gene 153 (GADD153). The accumulation of CHOP is a hallmark of ER stress and can lead to ER stress-associated apoptosis (Oyadomari et al., 2004). Studies have shown that CHOP levels become elevated in both the controlled cortical impact injury (CCI) rat model of TBI and in rat global cerebral ischemia (Krajewska et al., 2011, Paschen et al., 1998). As transcription factors of the UPR, the detection of ATF4 and XBP-1 can be used to measure the activity of the UPR (Valenzuela et al., 2012). Since ER stress and UPR activation are known to potentially lead to apoptosis, their association to activation of the caspase family of cysteine proteases has been studied (García de la Cadena and Massieu, 2016). In particular, Caspase-12 has been suggested to be activated by ER stress to initiate apoptosis (Nakagawa et al., 2000). Thus, ER stress has been shown to be involved in both TBI and ischemic stroke and can lead to cell damage and death. Targeting ER stress presents itself as a potential therapeutic option because it can prevent the accumulation of abnormal proteins to promote neuronal recovery.
4.2 Changes of autophagy after TBI and ischemic stroke
Like ER stress, autophagy is also affected after TBI and ischemic stroke. This controversial role of autophagy after TBI and ischemia is prevalent across the literature with some studies concluding that autophagy is neuroprotective and other studies suggesting that autophagy leads to cell death. Beclin 1, a member of the PI3K complex that is responsible for induction of autophagy, is a common marker of autophagic activity. In one study of ischemia in rats, inhibition of autophagy by 3-MA led to decreased Beclin 1 expression and increased cell death, but promotion of Beclin 1 and autophagy by rapamycin reduced cell death and brain injury, suggesting beneficial roles of autophagic activity (Carloni et al., 2008). Similarly in a study of the fluid percussion rat model of TBI, autophagy was stimulated after the injury through the accumulation of LC3-II, autophagosomes, and autolysosomes and deemed likely to serve a neuroprotective role (Liu et al., 2008). Indeed, promotors of autophagy such as rapamycin have been shown to provide neuroprotection after TBI in mice and reduce the rate of secondary TBI injuries like epilepsy (Erlich et al., 2007, Guo et al., 2013).
Conversely, other studies have shown that autophagy is neurotoxic and leads to cell death after TBI and ischemia. As an integral protein for the formation of the autophagosome, LC3-II is a common indicator of autophagy, and levels of LC3-II were elevated after permanent middle cerebral artery occlusion (pMCAO) in the rat model of cerebral ischemia (Wen et al., 2008). Studies have shown that the administration of the autophagy inhibitor 3-MA was actually neuroprotective and prevented neuronal death after ischemia induced by pMCAO and four-vessel occlusion (4-VO) in rats (Wen et al., 2008, Xin et al., 2011). Likewise, a study using the weight drop TBI mouse model and inhibition of autophagy with 3-MA found that autophagy contributed to increased cell death and neurological impairment (Luo et al., 2011).
Autophagy has also been found to have some relationship with ER stress. One study subjected cell culture to the ER stressors tunicamycin and thapsigargin found that autophagy was activated by the IRE1 arm of the UPR during ER stress (Ogata et al., 2006). In addition, when autophagy was blocked by 3-MA, those cells experienced higher cell death and activation of the apoptotic factor caspase-3. Ogata et al. showed that when autophagy was inhibited through ATG5 deficiency, the cells underwent ER stress-dependent death, and cells were more resistant to such activity when autophagy was induced by rapamycin. Thus, Ogata el al. suggest that autophagy protects cells from ER stress related death.
Additional research is warranted to better understand the complex role of autophagy in brain injury and tissue repair after TBI and ischemia. One explanation for the above controversial findings is that autophagy may serve a neuroprotective role if autophagy flux is unobstructed but is detrimental when autophagy flux is hindered (Lipinski et al., 2015). Indeed, a common measurement of autophagy for many of the previous studies cited relied on LC3-II levels, but elevated LC3-II concentrations may not necessarily equate to increased autophagy. In a study of CCI brain injury simulating TBI in mice, the increase of LC3-II was not from an increase in autophagy, but rather an impairment of autophagy flux evidenced by elevated levels of SQSTM1/p62, which is normally depleted during nominal autophagy flux (Sarkar et al., 2014). In other words, because p62 binds to LC3-II located in the inner membrane of the autophagosome and is degraded after formation of the autolysosome, its abundance shows that LC3-II accumulates after TBI due to an absence of autolysosome formation during autophagy flux. Since autophagy flux is necessary for the degradation of aberrant proteins and organelles, its disruption would cause their buildup instead of elimination. However, decreased autophagy flux was determined to be correlated with cell death and not necessarily causing the observed cell death in that study.
Further evidence supporting the theory that autophagy flux plays a role in determining the neuroprotection or destruction aspect of autophagy can be found in a study of the mouse focal cerebellar lesion TBI model, comparing wild-type mice and the mice with impaired autophagy flux pathways (Viscomi et al., 2012). In that study, autophagy-impaired beclin 1 heterozygous Becn1+/− mice experienced decreased autophagy flux through elevated p62 levels and decreased LC3-II levels compared to wild-type, but wild-type mice experienced the expected decrease in p62 levels paired with increased LC3-II levels, leading to significantly higher neuronal survivability and improved neurological recovery (NSS) score in wild-type mice. Thus, Viscomi et al. first concluded that both autophagy initiation and autophagy flux were impaired in the Becn1+/− mice, and that disruption of these pathways is detrimental to the brain. The researchers then studied the effects of autophagy enhancing rapamycin on both the wild type and Becn1+/− mice. They found that wild-type rapamycin treated mice produced significantly higher amounts of LC3-II and significantly lower amounts of p62 compared to wild-type mice that did not did not receive rapamycin to yield greater neuronal survivability and improved NSS score. However, Becn1+/− mice given rapamycin, which should have helped induce autophagy in these autophagy diminished mice, did not produce any change in LC3-II, p62, or neuronal survivability compared to untreated Becn1+/− mice. Therefore, Viscomi et al. concluded that when autophagy flux is unperturbed, induction of autophagy is neuroprotective, but weakened autophagy and autophagy flux pathways are conducive to cell death. No studies using the similar mouse line have been performed regarding autophagy flux after brain ischemia. But similar results can be found in studies of cardiac ischemia-reperfusion (I/R) injury in rats and mice. These studies have found that promoters of autophagy enhanced cardioprotection when autophagy flux was unimpaired but were not effective when autophagy flux was disrupted through lysosomal inhibition (Xie et al., 2015, Ling et al., 2016).
However, disrupted autophagy flux may be just one possible explanation. Another possibility could be the rupture or leakage of lysosomes after injury leading to cell death (Wen et al., 2008, Yamashima and Oikawa, 2009). It has been suggested that the severity and type of brain injury could influence the role of autophagy (Lipinski et al., 2015). Others have stated that the time at which autophagy is induced may determine the neuroprotective effects of autophagy (Chen et al., 2014). The studies cited in this review certainly contain a wide variety of injuries, many of which did not examine the effects of autophagy flux or lysosomes after injury. Hence, it is evident that more comprehensive studies are needed to conclude whether autophagy is beneficial or detrimental after TBI and ischemia.
5.0 DHA in aging brain
In the brain, docosahexaenoic acid (DHA, 22:6n-3) is the predominant omega-3 fatty acid. About 10–20% of gray matter, 2% of white matter, and 50% of the weight of a neuron’s plasma membrane are composed of DHA (McNamara, 2013, Gleissman et al., 2010). DHA supports brain development and protects neurological function. Although DHA plays a critical role in optimal brain functions, the dietary polyunsaturated fatty is not effectively converted to DHA. Thus, it has been recommended for people to obtain more DHA through food such as cold water fish or DHA supplement (Petraglia et al., 2011).
Many studies have documented the importance of DHA in promoting the healthy aging of the brain and how age-related neurodegenerative diseases are characterized by low levels of DHA in the brain. In human studies, numerous investigations have reported on the correlation between low levels of plasma DHA with cognitive decline and AD in elderly humans (Beydoun et al., 2007, Schaefer et al., 2006, Astarita et al., 2010). Conversely, populations that consume high dietary amounts of DHA experience reduced risk of cognitive impairment and neurodegenerative diseases in aging (Noel et al., 2006, Kalmijn et al., 2004, Morris et al., 2003). In 2010, a study concluded that supplementation of 900 mg/d DHA over a period of 24 weeks improved the learning and memory of healthy adults aged ≥55 (Yurko-Mauro et al., 2010). In 2016, a 12 month long study found that DHA improved participants’ results on the mini-mental state examination (MMSE) and increased the subjects’ apathy and reduced caregiver burden of those with cognitive impairment such as dementia (Hashimoto et al., 2016). Thus, DHA appears to have positive effects on those who experience mild age-related cognitive decline.
In mice, a DHA rich diet retains the learning and memory of aged senescence-accelerated prone 8 (SAMP8) mice that would normally experience cognitive decline (Petursdottir et al., 2008). Indeed, animal models have also shown that DHA treatment over a substantial period of time not only reduces neuronal loss in the brain but also improves the learning and memory of the animals as they age (Hooijmans et al., 2012). However, studies have asserted that DHA treatments do not have significant improvements of cognitive function in mice (Arendash et al., 2007) and humans that are already diagnosed with advanced dementia or AD (Fotuhi et al., 2009, Freund-Levi et al., 2006, Laurin et al., 2003, Stough et al., 2012).
The difficulty in treating dementia and AD with DHA can be explained by the lack of a definitive underlying mechanism relating DHA and neurodegenerative diseases. It is believed that neuronal loss from inflammation and oxidation leads to age-related cognitive decline. To date, there have been several proposed mechanisms linking DHA to the inhibition of neuronal inflammation and oxidation. First, the derivatives of omega-3 polyunsaturated fatty acids (PUFAs) such as DHA play a role in mediating the inflammatory response to prevent chronic inflammation (Dyall, 2015). One such class of derivatives is “specialized pro-resolving mediators” (SPMs) that form novel families of autacoids named resolvins, protectins, and maresins, which have potent tissue-protective and inflammatory resolution-stimulating functions (Bannenberg and Serhan, 2010). In particular, resolvins of the D series (RvD) and protectins are derived from DHA and strongly stimulate the resolution of inflammation (Serhan, 2007). This distinction is contrasted with another group of electrophilic omega-3 PUFA derivatives that promote the anti-inflammatory response. Electrophilic fatty acid oxo-derivatives (EFOXs) are produced from DHA, docosapentaenoic acd (DPA), and eicosapentaenoic acid (EPA) catalyzed by cyclooxygenase-2 (COX-2) to form 7-oxo-DHA, 7-oxo-DPA and 5-oxo-EPA, respectively (Cipollina et al., 2014). These molecules are anti-inflammatory by acting as agonists to peroxisome proliferator-activated receptor gamma (PPARγ), activating Nrf2-dependent antioxidant responses, and inhibiting cytokine production and inducible nitric oxide synthase (NOS-2) expression within activated macrophages (Groeger et al., 2010).
Although DHA has been demonstrated to improve membrane fluidity and increase mitochondrial activity in the 24-month aged female NMRI mouse brain (Afshordel et al., 2015), research has increasingly pointed to DHA as a precursor to products that regulate the gene expression of anti-inflammatory and protective pathways. It has been demonstrated that DHA inhibits p38 mitogen-activated protein kinase (p38 MAPK) phosphorylation, which consequently inhibits the expression of inflammatory molecules in microglia (Lu et al., 2013) as well as activating PPARγ to modulate microglial inflammatory response (Antonietta Ajimone-Cat et al., 2012). It is now known that such regulation comes not from DHA itself, but from its derivatives (Heras-Sandoval et al., 2016). Another DHA derivative involved in signal cascade that has been well studied is neuroprotectin D1 (NPD1). NPD1 induces cREL transcription and nuclear translocation, which promotes BIRC3 transcription for cell survival against oxidative stress (Calandria et al., 2015). In particular, NPD1 prevents oxidative stress induced cell death by up-regulating the anti-apoptotic proteins Bcl-2 and Bcl-xL while inhibiting pro-apoptotic Bax, Bad, and caspase-3 (Bazan, 2005).
Even though there is a wealth of evidence that links DHA and its derivatives to the reduction of inflammation and oxidation linked with brain aging, it is still important to keep in mind that there have been no definitive reports of DHA successfully reversing the effects of highly progressed dementia and AD. Thus, further research into a therapeutic treatment is needed.
Selective effects of DHA in ER stress and autophagy in cancer cells
DHA has also been reported to have anti-carcinogenic properties and acts through a variety of mechanisms. One such mechanism involves n-3 PUFA EPA and DHA inhibiting the synthesis of certain derivatives of arachidonic acid (AA), itself a n-3 PUFA eicosanoid, that cause inflammation (Larsson et al., 2004). Both n-3 PUFA and n-6 PUFA follow similar metabolic pathways and utilize similar enzymes, but n-3 PUFAs have greater affinity in particular with delta 6-desaturase to direct metabolic activity in favor of EPA over AA (Young et al., 2011). The prevalence of EPA and DHA over AA consequently leads to their increased presence in the membrane, where both types of PUFA are susceptible to oxidation by COX-2 (Burdge and Calder, 2005). Overexpression of COX-2 that causes increased eicosanoid derivatives of n-6 PUFA have been linked to cancer (Benoit et al., 2004), but reactions between COX-2 and DHA have the opposite effect (Benoit et al., 2004, Groeger et al., 2010).
A primary method of cancer formation is the activation of oncogenes. One such oncogene is epidermal growth factor receptor (EGFR) that causes cell proliferation when overexpressed, but its activation is reduced by EPA and DHA (Kang and Weylandt, 2008). Indeed, DHA was particularly effective in inducing cell apoptosis and reducing the expression of Bcl2 and procaspase-8 (Corsetto et al., 2011). Another similar factor involved with oncogenesis of a variety of cancers is human epidermal growth factor receptor-2 (HER-2) (Yang, W.J. et al., 2015). Once activated, HER-2 initiates a signal cascade that causes excess cell proliferation (Bollig-Fischer et al., 2010), but the pathway was inhibited by n-3 PUFA (Yee et al., 2013). DHA leads to the generation of mitochondrial reactive oxygen species (ROS) that inhibits the PI3K/Akt/mTOR pathway, which causes resistance to apoptosis (Morgan et al., 2009) and induces both apoptosis and autophagy (Shin et al., 2013). Administration of EPA and DHA through dietary fish oil supplementation was found to significantly increase expression of Bax, a proapoptotic protein, and reduce anti-apoptotic protein Bcl-2 (Manna et al., 2008).
In fact, a relatively recent discovery of anticarcinogenic propterties of DHA lies in its connection with ER stress. As mentioned previously in this review, eIF2α is a hallmark of ER stress, and when human colon adenocarcinoma cells, SW620, were exposed to DHA for 3 hours, increased levels of phosphorylated eIF2α were observed, indicating increased ER stress (Jakobsen, 2008). This study suggests that prolonged DHA-induced ER stress may lead to apoptosis in these cancer cells. However, at the time of this writing, there are very few other studies on the relationships between DHA, ER stress, and cancer to corroborate these findings. Still, it is abundantly clear that there are several promising avenues of further investigation for therapeutic roles of DHA in cancer.
6.0 Effects of DHA on ER stress and autophagy after brain injury
A growing number of studies suggest that DHA emerges as a neuroprotective agent against neurodegenerative diseases such as AD, PD, TBI and cerebrovascular disease (Calon and Cole, 2007, Mayurasakorn et al., 2011, Begum et al., 2014). However, the molecular mechanisms underlying DHA’s neuroprotective effects are not exactly known. DHA is thought to act on multiple pathways. As a component of neuronal cell membranes, DHA influences a variety of membrane-bound proteins including receptors, transporters and ion channels to maintain their normal conformation (Belayev et al., 2009). Additionally, DHA impacts neurogenesis, neurite growth, synaptogenesis, expression of synapsin, and synaptic connection (Dyall, 2015, Igarashi et al., 2015). Furthermore, through scavenging radicals, activating survival genes, and cell signaling pathways, DHA shows its neuroprotective effect (Bazinet and Layé, 2014).
Recent studies have shown that DHA possesses a neuroprotective effect via reducing ER stress. Under in vitro ischemic conditions, DHA reduced the exogenous IP3-mediated Ca2+ ER release rate and decreased the amount of ER Ca2+ release (~ 40%), but did not inhibit store-operated Ca2+ entry (SOCE) following depletion of Ca2+ ER stores in mouse astrocytes (Begum et al., 2012). Also, DHA reduced the expression of the other two ER stress markers, p-eIF2α and ATF-4, and ischemic cell death. These data suggest that DHA reduced ER stress.
In a CCI rat model, post-TBI administration of DHA (16 mg/kg in DMSO, at 5–15 min after onset of TBI and subsequent daily dose for 21d after TBI): i) decreased the expressions of UPR markers- including p-eIF2α, IRE1α, and ATF4 but not the UPR regulator protein GRP78- in the ipsilateral peri-lesion areas after TBI; ii) reduced expression of ER proapoptosis protein CHOP; iii) attenuated TBI-mediated protein ubiquitination, accumulation of amyloid precursor protein (APP) and phosphorylated tau (p-Tau) (Begum et al., 2014). In summary, administration of DHA reduces ER stress and toxic protein accumulation following TBI.
In a more recent study, DHA-treated brains showed significantly fewer proinflammatory CD16/32+ positive, but more anti-inflammatory CD206+ microglia or macrophages throughout 3 to 21 days post-TBI (Harvey et al., 2015). In addition, DHA treatment altered microglial or macrophage morphology from the activated, amoeboid-like state to the more surveillant, permissive state. These data suggest that the administration of DHA after TBI reduces neuronal ER stress and association with microglial or macrophage polarization, and decreases neurodegeneration.
7.0 Summary
Collectively, newly emerged evidence indicates that maintaining appropriate ER function and autophagy flux is important for cell survival in normal brains. However, ER stress and autophagy dysfunction are associated with aging, diverse cprotective hronic neurodegenerative diseases, and traumatic and ischemic brain injury. Therefore, restoring impaired ER function and autophagy flux represents a new target for novel therapies. In the brain, DHA is the predominant omega-3 fatty acid, supports brain development, protects neurological function. Although DHA shows effects against different types of neuronal disorders, brain aging and cancer, the underlying mechanisms have not yet been clearly defined. It has been shown that post-injury administration of DHA reduced ER stress and toxic protein accumulation following TBI and ischemic brain damage. However, whether DHA enhances autophagy flux and tissue repair remains to be tested.
Highlights.
Review of ER stress, UPR activation, autophagy, and autophagy flux processes
Changes of autophagy in the aging brain and neurodegenerative diseases
Review of ER stress and autophagy after TBI and ischemic stroke
Summary of effects of DHA on ER stress and autophagy after TBI and ischemia
Effects of DHA in the aging brain and cancer cells
Acknowledgments
Funding sources
This work was supported in part by the National Institutes of Health R01 NS038118, NS048216, and NS089051, and Veteran Affairs Merit Award I01BX002891 (D.S); the Chinese Dalian Municipal Science and Technology Plan Project [2014E14SF186], and Dalian Municipal Medical Training Project (Y.Y).
Footnotes
Disclosure of Potential Conflicts of Interest
No conflicts of interest are declared by the authors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Afshordel S, Hagl S, Werner D, Rohner N, Kogel D, Bazan NG, Eckert GP. Omega-3 polyunsaturated fatty acids improve mitochondrial dysfunction in brain aging--impact of Bcl-2 and NPD-1 like metabolites. Prostaglandins Leukot Essent Fatty Acids. 2015;92:23–31. doi: 10.1016/j.plefa.2014.05.008. [DOI] [PubMed] [Google Scholar]
- Antonietta Ajmone-Cat M, Lavinia Salvatori M, De Simone R, Mancini M, Biagioni S, Bernardo A, Cacci E, Minghetti L. Docosahexaenoic acid modulates inflammatory and antineurogenic functions of activated microglial cells. J Neurosci Res. 2012;90:575–587. doi: 10.1002/jnr.22783. [DOI] [PubMed] [Google Scholar]
- Aparicio IM, Martin Munoz P, Salido GM, Pena FJ, Tapia JA. The autophagy-related protein LC3 is processed in stallion spermatozoa during short-and long-term storage and the related stressful conditions. Animal. 2016:1–10. doi: 10.1017/S1751731116000240. [DOI] [PubMed] [Google Scholar]
- Arendash GW, Jensen MT, Salem N, Jr, Hussein N, Cracchiolo J, Dickson A, Leighty R, Potter H. A diet high in omega-3 fatty acids does not improve or protect cognitive performance in Alzheimer’s transgenic mice. Neuroscience. 2007;149:286–302. doi: 10.1016/j.neuroscience.2007.08.018. [DOI] [PubMed] [Google Scholar]
- Astarita G, Jung KM, Berchtold NC, Nguyen VQ, Gillen DL, Head E, Cotman CW, Piomelli D. Deficient liver biosynthesis of docosahexaenoic acid correlates with cognitive impairment in Alzheimer’s disease. PLoS One. 2010;5:e12538. doi: 10.1371/journal.pone.0012538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba M, Osumi M, Scott SV, Klionsky DJ, Ohsumi Y. Two distinct pathways for targeting proteins from the cytoplasm to the vacuole/lysosome. J Cell Biol. 1997;139:1687–1695. doi: 10.1083/jcb.139.7.1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442:916–919. doi: 10.1038/nature05016. [DOI] [PubMed] [Google Scholar]
- Bannenberg G, Serhan CN. Specialized pro-resolving lipid mediators in the inflammatory response: An update. Biochim Biophys Acta. 2010;1801:1260–1273. doi: 10.1016/j.bbalip.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazan NG. Neuroprotectin D1 NPD1: a DHA-derived mediator that protects brain and retina against cell injury-induced oxidative stress. Brain Pathol. 2005;15:159–166. doi: 10.1111/j.1750-3639.2005.tb00513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazinet RP, Laye S. Polyunsaturated fatty acids and their metabolites in brain function and disease. Nat Rev Neurosci. 2014;15:771–785. doi: 10.1038/nrn3820. [DOI] [PubMed] [Google Scholar]
- Begum G, Kintner D, Liu Y, Cramer SW, Sun D. DHA inhibits ER Ca2+ release and ER stress in astrocytes following in vitro ischemia. J Neurochem. 2012;120:622–630. doi: 10.1111/j.1471-4159.2011.07606.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begum G, Yan HQ, Li L, Singh A, Dixon CE, Sun D. Docosahexaenoic acid reduces ER stress and abnormal protein accumulation and improves neuronal function following traumatic brain injury. J Neurosci. 2014;34:3743–3755. doi: 10.1523/JNEUROSCI.2872-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belayev L, Khoutorova L, Atkins KD, Bazan NG. Robust docosahexaenoic acid-mediated neuroprotection in a rat model of transient, focal cerebral ischemia. Stroke. 2009;40:3121–3126. doi: 10.1161/STROKEAHA.109.555979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benussi L, Binetti G, Sina E, Gigola L, Bettecken T, Meitinger T, Ghidoni R. A novel deletion in progranulin gene is associated with FTDP-17 and CBS. Neurobiol Aging. 2008;29:427–435. doi: 10.1016/j.neurobiolaging.2006.10.028. [DOI] [PubMed] [Google Scholar]
- Benoit V, Relic B, Leval Xd X, Chariot A, Merville MP, Bours V. Regulation of HER-2 oncogene expression by cyclooxygenase-2 and prostaglandin E2. Oncogene. 2004;23:1631–1635. doi: 10.1038/sj.onc.1207295. [DOI] [PubMed] [Google Scholar]
- Bergamini E. Autophagy: a cell repair mechanism that retards ageing and age-associated diseases and can be intensified pharmacologically. Mol Aspects Med. 2006;27:403–410. doi: 10.1016/j.mam.2006.08.001. [DOI] [PubMed] [Google Scholar]
- Beydoun MA, Kaufman JS, Satia JA, Rosamond W, Folsom AR. Plasma n-3 fatty acids and the risk of cognitive decline in older adults: the Atherosclerosis Risk in Communities Study. Am J Clin Nutr. 2007;85:1103–1111. doi: 10.1093/ajcn/85.4.1103. [DOI] [PubMed] [Google Scholar]
- Bitto A, Lerner CA, Nacarelli T, Crowe E, Torres C, Sell C. P62/SQSTM1 at the interface of aging, autophagy, and disease. Age Dordr. 2014;36:9626. doi: 10.1007/s11357-014-9626-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollig-Fischer A, Dziubinski M, Boyer A, Haddad R, Giroux CN, Ethier SP. HER-2 signaling, acquisition of growth factor independence, and regulation of biological networks associated with cell transformation. Cancer Res. 2010;70:7862–7873. doi: 10.1158/0008-5472.CAN-10-1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo R, Parra V, Gatica D, Rodriguez AE, Torrealba N, Paredes F, Wang ZV, Zorzano A, Hill JA, Jaimovich E, et al. Endoplasmic reticulum and the unfolded protein response: dynamics and metabolic integration. Int Rev Cell Mol Biol. 2013;301:215–290. doi: 10.1016/B978-0-12-407704-1.00005-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdge GC, Calder PC. Conversion of alpha-linolenic acid to longer-chain polyunsaturated fatty acids in human adults. Reprod Nutr Dev. 2005;45:581–597. doi: 10.1051/rnd:2005047. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Lauderback CM. Lipid peroxidation and protein oxidation in Alzheimer’s disease brain: potential causes and consequences involving amyloid beta-peptide-associated free radical oxidative stress. Free Radic Biol Med. 2002;32:1050–1060. doi: 10.1016/s0891-5849(02)00794-3. [DOI] [PubMed] [Google Scholar]
- Button RW, Luo S, Rubinsztein DC. Autophagic activity in neuronal cell death. Neurosci Bull. 2015;31:382–394. doi: 10.1007/s12264-015-1528-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caballero B, Coto-Montes A. An insight into the role of autophagy in cell responses in the aging and neurodegenerative brain. Histol Histopathol. 2012;27:263–275. doi: 10.14670/HH-27.263. [DOI] [PubMed] [Google Scholar]
- Calandria JM, Asatryan A, Balaszczuk V, Knott EJ, Jun BK, Mukherjee PK, Belayev L, Bazan NG. NPD1-mediated stereoselective regulation of BIRC3 expression through cREL is decisive for neural cell survival. Cell Death Differ. 2015;22:1363–1377. doi: 10.1038/cdd.2014.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calon F, Cole G. Neuroprotective action of omega-3 polyunsaturated fatty acids against neurodegenerative diseases: evidence from animal studies. Prostaglandins Leukot Essent Fatty Acids. 2007;77:287–293. doi: 10.1016/j.plefa.2007.10.019. [DOI] [PubMed] [Google Scholar]
- Carloni S, Girelli S, Scopa C, Buonocore G, Longini M, Balduini W. Activation of autophagy and Akt/CREB signaling play an equivalent role in the neuroprotective effect of rapamycin in neonatal hypoxia-ischemia. Autophagy. 2008;6:366–377. doi: 10.4161/auto.6.3.11261. [DOI] [PubMed] [Google Scholar]
- Carvalho C, Santos MS, Oliveira CR, Moreira PI. Alzheimer’s disease and type 2 diabetes-related alterations in brain mitochondria, autophagy and synaptic markers. Biochim Biophys Acta. 2015;1852:1665–1675. doi: 10.1016/j.bbadis.2015.05.001. [DOI] [PubMed] [Google Scholar]
- Cenik B, Sephton CF, Kutluk Cenik B, Herz J, Yu G. Progranulin: A Proteolytically Processed Protein at the Crossroads of Inflammation and Neurodegeneration. The Journal of Biological Chemistry. 2012;287:32298–32306. doi: 10.1074/jbc.R112.399170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesen MH, Pegan K, Spes A, Turk B. Lysosomal pathways to cell death and their therapeutic applications. Exp Cell Res. 2012;318:1245–1251. doi: 10.1016/j.yexcr.2012.03.005. [DOI] [PubMed] [Google Scholar]
- Chen W, Sun Y, Liu K, Sun X. Autophagy: a double-edged sword for neuronal survival after cerebral ischemia. Neural Regen Res. 2014;9:1210–1216. doi: 10.4103/1673-5374.135329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipollina C, Salvatore SR, Muldoon MF, Freeman BA, Schopfer FJ. Generation and dietary modulation of anti-inflammatory electrophilic omega-3 fatty acid derivatives. PLoS One. 2014;9:e94836. doi: 10.1371/journal.pone.0094836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corsetto PA, Montorfano G, Zava S, Jovenitti IE, Cremona A, Berra B, Rizzo AM. Effects of n-3 PUFAs on breast cancer cells through their incorporation in plasma membrane. Lipids Health Dis. 2011;10:73. doi: 10.1186/1476-511X-10-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Villanueva JF, Diaz-Molina R, Garcia-Gonzalez V. Protein Folding and Mechanisms of Proteostasis. Int J Mol Sci. 2015;16:17193–17230. doi: 10.3390/ijms160817193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong W, Wang R, Ma LN, Xu BL, Zhang JS, Zhao ZW, Wang YL, Zhang X. Autophagy involving age-related cognitive behavior and hippocampus injury is modulated by different caloric intake in mice. Int J Clin Exp Med. 2015;8:11843–11853. [PMC free article] [PubMed] [Google Scholar]
- Delorenzo RJ, Sun DA, Deshpande LS. Cellular mechanisms underlying acquired epilepsy: the calcium hypothesis of the induction and maintenance of epilepsy. Pharmacol Ther. 2005;105:229–66. doi: 10.1016/j.pharmthera.2004.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan MJ, Stanley M, Parkhouse N, Cook K, Smith M. Acute caffeine ingestion enhances strength performance and reduces perceived exertion and muscle pain perception during resistance exercise. Eur J Sport Sci. 2013;13:392–399. doi: 10.1080/17461391.2011.635811. [DOI] [PubMed] [Google Scholar]
- Dunlop EA, Tee AR. mTOR and autophagy: a dynamic relationship governed by nutrients and energy. Semin Cell Dev Biol. 2014;36:121–129. doi: 10.1016/j.semcdb.2014.08.006. [DOI] [PubMed] [Google Scholar]
- Duraes FV, Niven J, Dubrot J, Hugues S, Gannage M. Macroautophagy in Endogenous Processing of Self- and Pathogen-Derived Antigens for MHC Class II Presentation. Front Immunol. 2015;6:459. doi: 10.3389/fimmu.2015.00459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta D, Chakraborty B, Sarkar A, Chowdhury C, Das P. A potent betulinic acid analogue ascertains an antagonistic mechanism between autophagy and proteasomal degradation pathway in HT-29 cells. BMC Cancer. 2016;16:23. doi: 10.1186/s12885-016-2055-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyall SC. Long-chain omega-3 fatty acids and the brain: a review of the independent and shared effects of EPA, DPA and DHA. Front Aging Neurosci. 2015;7:52. doi: 10.3389/fnagi.2015.00052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellgaard L, Helenius A. ER quality control: towards an understanding at the molecular level. Curr Opin Cell Biol. 2001;13:431–437. doi: 10.1016/s0955-0674(00)00233-7. [DOI] [PubMed] [Google Scholar]
- Erlich S, Alexandrovich A, Shohami E, Pinkas-Kramarski R. Rapamycin is a neuroprotective treatment for traumatic brain injury. Neurobiol Dis. 2007;26:86–93. doi: 10.1016/j.nbd.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Feng X, Yuan Y, Wang C, Feng J, Yuan Z, Zhang X, Sui W, Hu P, Zheng P, Ye J. Autophagy involved in lipopolysaccharide-induced foam cell formation is mediated by adipose differentiation-related protein. Lipids Health Dis. 2014;13:10. doi: 10.1186/1476-511X-13-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flamment M, Hajduch E, Ferre P, Foufelle F. New insights into ER stress-induced insulin resistance. Trends Endocrinol Metab. 2012;23:381–390. doi: 10.1016/j.tem.2012.06.003. [DOI] [PubMed] [Google Scholar]
- Flemming A, Noda T, Yoshimori T, Rubinsztein DC. Chemical modulators of autophagy as biological probes and potential therapeutics. Nature Chem Bio. 2011;7:9–17. doi: 10.1038/nchembio.500. [DOI] [PubMed] [Google Scholar]
- Fotuhi M, Mohassel P, Yaffe K. Fish consumption, long-chain omega-3 fatty acids and risk of cognitive decline or Alzheimer disease: a complex association. Nat Clin Pract Neurol. 2009;5:140–152. doi: 10.1038/ncpneuro1044. [DOI] [PubMed] [Google Scholar]
- Freund-Levi Y, Eriksdotter-Jonhagen M, Cederholm T, Basun H, Faxen-Irving G, Garlind A, Vedin I, Vessby B, Wahlund LO, Palmblad J. Omega-3 fatty acid treatment in 174 patients with mild to moderate Alzheimer disease: OmegAD study: a randomized double-blind trial. Arch Neurol. 2006;63:1402–1408. doi: 10.1001/archneur.63.10.1402. [DOI] [PubMed] [Google Scholar]
- Fujita E, Kouroku Y, Isoai A, Kumagai H, Misutani A, Matsuda C, Hayashi YK, Momoi T. Two endoplasmic reticulum-associated degradation ERAD systems for the novel variant of the mutant dysferlin: ubiquitin/proteasome ERADI and autophagy/lysosome ERADII. Hum Mol Genet. 2007;16:618–629. doi: 10.1093/hmg/ddm002. [DOI] [PubMed] [Google Scholar]
- Fujita N, Hayashi-Nishino M, Fukumoto H, Omori H, Yamamoto A, Noda T, Yoshimori T. An Atg4B mutant hampers the lipidation of LC3 paralogues and causes defects in autophagosome closure. Mol Biol Cell. 2008;19:4651–4659. doi: 10.1091/mbc.E08-03-0312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita N, Itoh T, Omori H, Fukuda M, Noda T, Yoshimori T. The Atg16L Complex Specifies the Site of LC3 Lipidation for Membrane Biogenesis in Autophagy. Mol Biol Cell. 2008;19:2092–2100. doi: 10.1091/mbc.E07-12-1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García de la Cadena S, Massieu L. Caspases and their role in inflammation and ischemic neuronal death. Focus on caspase-12. Apoptosis. 2016;21:763–77. doi: 10.1007/s10495-016-1247-0. [DOI] [PubMed] [Google Scholar]
- Garcia-Huerta P, Escudero-Troncoso P, Jerez C, Hetz C, Vidal RL. The intersection between growth factors, autophagy and ER stress: A new target to treat neurodegenerative diseases? Brain Res. 2016 doi: 10.1016/j.brainres.2016.02.052. in press. [DOI] [PubMed] [Google Scholar]
- Geng J, Baba M, Nair U, Klionsky DJ. Quantitative analysis of autophagy-related protein stoichiometry by fluorescence microscopy. J Cell Biol. 2008;182:129–140. doi: 10.1083/jcb.200711112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano S, Darley-Usmar V, Zhang J. Autophagy as an essential cellular antioxidant pathway in neurodegenerative disease. Redox Biol. 2014;2:82–90. doi: 10.1016/j.redox.2013.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleissman H, Johnsen JI, Kogner P. Omega-3 fatty acids in cancer, the protectors of good and the killers of evil? Exp Cell Res. 2010;316:1365–1373. doi: 10.1016/j.yexcr.2010.02.039. [DOI] [PubMed] [Google Scholar]
- Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. 2010;221:3–12. doi: 10.1002/path.2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Götzl JK, Mori K, Damme M, Fellerer K, Tahirovic S, Kleinberger G, et al. Common pathobiochemical hallmarks of progranulin-associated frontotemporal lobar degeneration and neuronal ceroid lipofuscinosis. Acta Neuropathol. 2014;127:845–860. doi: 10.1007/s00401-014-1262-6. [DOI] [PubMed] [Google Scholar]
- Greve MW, Zink BJ. Pathophysiology of traumatic brain injury. Mt Sinai J Med. 2009;76:97–104. doi: 10.1002/msj.20104. [DOI] [PubMed] [Google Scholar]
- Groeger AL, Cipollina C, Cole MP, Woodcock SR, Bonacci G, Rudolph TK, Rudolph V, Freeman BA, Schopfer FJ. Cyclooxygenase-2 generates anti-inflammatory mediators from omega-3 fatty acids. Nat Chem Biol. 2010;6:433–441. doi: 10.1038/nchembio.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo D, Zeng L, Brody DL, Wong M. Rapamycin Attenuates the Development of Posttraumatic Epilepsy in a Mouse Model of Traumatic Brain Injury. PLoS ONE. 2013;85:e64078. doi: 10.1371/journal.pone.0064078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–395. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey LD, Yin Y, Attarwala IY, Begum G, Deng J, Yan HQ, Dixon CE, Sun D. Administration of DHA Reduces Endoplasmic Reticulum Stress-Associated Inflammation and Alters Microglial or Macrophage Activation in Traumatic Brain Injury. ASN Neuro. 2015;7 doi: 10.1177/1759091415618969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto M, Kato S, Tanabe Y, Katakura M, Mamun AA, Ohno M, Hossain S, Onoda K, Yamaguchi S, Shido O. Beneficial effects of dietary docosahexaenoic acid intervention on cognitive function and mental health of the oldest elderly in Japanese care facilities and nursing homes. Geriatr Gerontol Int. 2016 doi: 10.1111/ggi.12691. in press. [DOI] [PubMed] [Google Scholar]
- Heras-Sandoval D, Pedraza-Chaverri J, Perez-Rojas JM. Role of docosahexaenoic acid in the modulation of glial cells in Alzheimer’s disease. J Neuroinflammation. 2016;13:61. doi: 10.1186/s12974-016-0525-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C, Martinon F, Rodriguez D, Glimcher LH. The unfolded protein response: integrating stress signals through the stress sensor IRE1alpha. Physiol Rev. 2011;91:1219–1243. doi: 10.1152/physrev.00001.2011. [DOI] [PubMed] [Google Scholar]
- Hetz C, Thielen P, Matus S, Nassif M, Court F, Kiffin R, et al. XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev. 2009;23:2294–2306. doi: 10.1101/gad.1830709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooijmans CR, Pasker-de Jong PC, de Vries RB, Ritskes-Hoitinga M. The effects of long-term omega-3 fatty acid supplementation on cognition and Alzheimer’s pathology in animal models of Alzheimer’s disease: a systematic review and meta-analysis. J Alzheimers Dis. 2012;28:191–209. doi: 10.3233/JAD-2011-111217. [DOI] [PubMed] [Google Scholar]
- Igarashi M, Santos RA, Cohen-Cory S. Impact of maternal n-3 polyunsaturated fatty acid deficiency on dendritic arbor morphology and connectivity of developing Xenopus laevis central neurons in vivo. J Neurosci. 2015;35:6079–6092. doi: 10.1523/JNEUROSCI.4102-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaber N, Dou Z, Chen JS, Catanzaro J, Jiang YP, Ballou LM, Selinger E, Ouyang X, Lin RZ, Zhang J, et al. Class III PI3K Vps34 plays an essential role in autophagy and in heart and liver function. Proc Natl Acad Sci U S A. 2012;109:2003–2008. doi: 10.1073/pnas.1112848109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobsen CH, Størvold GL, Bremseth H, Follestad T, Sand K, Mack M, et al. DHA induces ER stress and growth arrest in human colon cancer cells: associations with cholesterol and calcium homeostasis. Journal of Lipid Research. 2008;49:2089–2100. doi: 10.1194/jlr.M700389-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia G, Sowers JR. Autophagy: a housekeeper in cardiorenal metabolic health and disease. Biochim Biophys Acta. 2015;1852:219–224. doi: 10.1016/j.bbadis.2014.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalmijn S, van Boxtel MP, Ocke M, Verschuren WM, Kromhout D, Launer LJ. Dietary intake of fatty acids and fish in relation to cognitive performance at middle age. Neurology. 2004;62:275–280. doi: 10.1212/01.wnl.0000103860.75218.a5. [DOI] [PubMed] [Google Scholar]
- Kang C, Xu Q, Martin TD, Li MZ, Demaria M, Aron L, Lu T, Yankner BA, Campisi J, Elledge SJ. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science. 2015;349:aaa5612. doi: 10.1126/science.aaa5612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JX, Weylandt KH. Modulation of inflammatory cytokines by omega-3 fatty acids. Subcell Biochem. 2008;49:133–143. doi: 10.1007/978-1-4020-8831-5_5. [DOI] [PubMed] [Google Scholar]
- Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2008;7:1013–1030. doi: 10.1038/nrd2755. [DOI] [PubMed] [Google Scholar]
- Kirkin V, McEwan DG, Novak I, Dikic I. A role for ubiquitin in selective autophagy. Mol Cell. 2009;34:259–269. doi: 10.1016/j.molcel.2009.04.026. [DOI] [PubMed] [Google Scholar]
- Krajewska M, You Z, Rong J, Kress C, Huang X, Yang J, et al. Neuronal Deletion of Caspase 8 Protects against Brain Injury in Mouse Models of Controlled Cortical Impact and Kainic Acid-Induced Excitotoxicity. PLoS ONE. 2011;69:e24341. doi: 10.1371/journal.pone.0024341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larner SF, Hayes RL, Wang KK. Unfolded protein response after neurotrauma. J Neurotrauma. 2006;23:807–829. doi: 10.1089/neu.2006.23.807. [DOI] [PubMed] [Google Scholar]
- Larsson SC, Kumlin M, Ingelman-Sundberg M, Wolk A. Dietary long-chain n-3 fatty acids for the prevention of cancer: a review of potential mechanisms. Am J Clin Nutr. 2004;79:935–945. doi: 10.1093/ajcn/79.6.935. [DOI] [PubMed] [Google Scholar]
- Laurin D, Verreault R, Lindsay J, Dewailly E, Holub BJ. Omega-3 fatty acids and risk of cognitive impairment and dementia. J Alzheimers Dis. 2003;5:315–322. doi: 10.3233/jad-2003-5407. [DOI] [PubMed] [Google Scholar]
- Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol. 2003;23:7448–7459. doi: 10.1128/MCB.23.21.7448-7459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Sato Y, Nixon RA. Lysosomal proteolysis inhibition selectively disrupts axonal transport of degradative organelles and causes an Alzheimer’s-like axonal dystrophy. J Neurosci. 2011;31:7817–7830. doi: 10.1523/JNEUROSCI.6412-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenna S, Han R, Trojanowska M. Endoplasmic reticulum stress and endothelial dysfunction. IUBMB Life. 2014;66:530–537. doi: 10.1002/iub.1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li FJ, He CY. Acidocalcisome is required for autophagy in Trypanosoma brucei. Autophagy. 2014;10:1978–1988. doi: 10.4161/auto.36183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li WW, Li J, Bao JK. Microautophagy: lesser-known self-eating. Cell Mol Life Sci. 2012;69:1125–1136. doi: 10.1007/s00018-011-0865-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling Y, Chen G, Deng Y, et al. Polydatin post-treatment alleviates myocardial ischemia/reperfusion injury by promoting autophagic flux. Clin Sci Lond. 2016 doi: 10.1042/CS20160082. in press. [DOI] [PubMed] [Google Scholar]
- Lipinski MM, Wu J, Faden AI, Sarkar C. Function and Mechanisms of Autophagy in Brain and Spinal Cord Trauma. Antioxid Redox Signal. 2015;23:565–577. doi: 10.1089/ars.2015.6306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CL, Chen S, Dietrich D, Hu BR. Changes in autophagy after traumatic brain injury. Journal of Cerebral Blood Flow and Metabolism: Official Journal of the International Society of Cerebral Blood Flow and Metabolism. 2008;28:674–683. doi: 10.1038/sj.jcbfm.9600587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logsdon AF, Turner RC, Lucke-Wold BP, et al. Altering endoplasmic reticulum stress in a model of blast-induced traumatic brain injury controls cellular fate and ameliorates neuropsychiatric symptoms. Front Cell Neurosci. 2014;8:421. doi: 10.3389/fncel.2014.00421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Zhao LX, Cao DL, Gao YJ. Spinal injection of docosahexaenoic acid attenuates carrageenan-induced inflammatory pain through inhibition of microglia-mediated neuroinflammation in the spinal cord. Neuroscience. 2013;241:22–31. doi: 10.1016/j.neuroscience.2013.03.003. [DOI] [PubMed] [Google Scholar]
- Luo CL, Li BX, Li QQ, Chen XP, Sun YX, Bao HJ, Dai DK, Shen YW, Xu HF, Ni H, et al. Autophagy is involved in traumatic brain injury-induced cell death and contributes to functional outcome deficits in mice. Neuroscience. 2011;184:54–63. doi: 10.1016/j.neuroscience.2011.03.021. [DOI] [PubMed] [Google Scholar]
- Maday S, Wallace KE, Holzbaur EL. Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J Cell Biol. 2012;196:407–417. doi: 10.1083/jcb.201106120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J, Goldberg AL, Schiaffino S, Sandri M. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007;6:458–471. doi: 10.1016/j.cmet.2007.11.001. [DOI] [PubMed] [Google Scholar]
- Manna S, Chakraborty T, Ghosh B, Chatterjee M, Panda A, Srivastava S, Rana A, Chatterjee M. Dietary fish oil associated with increased apoptosis and modulated expression of Bax and Bcl-2 during 7,12-dimethylbenzalphaanthracene-induced mammary carcinogenesis in rats. Prostaglandins Leukot Essent Fatty Acids. 2008;79:5–14. doi: 10.1016/j.plefa.2008.05.005. [DOI] [PubMed] [Google Scholar]
- Mari M, Tooze SA, Reggiori F. The puzzling origin of the autophagosomal membrane. F1000 Biol Rep. 2011;3:25. doi: 10.3410/B3-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters CL, Simms G, Weinman NA. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proceedings of the National Academy of Sciences of the United States of America. 1985;82(12):4245–4249. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayurasakorn K, Williams JJ, Ten VS, Deckelbaum RJ. Docosahexaenoic acid: brain accretion and roles in neuroprotection after brain hypoxia and ischemia. Curr Opin Clin Nutr Metab Care. 2011;14:158–167. doi: 10.1097/MCO.0b013e328342cba5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy MK, Cookson MR. DJ-1 regulation of mitochondrial function and autophagy through oxidative stress. Autophagy. 2011;7:531–532. doi: 10.4161/auto.7.5.14684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh TK, Saatman KE, Raghupathi R. Calcium and the pathogenesis of traumatic CNS injury: cellular and molecular mechanisms. Neuroscientist. 1997;3:169–175. [Google Scholar]
- McNamara RK. Deciphering the role of docosahexaenoic acid in brain maturation and pathology with magnetic resonance imaging. Prostaglandins Leukot Essent Fatty Acids. 2013;88:33–42. doi: 10.1016/j.plefa.2012.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milisav I, Suput D, Ribaric S. Unfolded Protein Response and Macroautophagy in Alzheimer’s, Parkinson’s and Prion Diseases. Molecules. 2015;20:22718–22756. doi: 10.3390/molecules201219865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol. 2010;22:132–139. doi: 10.1016/j.ceb.2009.12.004. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Ohsumi Y, Yoshimori T. Autophagosome formation in mammalian cells. Cell Struct Funct. 2002;27:421–429. doi: 10.1247/csf.27.421. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollereau B, Rzechorzek NM, Roussel BD, Sedru M, Van den Brink DM, Bailly-Maitre B, Palladino F, Medinas DB, Domingos PM, Hunot S, et al. Adaptive preconditioning in neurological diseases - therapeutic insights from proteostatic perturbations. Brain Res. 2016 doi: 10.1016/j.brainres.2016.02.033. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan TM, Koreckij TD, Corey E. Targeted therapy for advanced prostate cancer: inhibition of the PI3K/Akt/mTOR pathway. Curr Cancer Drug Targets. 2009;9:237–249. doi: 10.2174/156800909787580999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto RI, Cuervo AM. Proteostasis and the aging proteome in health and disease. J Gerontol A Biol Sci Med Sci. 2014;69(Suppl 1):S33–38. doi: 10.1093/gerona/glu049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris MC, Evans DA, Bienias JL, Tangney CC, Bennett DA, Wilson RS, Aggarwal N, Schneider J. Consumption of fish and n-3 fatty acids and risk of incident Alzheimer disease. Arch Neurol. 2003;60:940–946. doi: 10.1001/archneur.60.7.940. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Zhu H, Morishima N, et al. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-β. Nature. 2000;403:98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat Rev Mol Cell Biol. 2009;10:458–467. doi: 10.1038/nrm2708. [DOI] [PubMed] [Google Scholar]
- Noel K, Hoffman J, Ellis L, Yurko-Mauro K, Cella C, Sercus B, Nalysnyk L. DHA and cognitive function in the elderly: A systematic review of the literature. Alzheimer’s & Dementia: The Journal of the Alzheimer’s Association. 2006;1:S62. [Google Scholar]
- Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, Imaizumi K. Autophagy Is Activated for Cell Survival after Endoplasmic Reticulum Stress. Mol Cell Biol. 2006;26:9220–9231. doi: 10.1128/MCB.01453-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004;11:381–389. doi: 10.1038/sj.cdd.4401373. [DOI] [PubMed] [Google Scholar]
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- Papp D, Kovacs T, Billes V, Varga M, Tarnoci A, Hackler L, Jr, Puskas LG, Liliom H, Tarnok K, Schlett K, et al. AUTEN-67, an autophagy-enhancing drug candidate with potent antiaging and neuroprotective effects. Autophagy. 2016;12:273–286. doi: 10.1080/15548627.2015.1082023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschen W, Gissel C, Linden T, Althausen S, Doutheil J. Activation of gadd153 expression through transient cerebral ischemia: evidence that ischemia causes endoplasmic reticulum dysfunction. Brain Res Mol Brain Res. 1998;60:115–122. doi: 10.1016/s0169-328x(98)00180-6. [DOI] [PubMed] [Google Scholar]
- Pereira L, Girardi JP, Bakovic M. Forms, crosstalks, and the role of phospholipid biosynthesis in autophagy. Int J Cell Biol. 2012;2012:931956. doi: 10.1155/2012/931956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perri ER, Thomas CJ, Parakh S, Spencer DM, Atkin JD. The Unfolded Protein Response and the Role of Protein Disulfide Isomerase in Neurodegeneration. Front Cell Dev Biol. 2016;3:80. doi: 10.3389/fcell.2015.00080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petraglia AL, Winkler EA, Bailes JE. Stuck at the bench: Potential natural neuroprotective compounds for concussion. Surg Neurol Int. 2011;2:146. doi: 10.4103/2152-7806.85987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petursdottir AL, Farr SA, Morley JE, Banks WA, Skuladottir GV. Effect of dietary n-3 polyunsaturated fatty acids on brain lipid fatty acid composition, learning ability, and memory of senescence-accelerated mouse. J Gerontol A Biol Sci Med Sci. 2008;63:1153–1160. doi: 10.1093/gerona/63.11.1153. [DOI] [PubMed] [Google Scholar]
- Raben N, Puertollano R. TFEB and TFE3: Linking lysosomes to cellular adaptation to stress. Annu Rev Cell Dev Biol. 2016;32:3.1–3.24. doi: 10.1146/annurev-cellbio-111315-125407. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghupathi R. Cell death mechanisms following traumatic brain injury. Brain Pathol. 2004;14:215–22. doi: 10.1111/j.1750-3639.2004.tb00056.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao RV, Bredesen DE. Misfolded proteins, endoplasmic reticulum stress and neurodegeneration. Curr Opin Cell Biol. 2004;16:653–662. doi: 10.1016/j.ceb.2004.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M, Korolchuk VI, Lichtenberg M, Luo S, et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev. 2010;90:1383–1435. doi: 10.1152/physrev.00030.2009. [DOI] [PubMed] [Google Scholar]
- Rubovitch V, Shachar A, Werner H, Pick CG. Does IGF-1 administration after a mild traumatic brain injury in mice activate the adaptive arm of ER stress? Neurochem Int. 2011;58:443–446. doi: 10.1016/j.neuint.2011.01.009. [DOI] [PubMed] [Google Scholar]
- Rzymski T, Milani M, Pike L, Buffa F, Mellor HR, Winchester L, Pires I, Hammond E, Ragoussis I, Harris AL. Regulation of autophagy by ATF4 in response to severe hypoxia. Oncogene. 2010;29:4424–4435. doi: 10.1038/onc.2010.191. [DOI] [PubMed] [Google Scholar]
- Sardiello M. Transcription factor EB: from master coordinator of lysosomal pathways to candidate therapeutic target in degenerative storage diseases. Ann N Y Acad Sci. 2016;1371:3–14. doi: 10.1111/nyas.13131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar C, Zhao Z, Aungst S, Sabirzhanov B, Faden AI, Lipinski MM. Impaired autophagy flux is associated with neuronal cell death after traumatic brain injury. Autophagy. 2014;10:2208–2222. doi: 10.4161/15548627.2014.981787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer EJ, Bongard V, Beiser AS, Lamon-Fava S, Robins SJ, Au R, Tucker KL, Kyle DJ, Wilson PW, Wolf PA. Plasma phosphatidylcholine docosahexaenoic acid content and risk of dementia and Alzheimer disease: the Framingham Heart Study. Arch Neurol. 2006;63:1545–1550. doi: 10.1001/archneur.63.11.1545. [DOI] [PubMed] [Google Scholar]
- Schonthal AH. Endoplasmic reticulum stress: its role in disease and novel prospects for therapy. Scientifica Cairo. 2012;2012:857516. doi: 10.6064/2012/857516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder M. Endoplasmic reticulum stress responses. Cell Mol Life Sci. 2008;65:862–894. doi: 10.1007/s00018-007-7383-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN. Resolution phase of inflammation: novel endogenous anti-inflammatory and proresolving lipid mediators and pathways. Annu Rev Immunol. 2007;25:101–137. doi: 10.1146/annurev.immunol.25.022106.141647. [DOI] [PubMed] [Google Scholar]
- Sharifi MN, Mowers EE, Drake LE, Macleod KF. Measuring autophagy in stressed cells. Methods Mol Biol. 2015;1292:129–150. doi: 10.1007/978-1-4939-2522-3_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin S, Jing K, Jeong S, Kim N, Song KS, Heo JY, Park JH, Seo KS, Han J, Park JI, et al. The omega-3 polyunsaturated fatty acid DHA induces simultaneous apoptosis and autophagy via mitochondrial ROS-mediated Akt-mTOR signaling in prostate cancer cells expressing mutant p53. Biomed Res Int. 2013;2013:568671. doi: 10.1155/2013/568671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KR, Damiano J, Franceschetti S, Carpenter S, Canafoglia L, Morbin M, Rossi G, Pareyson D, Mole SE, Staropoli JF, Sims KB, Lewis J, Lin WL, Dickson DW, Dahl HH, Bahlo M, Berkovic SF. Strikingly different clinicopathological phenotypes determined by progranulin-mutation dosage. Am J Hum Genet. 2012;90:1102–1107. doi: 10.1016/j.ajhg.2012.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spilman P, Podlutskaya N, Hart MJ, et al. Inhibition of mTOR by Rapamycin Abolishes Cognitive Deficits and Reduces Amyloid-β Levels in a Mouse Model of Alzheimer’s Disease. PLoS ONE. 2010;54:e9979. doi: 10.1371/journal.pone.0009979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoica BA, Faden AI. Cell death mechanisms and modulation in traumatic brain injury. Neurotherapeutics. 2010;7:3–12. doi: 10.1016/j.nurt.2009.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stough C, Downey L, Silber B, Lloyd J, Kure C, Wesnes K, Camfield D. The effects of 90-day supplementation with the omega-3 essential fatty acid docosahexaenoic acid DHA on cognitive function and visual acuity in a healthy aging population. Neurobiol Aging. 2012;33:824e1–824.e3. doi: 10.1016/j.neurobiolaging.2011.03.019. [DOI] [PubMed] [Google Scholar]
- Sun DA, Deshpande LS, Sombati S, Baranova A, Wilson MS, Hamm RJ, DeLorenzo RJ. Traumatic brain injury causes a long-lasting calcium Ca2+-plateau of elevated intracellular Ca levels and altered Ca2+ homeostatic mechanisms in hippocampal neurons surviving brain injury. Eur J Neurosci. 2008;27:1659–1672. doi: 10.1111/j.1460-9568.2008.06156.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanik SA, Schultheiss CE, Volpicelli-Daley LA, Brunden KR, Lee VM. Lewy body-like alpha-synuclein aggregates resist degradation and impair macroautophagy. J Biol Chem. 2013;288:15194–15210. doi: 10.1074/jbc.M113.457408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorell WE, Leibrock LG, Agrawal SK. Role of RyRs and IP3 Receptors after Traumatic Injury to Spinal Cord White Matter. J Neurotrauma. 2002;19:335–343. doi: 10.1089/089771502753594909. [DOI] [PubMed] [Google Scholar]
- Thuerauf DJ, Hoover H, Meller J, et al. Sarco/endoplasmic reticulum calcium ATPase-2 expression is regulated by ATF6 during the endoplasmic reticulum stress response: intracellular signaling of calcium stress in a cardiac myocyte model system. J Biol Chem. 2001;276:48309–48317. doi: 10.1074/jbc.M107146200. [DOI] [PubMed] [Google Scholar]
- Todd DJ, Lee AH, Glimcher LH. The endoplasmic reticulum stress response in immunity and autoimmunity. Nat Rev Immunol. 2008;8:663–674. doi: 10.1038/nri2359. [DOI] [PubMed] [Google Scholar]
- Triplett JC, Tramutola A, Swomley A, Kirk J, Grimes K, Lewis K, Orr M, Rodriguez K, Cai J, Klein JB, et al. Age-related changes in the proteostasis network in the brain of the naked mole-rat: Implications promoting healthy longevity. Biochim Biophys Acta. 2015;1852:2213–2224. doi: 10.1016/j.bbadis.2015.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truettner JS, Hu K, Liu CL, Dietrich WD, Hu B. Subcellular stress response and induction of molecular chaperones and folding proteins after transient global ischemia in rats. Brain Res. 2009;1249:9–18. doi: 10.1016/j.brainres.2008.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai YC, Weissman AM. The Unfolded Protein Response, Degradation from Endoplasmic Reticulum and Cancer. Genes Cancer. 2010;1:764–778. doi: 10.1177/1947601910383011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenzuela V, Collyer E, Armentano D, Parsons GB, Court FA, Hetz C. Activation of the unfolded protein response enhances motor recovery after spinal cord injury. Cell Death & Disease. 2012;3:272. doi: 10.1038/cddis.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Kant R, Neefjes J. Small regulators, major consequences - Ca2+ and cholesterol at the endosome-ER interface. J Cell Sci. 2014;127:929–938. doi: 10.1242/jcs.137539. [DOI] [PubMed] [Google Scholar]
- Viscomi MT, D’Amelio M, Cavallucci V, Latini L, Bisicchia E, Nazio F, Fanelli F, Maccarrone M, Moreno S, Cecconi F, Molinari M. Stimulation of autophagy by rapamycin protects neurons from remote degeneration after acute focal brain damage. Autophagy. 2012;8:222–235. doi: 10.4161/auto.8.2.18599. [DOI] [PubMed] [Google Scholar]
- Wang B, Cai Z, Tao K, Zeng W, Lu F, Yang R, Feng D, Gao G, Yang Q. Essential Control of Mitochondrial Morphology and Function by Chaperone-mediated Autophagy through Degradation of PARK7. Autophagy. 2016 doi: 10.1080/15548627.2016.1179401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber-Boyvat M, Zhong W, Yan D, Olkkonen VM. Oxysterol-binding proteins: functions in cell regulation beyond lipid metabolism. Biochem Pharmacol. 2013;86:89–95. doi: 10.1016/j.bcp.2013.02.016. [DOI] [PubMed] [Google Scholar]
- Wei K, Wang P, Miao CY. A double-edged sword with therapeutic potential: an updated role of autophagy in ischemic cerebral injury. CNS Neurosci Ther. 2012;18:879–886. doi: 10.1111/cns.12005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidberg H, Shvets E, Elazar Z. Biogenesis and cargo selectivity of autophagosomes. Annu Rev Biochem. 2011;80:125–156. doi: 10.1146/annurev-biochem-052709-094552. [DOI] [PubMed] [Google Scholar]
- Wen YD, Sheng R, Zhang LS, Han R, Zhang X, Zhang XD, Han F, Fukunaga K, Qin ZH. Neuronal injury in rat model of permanent focal cerebral ischemia is associated with activation of autophagic and lysosomal pathways. Autophagy. 2008;4:762–769. doi: 10.4161/auto.6412. [DOI] [PubMed] [Google Scholar]
- Wong YC, Holzbaur EL. The regulation of autophagosome dynamics by huntingtin and HAP1 is disrupted by expression of mutant huntingtin, leading to defective cargo degradation. J Neurosci. 2014;34:1293–1305. doi: 10.1523/JNEUROSCI.1870-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie H, Xu Q, Jia J, Ao G, Sun Y, Hu L, et al. Hydrogen sulfide protects against myocardial ischemia and reperfusion injury by activating AMP-activated protein kinase to restore autophagic flux. Biochem Biophys Res Commun. 2015;458:632–8. doi: 10.1016/j.bbrc.2015.02.017. [DOI] [PubMed] [Google Scholar]
- Xin XY, Pan J, Wang XQ, Ma JF, Ding JQ, Yang GY, Chen SD. 2-methoxyestradiol attenuates autophagy activation after global ischemia. Can J Neurol Sci. 2011;38:631–638. doi: 10.1017/s031716710001218x. [DOI] [PubMed] [Google Scholar]
- Yamashima T, Oikawa S. The role of lysosomal rupture in neuronal death. Prog Neurobiol. 2009;89:343–358. doi: 10.1016/j.pneurobio.2009.09.003. [DOI] [PubMed] [Google Scholar]
- Yan WJ, Dong HL, Xiong LZ. The protective roles of autophagy in ischemic preconditioning. Acta Pharmacol Sin. 2013;34:636–643. doi: 10.1038/aps.2013.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WJ, Shen XJ, Ma XX, Tan ZG, Song Y, Guo YT, Yuan M. Correlation of human epidermal growth factor receptor protein expression and colorectal cancer. World J Gastroenterol. 2015;21:8687–8696. doi: 10.3748/wjg.v21.i28.8687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YP, Hu LF, Zheng HF, Mao CJ, Hu WD, Xiong KP, Wang F, Liu CF. Application and interpretation of current autophagy inhibitors and activators. Acta Pharmacol Sin. 2013;34:625–635. doi: 10.1038/aps.2013.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yankner BA, Caceres A, Duffy LK. Nerve growth factor potentiates the neurotoxicity of beta amyloid. Proc Natl Acad Sci USA. 1990;87:9020–9023. doi: 10.1073/pnas.87.22.9020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee LD, Agarwal D, Rosol TJ, Lehman A, Tian M, Hatton J, Heestand J, Belury MA, Clinton SK. The inhibition of early stages of HER-2/neu-mediated mammary carcinogenesis by dietary n-3 PUFAs. Mol Nutr Food Res. 2013;57:320–327. doi: 10.1002/mnfr.201200445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin XM, Ding WX, Gao W. Autophagy in the liver. Hepatology. 2008;47:1773–1785. doi: 10.1002/hep.22146. [DOI] [PubMed] [Google Scholar]
- Yoneda T, Imaizumi K, Oono K, Yui D, Gomi F, Katayama T, Tohyama M. Activation of caspase-12, an endoplastic reticulum ER resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J Biol Chem. 2001;276:13935–13940. doi: 10.1074/jbc.M010677200. [DOI] [PubMed] [Google Scholar]
- Young LR, Kurzer MS, Thomas W, Redmon JB, Raatz SK. Effect of dietary fat and omega-3 fatty acids on urinary eicosanoids and sex hormone concentrations in postmenopausal women: a randomized controlled feeding trial. Nutr Cancer. 2011;63:930–939. doi: 10.1080/01635581.2011.589957. [DOI] [PubMed] [Google Scholar]
- Yu WH, Cuervo AM, Kumar A, Peterhoff CM, Schmidt SD, Lee JH. Macroautophagy--a novel Beta-amyloid peptide-generating pathway activated in Alzheimer’s disease. J Cell Biol. 2005;171:87–98. doi: 10.1083/jcb.200505082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z, Sheng H, Liu S, Zhao S, Glembotski CC, Warner DS, Paschen W, Yang W. Activation of the ATF6 branch of the unfolded protein response in neurons improves stroke outcome. J Cereb Blood Flow Metab. 2016 doi: 10.1177/0271678X16650218. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yurko-Mauro K, McCarthy D, Rom D, Nelson EB, Ryan AS, Blackwell A, Salem N, Jr, Stedman M Investigators M. Beneficial effects of docosahexaenoic acid on cognition in age-related cognitive decline. Alzheimers Dement. 2010;6:456–464. doi: 10.1016/j.jalz.2010.01.013. [DOI] [PubMed] [Google Scholar]
- Zhang X, Yuan Y, Jiang L, Zhang J, Gao J, Shen Z, Zheng Y, Deng T, Yan H, Li W, et al. Endoplasmic reticulum stress induced by tunicamycin and thapsigargin protects against transient ischemic brain injury: Involvement of PARK2-dependent mitophagy. Autophagy. 2014;10:1801–1813. doi: 10.4161/auto.32136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XJ, Chen S, Huang KX, Le WD. Why should autophagic flux be assessed? Acta Pharmacol Sin. 2013;34:595–599. doi: 10.1038/aps.2012.184. [DOI] [PMC free article] [PubMed] [Google Scholar]