

Summary
Type 2 Diabetes is a highly heritable disease, but only ~15 % of this heritability can be explained by known genetic variant loci. In fact, body mass index is more predictive of diabetes than any of the common risk alleles identified by genome-wide association studies. This discrepancy may be explained by epigenetic inheritance, whereby changes in gene regulation can be passed along to offspring. Epigenetic changes throughout an organism’s lifetime, based on environmental factors such as chemical exposures, diet, physical activity, and age, can also affect gene expression and susceptibility to diabetes. Recently, novel genome-wide assays of epigenetic marks have resulted in a greater understanding of how genetics, epigenetics, and the environment interact in the development and inheritance of diabetes.
1. Introduction
1.1. Epigenetics and its relevance to T2D
1.1.1. Definition of epigenetics
Each cell in a multicellular organism is equipped with the same set of genomic instructions, and hence diversity in cell type and function must be imparted by regulatory mechanisms that reinterpret genomic information using developmental and environmental cues to dictate specific gene expression profiles. Such regulatory mechanisms include both acute changes in gene activation status, achieved in large part through direct control of gene transcription, and ‘heritable’ changes to the gene activation program that are maintained at least through one mitotic cell division in any of the several hundred cell types in our bodies. The latter events are the subject of study in the field of epigenetics.
Epigenetic modifications include DNA methylation and histone modifications, which collectively determine chromatin compaction and accessibility of transcriptional machinery to the genome. Non-coding RNAs are also considered to be epigenetic modifiers, as they can direct chromatin remodeling in addition to regulating gene expression post-transcriptionally, although strictly speaking, the regulation of these non-coding RNAs itself is again under genetic control through the design of the regulatory elements in the DNA sequences that govern their distribution. Beyond their role in cellular differentiation and maintenance of cellular identity, epigenetic mechanisms also regulate interactions between the genome and the environment to facilitate permanent or semi-permanent changes in gene expression programs in response to environmental cues. Thus, epigenetic modifications must be stable yet flexible to maintain appropriate cell function while permitting adaptation. Disruption of epigenetic regulation can have pathophysiological consequences and likely contributes to the etiology of diabetes as well.
1.1.2. The contribution of the endocrine pancreas to glucose homeostasis
The pancreas is a dual function organ composed of both exocrine and endocrine glands [1]. The exocrine portion of the pancreas supports digestion by secreting pancreatic juices containing digestive enzymes into the small intestine. The endocrine portion of the pancreas, composed of the islets of Langerhans, is a nutrient-sensing organ critical in the maintenance of glucose homeostasis that secretes hormones as a rapid response to changing blood glucose levels. These pancreatic islets are vascularized spheroid clusters of endocrine cells scattered throughout the pancreas, comprising only about 1% of its mass. They are composed of several main endocrine cell types, including α-, β-, δ-, ε- and PP-cells, which work in concert to regulate blood glucose [2]. β-cells secrete insulin in response to elevated blood sugar levels to promote glucose uptake in peripheral tissues such as skeletal muscle and adipose tissue, while suppressing hepatic glucose output. α-cells fulfill the opposing function by secreting glucagon during periods of low blood glucose to mobilize alternative fuel sources, primarily by stimulating hepatic glycogenolysis and gluconeogenesis. δ-cells secrete somatostatin to suppress hormone secretion of neighboring α- and β-cells, thus fine-tuning the glucose response [3]. Finally, PP-cells secrete pancreatic polypeptide and ε-cells produce ghrelin, the latter of which plays a role in regulating food intake and glucose-stimulated somatostatin secretion [4, 5].
β-cell dysfunction is at the core of the pathogenesis of diabetes mellitus, a group of metabolic diseases affecting over 347 million patients world-wide [6]. The two main forms of this disease are type 1 and type 2 diabetes. Type 1 diabetes usually presents early in life and is a consequence of autoimmune destruction of β-cells. Type 2 diabetes is an age-associated disorder in which peripheral insulin resistance leads to β-cell exhaustion and ultimately β-cell death or dysfunction. Type 1 diabetes accounts for only about 5% of all cases while type 2 diabetes patients make up the remaining 95% [2]. Type 2 diabetes is a complex multifactorial disease with both genetic and environmental underpinnings, and epigenetics may couple their interactions. Thus, understanding the mechanism by which epigenetics mediates β-cell function and dysfunction could lead to improved treatment strategies.
1.1.3. Epigenetic implications for T2D
Type 2 diabetes has been called a “nightmare for geneticists” [7]. This difficulty is because type 2 diabetes has only a weak genetic component compared to other common traits, with a “sibling relative risk” (i.e. the chance of being affected by a condition if a sibling is affected relative to the risk in the general population) of only 3 to 4, compared to 17 to 35 for Crohn’s disease, for instance. The concomitant dramatic increase in type 2 diabetes prevalence over the past 100 years with the rise in availability of plentiful food and an increasingly sedentary lifestyle points to a strong epigenetic contribution to the disease.
Nevertheless, over 90 genes associated with diabetes have been identified over the past 30 years. The first wave of progress came from the analysis of a small subset of type 2 diabetes caused by autosomal dominant mutations (monogenic diabetes). Maturity onset diabetes of the young, or MODY, was originally described by Tattersall and Fajans [8], who identified the first families with monogenic diabetes. MODY is characterized by onset before the age of 25, and to date, eleven MODY genes have been identified. The importance of transcription factors in the pathophysiology of diabetes is highlighted by the fact that seven of the MODY genes encode transcription factors [9–13].
The second wave of progress in our understanding of the genetic component of type 2 diabetes has come from genome-wide association studies. Using single nucleotide polymorphism (SNP) genotyping of tens of thousands of cases and controls, more than 80 loci have been found to be associated with type 2 diabetes risk. However, the effect size is very small for all of them, with the largest odds ratio of only 1.38 reported for a SNP near TCF7L2 [14, 15]. Interestingly, the suggested function of many of these genes point to the pancreatic β-cell as the major site of action, consistent with a role of failed β-cell compensation in the etiology of type 2 diabetes [16, 17], although at least one of the genes at a type 2 diabetes risk locus is required for the differentiation of somatostatin-secreting δ-cells with little or no direct effect on β-cells [18]. The novel loci identified by this enormous effort will certainly be useful in formulating new hypotheses regarding the pathways that contribute to β-cell function. On a more global level, however, the genetic analyses thus far have been disappointing in the sense that, combined, all loci account for only ~15% of the heritable risk of type 2 diabetes [14, 15]. Thus, it is likely that epigenetic changes that occur in response to the metabolic demands brought about by life-style changes and the micro-environment of the patient contribute to the heritability of T2D as well.
Diabetes is a disease of the interplay between environment and genes, or more specifically, the increased caloric intake and decreased energy expenditure that has occurred in much of the Western world over the past two generations and the genetic susceptibility to these lifestyle changes. The risk of type 2 diabetes increases with obesity, or with body mass index, as shown in Figure 1. Very strong evidence for the central role of epigenetic factors in the pathogenesis of diabetes is the highly reproducible finding that intrauterine malnutrition leads to diabetes in adulthood. Originally observed by Barker [19], an inverse relationship between birth weight and adult incidence of diabetes and co-morbid conditions has been confirmed in multiple human populations [20–22], as well as in rodent models [23, 24]. The “memory” of the inadequate intrauterine environment later in adult life is widely recognized to have an epigenetic basis. However, critical details of the underlying mechanism are poorly understood.
Figure 1. Obesity contributes to Type 2 Diabetes.
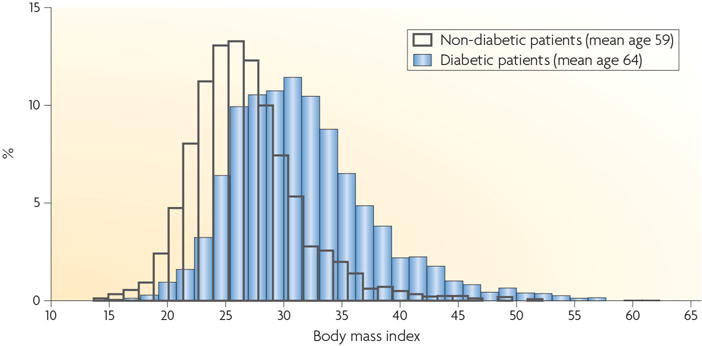
A higher BMI correlates with a higher incidence of diabetes. Reprint from [7].
1.2. The major epigenetic marks
1.2.1. DNA methylation
DNA methylation is the covalent addition of a methyl group to the C5 position of cytosine residues, typically in the context of CpG dinucleotides. DNA methylation was initially thought to function solely in transcriptional repression, but a more complex role has emerged over the past decade: at some loci, DNA methylation results in gene activation [25]. In addition to transcriptional regulation, DNA methylation is critical for maintaining genome integrity through inactivating transposable elements in repeat regions, for X chromosome inactivation in females, and for genomic imprinting [26]. The landscape of DNA methylation throughout the genome largely reflects these divergent roles.
There are approximately 30 million CpG dinucleotides in the human genome, constituting about 1% of total nucleotide content. A fraction of these CpGs, 1–2%, are clustered in highly dense areas known as CpG islands (CGIs), while the majority of CpGs are dispersed sparsely throughout the genome. A CGI is defined as a region greater than 500 bps with more than 50% CpG content [27]. More than half of all CGIs are associated with coding gene promoters, directly regulating gene expression by determining whether or not the basal transcriptional machinery can bind. The vast majority of the genome is highly methylated, which promotes genomic stability by limiting the activity of transposable elements, while certain regulatory elements, including CGIs and enhancers, are usually lowly methylated [28–30].
DNA methylation is catalyzed by DNA methyltransferases (DNMTs), including the de novo methyltransferases, DNMT3a and DNMT3b, and the maintenance methyltransferase DNMT1. DNMT1 has a higher affinity for hemimethylated DNA and thus is able to maintain DNA methylation patterns across subsequent rounds of DNA replication. S-adenosylmethionine (SAM) is the major methyl donor for all methyltransferase reactions in the cell, and its continuous availability is therefore critical to maintaining and propagating epigenetic status. Under certain conditions, DNMT3 enzymes can also compensate for loss of DNMT1 in maintenance methylation [31].
The recent discovery of the Ten-eleven translocation (TET) gene family, which can catalyze the oxidation of 5mC to 5-hydroxymethylcytosine (5hmC), presents a novel path for the removal of 5mC [32, 33]. Two non-mutually exclusive mechanisms have been suggested to mediate demethylation. First, Tet enzymes can further oxidize 5hmC to 5-formlyctosine (5fC) and then 5-carboxylcytosine (5caC), although at much reduced efficacy than oxidation from 5mC to 5hmC [34]. 5fC and 5caC can be further metabolized and eventually replaced by an un-modified cytosine via tyrosine DNA glycosylase (TDG)-mediated base excision repair [35, 36]. Second, and perhaps more plausibly, hydroxymethylation of specific sites leads to their targeted, passive demethylation in replicating cells, because hemimethylated 5hmC nucleotides are not recognized by the maintenance DNA methyltransferases following DNA replication during S-phase (See Figure 2). Either way, hydroxymethylation via the Tet enzymes offers a pathway to decreased DNA methylation even in fully lineage-committed cells.
Figure 2. Models of demethylation mediated by the TET enzymes.
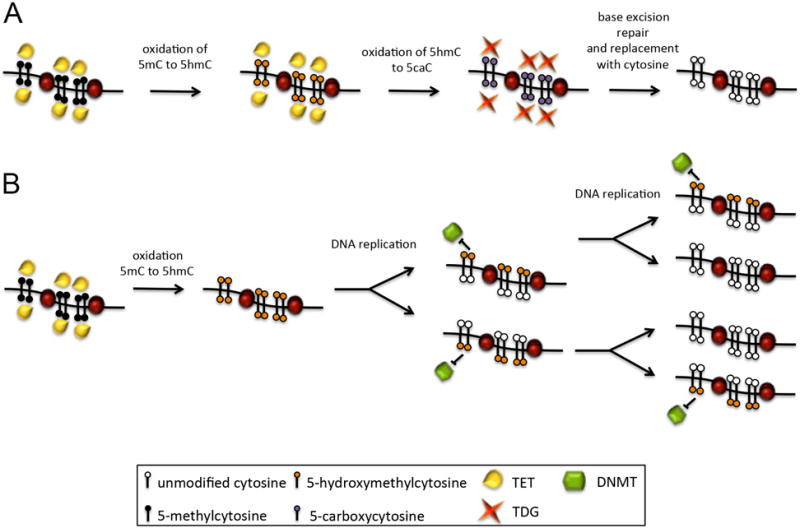
A) TET-assisted active demethylation. The TET enzymes sequentially oxidize methylated cytosines (5mC) into 5-hydroxymethylcytosine, then 5-formylcytosine (5fC) and finally 5-carboxycytosine (5caC). These altered cytosines can then be excised by the thymine DNA glycosylase enzyme (TDG) and replaced with an unmodified cytosine by part of the base-excision repair pathway. B) TET-assisted passive demethylation. The TET enzymes oxidize methylated cytosines into 5hmC, which are a poor substrate for DNMT activity after DNA replication. With each cycle of replication, the methylation status is further diluted.
Although DNA methylation is considered to be a generally stable epigenetic modification, dynamic DNA methylation remodeling is crucial to embryonic development in mammals. Post-fertilization, the preimplantation embryo undergoes genome-wide DNA de-methylation in order to maintain cellular totipotency. After implantation, DNA methylation patterns are re-established to direct cellular differentiation. A similar process of de-methylation followed by re-methylation is observed in the development of primordial germ cells [27]. In addition, even in adults, the rapid transition from intestinal stem cell to differentiated enterocyte is accompanied by gain and loss of DNA methylation at specific sites [29]. Even within the established β-cell lineage, DNA methylation is dynamic with aging [28], as will be discussed in detail below. This point is particularly relevant since changes in methylation often accompany alterations in gene function and suggests a mechanism by which cells can be locked into an “aged” phenotype.
1.2.2. Histone modifications
Genomic material in the cell is packaged into chromatin, which is a DNA-protein complex consisting of linear DNA wrapped around a histone core. A nucleosome is a unit of chromatin defined by 147 base pairs wound around each histone core, which includes four dimers of histones H2A, H2B, H3, and H4. The N-terminal tails of these histones can be covalently modified to alter chromatin compaction and recruit transcriptional regulators to modify gene expression in a multitude of ways [37, 38]. Histone acetylation and methylation are the most well studied modifications, although several others have been identified, such as ubiquitnation, sumoylation, and phosphorylation. In general, histone acetylation is associated with increased gene expression. Histone acetylation is accomplished by histone acetyltransferases (HATs), and acetyl groups are removed by histone deacetylases (HDACs) [39]. The effect of methylation is context-dependent, varying by which residue is methylated and the number of methyl groups present [40].
2. Epigenetic programming of β-cell development and function
2.1. Cell fate determination
Multicellular organisms that reproduce sexually start as a single, undifferentiated cell after fertilization. To become multicellular organisms with many distinct cells types, cells must divide while undergoing a series of choices that lock cells into a specific fate, an idea first proposed by Waddington in 1957 [41]. Cell-fate specification occurs through the interplay of extrinsic and intrinsic factors: signaling molecules, transcription factors, and epigenetic modifications of chromatin. When these factors work together correctly to activate some genes and silence others, cell fate is stable. In disease states, however, cells can dedifferentiate, becoming more progenitor-like, or even trans-differentiate into a related cell fate. An example of trans-differentiation has been observed in a transition from the β- to the α-cell fate in T2D [42].
2.1.1. Pancreas specification
The pancreas is derived from the foregut endoderm, from which the liver, lungs, esophagus, and stomach also arise. Permissive and instructive signals from the surrounding mesenchyme control the evagination of the pancreatic buds, a process that occurs in midgestation. During pancreatogenesis, a well-characterized transcription factor cascade (reviewed in [43]) controls the development from this undifferentiated pancreatic plexus into ductal, acinar, and endocrine cells types (reviewed in [44]). As predicted by Waddington’s model, both gene activation and silencing are essential for the differentiation process.
Embryonic stem cells from mouse and human can be differentiated into foregut endoderm, multipotent pancreatic progenitor cells (MPCs), endocrine precursors, and immature endocrine cells. The pioneer transcription factors FoxA1 and FoxA2 bind and open enhancer chromatin by displacing a linker histone and even causing eviction of entire nucleosomes [45]. A study of in vitro-differentiated cells revealed that at the foregut endoderm stage, FoxA1/2 bind enhancers throughout the genome. As cells become more like pancreatic progenitors, the pancreas-specific factor Pdx1 is expressed and binds pancreas-specific enhancers, and FoxA1/2 remain bound to pancreas-specific enhancers while its presence at liver foregut endoderm enhancers is lost [46]. Simultaneously, the chromatin at these sites in the pancreas goes from a poised state having both active and inactive enhancer histone marks to having only active marks. This work is supported by results from FACS-sorted E8.25 endoderm, a time before pancreas cells have differentiated, demonstrating a poised state at pancreas but not hepatic genes [47]. Together, these findings confirm earlier discoveries of the FoxA factors and Pdx1 as critical determinants of early pancreas development [48–50].
In addition to the transition from poised to active histone marks that accompanies pancreatic differentiation, the importance of both gene activation and silencing in pancreas development is highlighted by two studies employing mouse genetics. Pancreas-specific deletion of Brg1, a component of the nucleosomal remodeling SWI/SNF complex, an activator of gene activity and a Pdx1 co-regulator, results in a hypoplastic pancreas [51]. When the DNA methyltransferase Dnmt1a is deleted within MPCs, mice display pancreatic agenesis due to aberrant p53 gene activation [52].
Finally, non-coding RNAs are essential in pancreas development. When the double-stranded RNA-processing enzyme Dicer is deleted in MPCs, a hypoplastic, undifferentiated pancreas results [53]. However, despite the expression of 1,128 long non-coding RNAs in human islets [54], of which 148 were β-cell specific [55], and a series of articles investigating the role of microRNAs in pancreas or endocrine cell differentiation and function, no role in pancreas specification has been found for a non-coding RNA molecule acting alone, although both lncRNAs and miRNAs contribute individually to β-cell function [56–58], and deletion of Dicer specifically within β-cells leads to diabetes in mice [55].
2.1.2. Endocrine cell differentiation
While epigenetic changes in differentiating pancreatic cells at specified stages have not been examined directly, studies using inhibitors of epigenetic enzymes or employing mice deficient for different classes of epigenetic modifiers indicate that these chromatin-modifying factors act specifically and sequentially to drive and lock cells into a differentiated state. Histone deactylases (HDACs) and the polycomb repressive complexes (PRCs) all play important roles in endocrine cell differentiation. These different classes of molecules are all epigenetic modifiers normally associated with gene silencing.
Use of HDAC inhibitors on pancreas explants or deletion of HDACs within the pancreatic domain results in increased endocrine progenitors, which eventually leads to an increased number of β- and δ-cells. Conversely, pancreatic overexpression of HDAC yields reduced β- and δ-cell mass. The PRCs act in succession to coordinate gene silencing. PRC2 suppresses transcription by depositing histone 3 lysine 27 trimethylation (H3K27me3), a repressive histone modification. PRC1 subsequently recognizes H3K27 trimethylation and facilitates chromatin compaction by histone 2A (H2A) ubiquitylation, which in turn blocks recruitment of the histone 3 lysine 4 (H3K4) methyltransferase MLL1, an activator of gene expression [59]. As with HDACs, deletion of the PRC2 component Ezh2 throughout the embryonic pancreas results in more endocrine progenitors and subsequent increased β-cell mass, while deletion of the PRC1 component Ring1b in multipotent progenitors but not in adult β-cells result in re-expression of genes normally repressed in β-cells such as the α-cell genes aristaless-related homeobox (Arx), Inhibitor of DNA binding 3 (Id3), and neuropeptide Y (Npy).
2.2. Epigenetics in maintaining β-cell identity and function
2.2.1. β-cell identity
Following cellular differentiation, epigenetic signatures also play a critical role in maintaining β-cell identity and function. Genome-wide studies have begun to elucidate the epigenetic landscape of islet endocrine cell subtypes and the mechanisms by which they are maintained. For example, thousands of genes that are bivalently marked in human α-cells by both activating H3K4me3 and repressive H3K27me3 histone modifications are solely monovalently marked in human β-cells, suggesting greater cellular plasticity in α-cells [60]. Indeed, treatment of human islets with a histone methyltransferase inhibitor resulted in the appearance of bihormonal cells expressing both insulin and glucagon, suggesting partial conversion between the endocrine subtypes.
A common theme in epigenetic regulation of β-cell identity is maintaining appropriate expression of key β-cell signature genes, particularly transcription factors, while disallowing non-β-cell genes [61, 62]. Additionally, epigenetic mechanisms do not work in isolation, but rather are part of complex networks of diverse epigenetic regulators guided by cell-type specific transcription factors. For instance, maintenance of β-cell identity requires ongoing epigenetic repression of Arx, a transcription factor critical to α-cell fate. Arx is repressed in β-cells by DNA methylation of the promoter and subsequent binding of the methyl-binding protein MeCP2, which recruits the histone 3 arginine 2 methyltransferase PRMT6 [63]. Additionally, in β-cells, the transcription factor Nkx2.2 occupies the Arx promoter in complex with the DNA methyltransferase Dnmt3a, the repressor cofactor Grg3, and the histone deacetyalse HDAC1 to further repress Arx expression [64]. Interestingly, Dnmt3a interacts specifically with Nkx2.2 but not with other β-cell transcription factors such as Pax4 and Isl1 [64]. Disruption of this epigenetic signature by deletion of Dnmt1 or Dnmt3 results in expression of α-cell genes and α- to β-cell conversion [63, 64].
Several other lineage-specific transcription factors and ubiquitously expressed epigenetic modifiers impact maintenance of the β-cell identity. For instance, one of the MODY transcription factors, HNF1A, directs hyperacetylation of β-cell identity genes, increasing their gene expression [65, 66]. The transcription factor PAX4, which depending on the type of mutation or gene variant has been associated with T1D, T2DI or MODY, functions in controlling proliferation in the adult pancreas and in the response to chronic hyperglycemia. Strikingly, exogenous Pax4 over-expression in α-cells promotes conversion towards the β-cell phenotype (reviewed in [67]). Finally, synthetic, broad-acting inhibitors of histone deacetylases, DNA methyltransferases, and histone lysine methyl transferases have varying effects on α– and β-cell identity when administered to endocrine pancreas cell lines [68]. However, these latter studies suffer from the lack of specificity of the small molecules employed and should therefore be interpreted with caution.
2.2.2. Glucose responsiveness
In addition to restricting cell fate to the β-cell lineage, epigenetics is also implicated in dynamic regulation of β-cell function. The β-cell is the sole insulin-producing cell in the body, and thus organismal glucose homeostasis is highly dependent upon the ability of the β-cell to produce and secrete insulin in response to adequate stimuli. The insulin gene is epigenetically silenced in non-β-cell types by DNA methylation, and in β-cells is dynamically regulated by epigenetic mechanisms in response to changes in glucose levels. Thus, at low glucose levels the MODY transcription factor Pdx1 associates with histone deacetylases to decrease insulin transcription, while at high glucose levels Pdx1 facilitates recruitment of the histone acetyltransferase p300, which catalyzes histone 4 acetylation, and histone methyltransferase Set7/9, which catalyzes H3K4 dimethylation, to create an active chromatin structure [69].
The histone methyltransferase Set7/9 also promotes expression of other important glucose responsiveness genes such as MafA, Slc2a2, and Ins2 [70, 71]. Depletion of Set7/9 in primary mouse islets resulted in decreased H3K4 dimethylation of these targets, decreased gene expression, and impaired glucose-stimulated insulin secretion [70]. Interestingly, Set7/9 and Pdx1 also appear to directly regulate one another. Setd7, the gene encoding Set7/9, has an islet-specific enhancer occupied by Pdx1, and Set7/9 was recently shown to directly methylate the N-terminal lysine residues of Pdx1, enhancing transcriptional activity and β-cell glucose responsiveness [70, 72].
Type 2 diabetes is an age-related disorder, and therefore, it has been postulated previously that β-cell function deteriorates with age regardless of overall metabolic health. However, recent evidence in mouse models and in human samples suggests that β-cell function actually improves as β-cells mature, and that epigenetics may facilitate functional maturity. An integrative analysis of DNA methylation, histone modifications, gene expression and β-cell function in prepubescent and post-fertile aged murine β-cells demonstrated both a general genome-wide epigenetic drift as well as specific, targeted changes in distal regulatory elements [28]. The targeted changes in methylation status were associated both with cell cycle and β-cell function genes. Indeed, this study identified age-related reductions in Cdkn2a DNA methylation at distal regulatory elements that correlate with increased Cdkn2a expression in aged mouse β-cells. Conversely, maturation-related loss of methylation was associated with activation of metabolic regulators, and increased expression of these same key β-cell transcription factors correlated with improved β-cell function. Additionally, the methylation status of many genes important for islet function or associated with T2D changes with age in human islets [73]. In a recent study of human islets from juvenile and adult organ donors, enhanced insulin secretion was also seen in the adult samples [74]. These results challenge the current paradigm of β-cell failure as purely a phenomenon of aging.
Further evidence for the contribution of epigenetic mechanisms comes from the discovery that the insulin promoter itself is hypermethylated in T2D patients [75, 76]. A very exciting, practical use from the site-specific methylation status of the insulin promoter is its use as a screening tool of human blood samples to detect recent β-cell death [77–82]. Dying cells release genomic DNA into the bloodstream. Since the insulin promoter overall is only lowly methylated in endocrine cells but very highly methylated in all other cell types in the body, DNA in the blood stream coming from β-cells will be unmethylated at the insulin promoter, but DNA coming from other cells will be highly methylated (see Figure 3). Thus, a relatively high proportion of unmethylated insulin promoter DNA in the blood stream is a biomarker of recent β-cell death. This novel technology promises to be useful for assessing β-cell survival after islet transplantation and in the detection of very early onset type 1 diabetes.
Figure 3. Using the methylation status of the insulin promoter in blood to assess β-cell death.
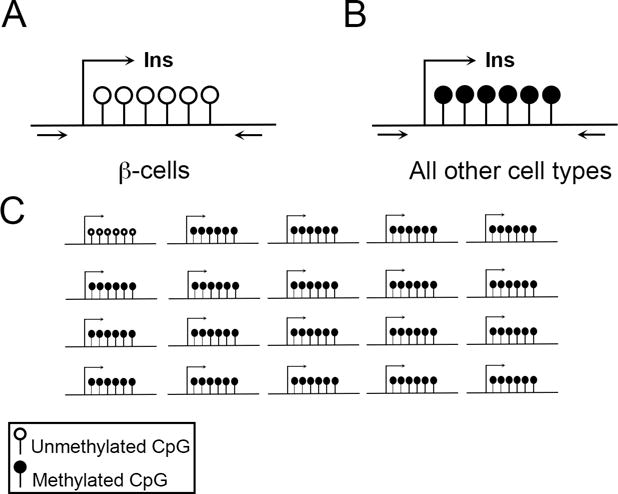
A–B) The insulin promoter is differentially methylated in β-cell compared to other cell types. The methylation status of the insulin promoter can be detected using bisulfite sequencing. Arrows represent primers used for DNA amplification. (A) depicts the unmethylated insulin promoter in β-cells while (B) depicts the methylated and silenced insulin promoter in all other cells types. C) Massive β-cell death can be detected with bisulfite sequencing with a simple blood test, because in those circumstances, up to 5% of cell-free DNA in the blood comes from β-cells.
2.2.3. Proliferation
Pancreatic β-cell mass is established prior to adulthood but can reversibly expand in response to instances of increased metabolic demand such as in pregnancy and obesity, at least in rodents. Failure to compensate can result in gestational diabetes or type 2 diabetes, respectively [83, 84]. Epigenetics mediates the β-cell proliferative response to these metabolic stressors and also plays a role in the age-related decline in the ability of β-cells to proliferate.
In mouse β-cells, the cell cycle inhibitor p16, encoded by the Cdkn2a gene, is a well-established mediator of the age-related decline in proliferative capacity. Decreasing p16 levels through germline gene inactivation can rescue the proliferative capacity of mouse adult β-cells [85]. Increased expression of Cdkn2a with age appears to be mediated by epigenetic mechanisms involving PRC1 and PRC2, which also play a role in endocrine specification, as mentioned above. Expression of Ezh2, an H3K27 histone methyltransferase associated with PRC2, decreases with age in murine β-cells, leading to de-repression of the Cdkn2a locus [86]. The age-related decrease in expression of Bmi-1, which encodes a polycomb group protein that is critical for stability of PRC1, ultimately leads to transcriptional activation of Cdkn2a by permitting MLL1 recruitment [70].
Innovative mouse genetic models were also employed to demonstrate the interplay of multiple epigenetic modifiers in controlling Cdkn2a/p16 expression and how they can be exploited to maintain the high proliferative capacity of young β-cells. Using transgenic mouse models, the Bushan group showed that overexpressing Ezh2, the key histone-methyltransferase enzyme in the aforementioned PRC2 complex, limits Cdkn2a/p16 expression in young adult β-cells, thus allowing for increased replication [87]. However, activation of Ezh2 alone was not effective in β-cells of older mice, in which the activating histone marks have been firmly established. Thus, in older mice, simultaneous suppression of the entire enzymatic complex responsible for activating histone marks and activation of a complex responsible for gene silencing was required to reduce Cdkn2a/p16 expression and enable β-cell replication. Recently, a novel targeted approach was developed that allows for the gene-specific targeting of epigenetic modifiers, in this case DNA methyltransferases, to the promoter of the human CDKN2A gene, reducing its expression and stimulating replication of human fibroblasts [88]. If this targeted technology could be transferred to human β-cells in vivo, therapeutic expansion of β-cell mass might become a reality.
3. Role of epigenetics in the development of type 2 diabetes
Provided the critical role of epigenetics in the development and maintenance of β-cell identity and function, it follows that disruption of epigenetic programming may have severe pathophysiological consequences. Epigenetic alterations leading to β-cell dysfunction can be introduced by lifestyle risk factors such as obesity and limited physical activity. However, epigenetic alterations predisposing individuals to T2D may also be introduced even before birth through direct in utero exposure or transgenerational inheritance. Epigenetics provides a potential means by which the effects of transient exposure can have a lasting effect, even transgenerationally. While the precise molecular mechanisms are incompletely understood, several lines of evidence provide support for the critical contribution of epigenetic regulation to T2D susceptibility.
3.1. Alterations in DNA Methylation
The methylation status of the promoter and corresponding gene expression of many candidate factors with a known role in β-cell function have been examined. Hypermethylation of the glucagon-like peptide 1 receptor (GLP1R) promoter and the associated reduced gene expression correlates with an increased body mass index (BMI) and hemoglobin A1c (HbA1c) [89]. The transcriptional coactivator peroxisome proliferator activated receptor gamma coactivator-1 alpha (PGC-1α, encoded by PPARGC1A) is an important regulator of mitochondrial function and metabolism [90]. Compared to control donors, in T2D islets, the PPARGC1A promoter is hypermethylated and its expression reduced by 90%. While neither of these studies conclusively demonstrates that an aberrant methylation status of any single gene leads to the onset of diabetes, together they suggest that abnormal epigenetic regulation can contribute to the disease.
Recent advances in genome-wide techniques have permitted more systematic screens to identify epigenetic changes in T2D patients. Array-based techniques are convenient for assessing changes in DNA methylation with the caveat that they only cover about 0.5% of all CpG sites in the genome. Volkmar and colleagues compared five T2D methylomes to eleven control methylomes and found 273 differentially methylated CpGs [91]. The T2D methylomes were more similar to each other than to any of the control methylomes, but the differential methylation was specific to certain genes and not a genome-wide phenomenon. A second group using the same approach with 87 control donors showed that, as in mice [28], DNA methylation in islets changes with age, and these changes often occurred in genes known to have roles in β-cell function or proliferation [73].
3.2. Epigenetic and Genetic Interactions
Changes in epigenetic regulation can occur for a variety of reasons. In some cases, they occur because of changes in gene regulatory sequences, such as those that alter transcription factor binding of islet enhancers [92, 93] or which add CpGs that can contribute to gene silencing [94]. In both of these cases, SNPs associated with alterations in gene expression were also T2D variants.
3.3. Environmental factors during an organism’s lifetime
Environmental exposures are also a large source of epigenetic variation. Diet, physical activity, and BMI all play a role in epigenetic regulation of gene expression. As previously reviewed, certain metabolites are required for the activity of genetic modifiers [95], and diet has been postulated to limit the availability of these factors [96]. Indeed, alterations of diet in rodent studies have led to epigenetic changes in diabetes-associated genes in adipose tissue [97, 98] and islets [99], and a post-weaning methyl-deficient diet resulted in decreased insulin secretion [100]. Exercise also leads to modifications in the methylation status of gene regulatory regions in muscle and adipose tissue [101, 102]; thus, epigenetic modifications may mediate and lock in the insulin resistance that can result from reduced physical activity [103]. While epigenetic regulation clearly differs in obese and lean humans [104], whether the change is causative or derivative of obesity is unclear.
Aging is also a factor in epigenetic alterations. In addition to the aging mouse and human studies demonstrating that alterations in islet regulatory region methylation correspond to changing gene expression mentioned above [28, 73], in humans, increased methylation and decreased gene expression of several genes important for mitochondrial function has been observed with age [105–107].
3.4. Parental transmission of T2D risk
Offspring can be subject to aberrant epigenetic alterations either through in utero exposure or through inheritance of these alterations from parental gametes. The transmission of epigenetic marks from parental gametes is particularly interesting since the epigenome undergoes widespread erasure during gametogenesis and again post-fertilization in the preimplantation embryo to maintain cellular pluripotency. Thus, certain environmental cues must permit epigenetic changes that escape these erasures. However, exposure and true epigenetic alteration inheritance must be carefully distinguished from one another. For instance, if a F1 fetus is exposed in utero, the gametes of that fetus (F2) are also exposed. In this example, transmission is only transgenerational if passed on to the F3 generation (Figure 4). Indeed, mounting evidence suggests that both multigenerational and transgenerational epigenetic inheritance can contribute to β-cell dysfunction.
Figure 4. Epigenetic inheritance can be multigenerational or transgenerational.
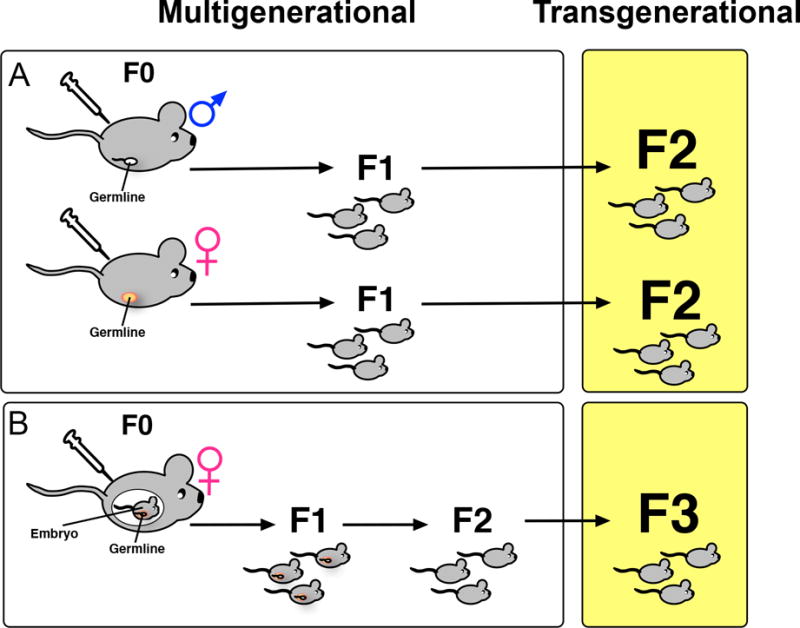
A) Multigenerational inheritance refers to a change in a trait or phenotype in the F1 offspring of males or non-pregnant females (F0) exposed to a stimulus that impacts the epigenome without changing the DNA sequence. B) In the case of a pregnant female exposed to an environmental toxin, for instance the F0 parent, the F1 fetus, and the F2 germline within the fetus are all exposed. Therefore, in this case, only if the F3 generation also shows an epigenetically altered phenotype is it considered transgenerational inheritance.
3.4.1. Intrauterine environment
Early observations in human studies suggested that maternal diet plays a role in predisposition to diabetes. The Dutch hunger famine that occurred at the end of World War II is perhaps the most famous example, where offspring of mothers that experienced short-term malnutrition during pregnancy were born with low birth weight and were more likely to develop metabolic syndrome later in life [108]. Another study similarly found that low birth-weight individuals were most likely to develop T2D or insulin resistance, while individuals with the highest birth weight were least likely [20].
Intrauterine growth restriction (IUGR) animal models performed by inducing poor maternal nutrition, or uteroplacental insufficiency induced by maternal uterine artery ligation have prompted researchers to investigate the mechanisms by which maternal nutritional status during pregnancy can affect the long-term metabolic health of their offspring [24, 109, 110]. A common result among these models is that altered expression of key β-cell transcription factors accompanied by epigenetic changes. For example, a rat model of maternal isocaloric protein-restriction resulted in decreased HNF4a expression, promoter hypermethylation, and histone marks indicative of an inactive promoter, a finding that was exacerbated with age [111]. Many examples of epigenetic silencing of important β-cell genes, including transcription factors, due to maternal uterine artery ligation also exist [111–114].
Exposure to environmental factors such as endocrine-disrupting chemicals may also result in aberrant epigenetic alterations that are passed down transgenerationally, resulting in impaired β-cell function. While it was already known that in rodent models, fetal exposure to bisphenol A (BPA) results in disrupted glucose homeostasis [115], a recent study in rats demonstrated that these effects correlated with changes in histone acetylation and methylation at the Pdx1 locus [116]. This impaired glucose tolerance and β-cell function is also observed in the F2 generation and is associated with increased DNA methylation and decreased expression of Igf2, a well-established regulator of β-cell development and function [117]. Interestingly, these epigenetic changes in Igf2 were also observed in sperm of F1 offspring, and in a model of maternal calorie restriction, more global methylation changes in metabolic genes were observed in F1 sperm [118], suggesting that environmentally induced epigenetic changes could be inherited paternally as well as maternally.
3.4.2. Paternal transmission
In contrast to in utero exposure, paternal contribution to offspring predisposition to β-cell dysfunction and T2D can be isolated to the genetic and epigenetic information carried in sperm. A study in Sprague-Dawley (SD) rats showed that paternal chronic high fat diet (HFD) results in impaired insulin secretion and glucose tolerance in female offspring that worsened with age. While β-cell mass was not significantly affected, approximately 2,500 genes were differentially expressed in adult female HFD offspring compared to controls, many of which are implicated in insulin granule exocytosis and cell survival [119]. Another study assessing the transgenerational impact of paternal HFD in SD rats similarly found impaired glucose metabolism in female F1 and F2 offspring, although it was due to peripheral glucose metabolism rather than β-cell dysfunction [120]. Finally, a recent study demonstrated that DNA methylation of sperm from obese men was significantly different from that of lean men [121], and this observation combined with the observations of Igf2 in F1 sperm mentioned above [117] lends credence to the idea that a propensity to become obese could be transmitted epigenetically.
4. Conclusions
Epigenetic regulation clearly contributes to T2D risk and development, at least partially through changes in the β-cell. Therefore, a crucial goal for many researchers has been to alter epigenetic marks to improve β-cell health and function. Once approach has been to use drugs, such as the treatment of isolated islets with HDAC inhibitors to prevent apoptosis in the presence of cytokines [122]. Future tactics will likely employ mutated TALE and CRISPR-Cas proteins that are tethered to HDACs, HATs, DNA methyltransferases, and other epigenetic-altering molecules. Whatever the method used, to treat diabetes, these molecules will need to be delivered either to β-cells ex vivo before transplantation or directly to β-cells in vivo to prevent potential harmful effects on other cell types.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chandra R, Liddle RA. Modulation of pancreatic exocrine and endocrine secretion. Current opinion in gastroenterology. 2013;29(5):517–522. doi: 10.1097/MOG.0b013e3283639326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vetere A, Choudhary A, Burns SM, Wagner BK. Targeting the pancreatic beta-cell to treat diabetes. Nature Reviews Drug Discovery. 2014;13(4):278–289. doi: 10.1038/nrd4231. [DOI] [PubMed] [Google Scholar]
- 3.Hauge-Evans AC, King AJ, Carmignac D, Richardson CC, Robinson ICAF, Low MJ, et al. Somatostatin Secreted by Islet delta-Cells Fulfills Multiple Roles as a Paracrine Regulator of Islet Function. Diabetes. 2009;58(2):403–411. doi: 10.2337/db08-0792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Batterham RL, Le Roux CW, Cohen MA, Park AJ, Ellis SM, Patterson M, et al. Pancreatic polypeptide reduces appetite and food intake in humans. Journal of Clinical Endocrinology & Metabolism. 2003;88(8):3989–3992. doi: 10.1210/jc.2003-030630. [DOI] [PubMed] [Google Scholar]
- 5.DiGruccio MR, Mawla AM, Donaldson CJ, Noguchi GM, Vaughan J, Cowing-Zitron C, et al. Comprehensive alpha, beta and delta cell transcriptomes reveal that ghrelin selectively activates delta cells and promotes somatostatin release from pancreatic islets. Mol Metab. 2016;5(7):449–58. doi: 10.1016/j.molmet.2016.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang P, Fiaschi-Taesch NM, Vasavada RC, Scott DK, Garcia-Ocana A, Stewart AF. Diabetes mellitus-advances and challenges in human beta-cell proliferation. Nature Reviews Endocrinology. 2015;11(4):201–212. doi: 10.1038/nrendo.2015.9. [DOI] [PubMed] [Google Scholar]
- 7.Frayling TM. Genome-wide association studies provide new insights into type 2 diabetes aetiology. Nat Rev Genet. 2007;8(9):657–62. doi: 10.1038/nrg2178. [DOI] [PubMed] [Google Scholar]
- 8.Tattersall RB, Fajans SS. A difference between the inheritance of classical juvenile-onset and maturity-onset type diabetes of young people. Diabetes. 1975;24(1):44–53. doi: 10.2337/diab.24.1.44. [DOI] [PubMed] [Google Scholar]
- 9.Yamagata K, Furuta H, Oda N, Kaisaki PJ, Menzel S, Cox NJ, et al. Mutations in the hepatocyte nuclear factor-4alpha gene in maturity-onset diabetes of the young (MODY1) Nature. 1996;384(6608):458–60. doi: 10.1038/384458a0. [DOI] [PubMed] [Google Scholar]
- 10.Froguel P, Vaxillaire M, Sun F, Velho G, Zouali H, Butel MO, et al. Close linkage of glucokinase locus on chromosome 7p to early-onset non- insulin-dependent diabetes mellitus [published erratum appears in Nature 1992 Jun 18;357(6379):607] Nature. 1992;356(6365):162–4. doi: 10.1038/356162a0. [DOI] [PubMed] [Google Scholar]
- 11.Malecki MT, Jhala US, Antonellis A, Fields L, Doria A, Orban T, et al. Mutations in NEUROD1 are associated with the development of type 2 diabetes mellitus. Nat Genet. 1999;23(3):323–8. doi: 10.1038/15500. [DOI] [PubMed] [Google Scholar]
- 12.Neve B, Fernandez-Zapico ME, Ashkenazi-Katalan V, Dina C, Hamid YH, Joly E, et al. Role of transcription factor KLF11 and its diabetes-associated gene variants in pancreatic beta cell function. Proc Natl Acad Sci U S A. 2005;102(13):4807–12. doi: 10.1073/pnas.0409177102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Plengvidhya N, Kooptiwut S, Songtawee N, Doi A, Furuta H, Nishi M, et al. PAX4 mutations in Thais with maturity onset diabetes of the young. J Clin Endocrinol Metab. 2007;92(7):2821–6. doi: 10.1210/jc.2006-1927. [DOI] [PubMed] [Google Scholar]
- 14.Mahajan A, Go MJ, Zhang W, Below JE, Gaulton KJ, Ferreira T, et al. Genome-wide trans-ancestry meta-analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nature genetics. 2014;46(3):234–+. doi: 10.1038/ng.2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morris AP, Voight BF, Teslovich TM, Ferreira T, Segre AV, Steinthorsdottir V, et al. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet. 2012;44(9):981–90. doi: 10.1038/ng.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang-Chen KJ, Mullur R, Bernal-Mizrachi E. beta-cell failure as a complication of diabetes. Rev Endocr Metab Disord. 2008 doi: 10.1007/s11154-008-9101-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leahy JL. Mary, Mary, quite contrary, how do your beta-cells fail? Diabetes. 2008;57(10):2563–4. doi: 10.2337/db08-0869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang J, McKenna LB, Bogue CW, Kaestner KH. The diabetes gene Hhex maintains delta-cell differentiation and islet function. Genes Dev. 2014;28(8):829–34. doi: 10.1101/gad.235499.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barker DJ, Osmond C. Infant mortality, childhood nutrition, and ischaemic heart disease in England and Wales. Lancet. 1986;1(8489):1077–81. doi: 10.1016/s0140-6736(86)91340-1. [DOI] [PubMed] [Google Scholar]
- 20.Hales CN, Barker DJ, Clark PM, Cox LJ, Fall C, Osmond C, et al. Fetal and infant growth and impaired glucose tolerance at age 64. BMJ. 1991;303(6809):1019–22. doi: 10.1136/bmj.303.6809.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rich-Edwards JW, Colditz GA, Stampfer MJ, Willett WC, Gillman MW, Hennekens CH, et al. Birthweight and the risk for type 2 diabetes mellitus in adult women. Ann Intern Med. 1999;130(4 Pt 1):278–84. doi: 10.7326/0003-4819-130-4_part_1-199902160-00005. [DOI] [PubMed] [Google Scholar]
- 22.Valdez R, Athens MA, Thompson GH, Bradshaw BS, Stern MP. Birthweight and adult health outcomes in a biethnic population in the USA. Diabetologia. 1994;37(6):624–31. doi: 10.1007/BF00403383. [DOI] [PubMed] [Google Scholar]
- 23.Jimenez-Chillaron JC, Hernandez-Valencia M, Lightner A, Faucette RR, Reamer C, Przybyla R, et al. Reductions in caloric intake and early postnatal growth prevent glucose intolerance and obesity associated with low birthweight. Diabetologia. 2006;49(8):1974–84. doi: 10.1007/s00125-006-0311-7. [DOI] [PubMed] [Google Scholar]
- 24.Simmons RA, Templeton LJ, Gertz SJ. Intrauterine growth retardation leads to the development of type 2 diabetes in the rat. Diabetes. 2001;50(10):2279–2286. doi: 10.2337/diabetes.50.10.2279. [DOI] [PubMed] [Google Scholar]
- 25.Hark AT, Schoenherr CJ, Katz DJ, Ingram RS, Levorse JM, Tilghman SM. CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature. 2000;405(6785):486–9. doi: 10.1038/35013106. [DOI] [PubMed] [Google Scholar]
- 26.Reik W, Walter J. Genomic imprinting: parental influence on the genome. Nat Rev Genet. 2001;2(1):21–32. doi: 10.1038/35047554. [DOI] [PubMed] [Google Scholar]
- 27.Zampieri M, Ciccarone F, Calabrese R, Franceschi C, Burkle A, Caiafa P. Reconfiguration of DNA methylation in aging. Mech Ageing Dev. 2015;151:60–70. doi: 10.1016/j.mad.2015.02.002. [DOI] [PubMed] [Google Scholar]
- 28.Avrahami D, Li C, Zhang J, Schug J, Avrahami R, Rao S, et al. Aging-Dependent Demethylation of Regulatory Elements Correlates with Chromatin State and Improved beta Cell Function. Cell metabolism. 2015;22(4):619–32. doi: 10.1016/j.cmet.2015.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sheaffer KL, Kim R, Aoki R, Elliott EN, Schug J, Burger L, et al. DNA methylation is required for the control of stem cell differentiation in the small intestine. Genes & development. 2014;28(6):652–664. doi: 10.1101/gad.230318.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stadler MB, Murr R, Burger L, Ivanek R, Lienert F, Schoeler A, et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature. 2011;480(7378):490–495. doi: 10.1038/nature10716. [DOI] [PubMed] [Google Scholar]
- 31.Elliott EN, Sheaffer KL, Kaestner KH. The ‘de novo’ DNA methyltransferase Dnmt3b compensates the Dnmt1-deficient intestinal epithelium. Elife. 2016;5 doi: 10.7554/eLife.12975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324(5929):929–930. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324(5929):930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333(6047):1300–1303. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maiti A, Drohat AC. Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine potential implications for active demethylation of CpG sites. J Biol Chem. 2011 doi: 10.1074/jbc.C111.284620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weber AR, Krawczyk C, Robertson AB, Kuśnierczyk A, Vågbø CB, Schuermann D, et al. Biochemical reconstitution of TET1-TDG-BER-dependent active DNA demethylation reveals a highly coordinated mechanism. Nat Commun. 2016;7:10806. doi: 10.1038/ncomms10806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447(7143):407–12. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- 38.Gurard-Levin ZA, Almouzni G. Histone modifications and a choice of variant: a language that helps the genome express itself F1000Prime Rep. 2014;6:76. doi: 10.12703/P6-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang XJ, Seto E. HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene. 2007;26(37):5310–8. doi: 10.1038/sj.onc.1210599. [DOI] [PubMed] [Google Scholar]
- 40.Zhang T, Cooper S, Brockdorff N. The interplay of histone modifications - writers that read. EMBO Rep. 2015;16(11):1467–81. doi: 10.15252/embr.201540945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Waddington CH. The Strategy of the Genes. London: Allen and Unwin; 1957. [Google Scholar]
- 42.Spijker HS, Song H, Ellenbroek JH, Roefs MM, Engelse MA, Bos E, et al. Loss of beta-Cell Identity Occurs in Type 2 Diabetes and Is Associated With Islet Amyloid Deposits. Diabetes. 2015;64(8):2928–38. doi: 10.2337/db14-1752. [DOI] [PubMed] [Google Scholar]
- 43.Cano DA, Soria B, Martin F, Rojas A. Transcriptional control of mammalian pancreas organogenesis. Cell Mol Life Sci. 2014;71(13):2383–402. doi: 10.1007/s00018-013-1510-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pan FC, Wright C. Pancreas organogenesis: from bud to plexus to gland. Dev Dyn. 2011;240(3):530–65. doi: 10.1002/dvdy.22584. [DOI] [PubMed] [Google Scholar]
- 45.Li Z, Gadue P, Chen K, Jiao Y, Tuteja G, Schug J, et al. Foxa2 and H2A.Z mediate nucleosome depletion during embryonic stem cell differentiation. Cell. 2012;151(7):1608–16. doi: 10.1016/j.cell.2012.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang A, Yue F, Li Y, Xie R, Harper T, Patel NA, et al. Epigenetic priming of enhancers predicts developmental competence of hESC-derived endodermal lineage intermediates. Cell Stem Cell. 2015;16(4):386–99. doi: 10.1016/j.stem.2015.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xu CR, Cole PA, Meyers DJ, Kormish J, Dent S, Zaret KS. Chromatin “prepattern” and histone modifiers in a fate choice for liver and pancreas. Science. 2011;332(6032):963–6. doi: 10.1126/science.1202845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gao N, LeLay J, Vatamaniuk MZ, Rieck S, Friedman JR, Kaestner KH. Dynamic regulation of Pdx1 enhancers by Foxa1 and Foxa2 is essential for pancreas development. Genes Dev. 2008;22(24):3435–48. doi: 10.1101/gad.1752608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jonsson J, Carlsson L, Edlund T, Edlund H. Insulin-promoter-factor 1 is required for pancreas development in mice. Nature. 1994;371(6498):606–9. doi: 10.1038/371606a0. [DOI] [PubMed] [Google Scholar]
- 50.Offield MF, Jetton TL, Labosky PA, Ray M, Stein RW, Magnuson MA, et al. PDX-1 is required for pancreatic outgrowth and differentiation of the rostral duodenum. Development. 1996;122(3):983–95. doi: 10.1242/dev.122.3.983. [DOI] [PubMed] [Google Scholar]
- 51.McKenna B, Guo M, Reynolds A, Hara M, Stein R. Dynamic recruitment of functionally distinct Swi/Snf chromatin remodeling complexes modulates Pdx1 activity in islet beta cells. Cell Rep. 2015;10(12):2032–42. doi: 10.1016/j.celrep.2015.02.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Georgia S, Kanji M, Bhushan A. DNMT1 represses p53 to maintain progenitor cell survival during pancreatic organogenesis. Genes Dev. 2013;27(4):372–7. doi: 10.1101/gad.207001.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lynn FC, Skewes-Cox P, Kosaka Y, McManus MT, Harfe BD, German MS. MicroRNA expression is required for pancreatic islet cell genesis in the mouse. Diabetes. 2007;56(12):2938–45. doi: 10.2337/db07-0175. [DOI] [PubMed] [Google Scholar]
- 54.Moran I, Akerman I, van de Bunt M, Xie R, Benazra M, Nammo T, et al. Human beta cell transcriptome analysis uncovers lncRNAs that are tissue-specific, dynamically regulated, and abnormally expressed in type 2 diabetes. Cell Metab. 2012;16(4):435–48. doi: 10.1016/j.cmet.2012.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kalis M, Bolmeson C, Esguerra JL, Gupta S, Edlund A, Tormo-Badia N, et al. Beta-cell specific deletion of Dicer1 leads to defective insulin secretion and diabetes mellitus. PLoS One. 2011;6(12):e29166. doi: 10.1371/journal.pone.0029166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Arnes L, Akerman I, Balderes DA, Ferrer J, Sussel L. betalinc1 encodes a long noncoding RNA that regulates islet beta-cell formation and function. Genes Dev. 2016;30(5):502–7. doi: 10.1101/gad.273821.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Baroukh N, Ravier MA, Loder MK, Hill EV, Bounacer A, Scharfmann R, et al. MicroRNA-124a regulates Foxa2 expression and intracellular signaling in pancreatic beta-cell lines. J Biol Chem. 2007;282(27):19575–88. doi: 10.1074/jbc.M611841200. [DOI] [PubMed] [Google Scholar]
- 58.Joglekar MV, Parekh VS, Mehta S, Bhonde RR, Hardikar AA. MicroRNA profiling of developing and regenerating pancreas reveal post-transcriptional regulation of neurogenin3. Dev Biol. 2007;311(2):603–12. doi: 10.1016/j.ydbio.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 59.van Kruijsbergen I, Hontelez S, Veenstra GJ. Recruiting polycomb to chromatin. Int J Biochem Cell Biol. 2015;67:177–87. doi: 10.1016/j.biocel.2015.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bramswig NC, Everett LJ, Schug J, Dorrell C, Liu C, Luo Y, et al. Epigenomic plasticity enables human pancreatic alpha to beta cell reprogramming. Journal of Clinical Investigation. 2013;123(3):1275–1284. doi: 10.1172/JCI66514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lemaire K, Thorrez L, Schuit F. Disallowed and Allowed Gene Expression: Two Faces of Mature Islet Beta Cells. Annu Rev Nutr. 2016 doi: 10.1146/annurev-nutr-071715-050808. [DOI] [PubMed] [Google Scholar]
- 62.Thorrez L, Laudadio I, Van Deun K, Quintens R, Hendrickx N, Granvik M, et al. Tissue-specific disallowance of housekeeping genes: the other face of cell differentiation. Genome Res. 2011;21(1):95–105. doi: 10.1101/gr.109173.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dhawan S, Georgia S, Tschen S-i, Fan G, Bhushan A. Pancreatic beta Cell Identity Is Maintained by DNA Methylation-Mediated Repression of Arx. Developmental Cell. 2011;20(4):419–429. doi: 10.1016/j.devcel.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Papizan JB, Singer RA, Tschen SI, Dhawan S, Friel JM, Hipkens SB, et al. Nkx2.2 repressor complex regulates islet beta-cell specification and prevents beta-to-alpha-cell reprogramming. Genes Dev. 2011;25(21):2291–305. doi: 10.1101/gad.173039.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Luco RF, Maestro MA, Sadoni N, Zink D, Ferrer J. Targeted deficiency of the transcriptional activator Hnf1alpha alters subnuclear positioning of its genomic targets. PLoS Genet. 2008;4(5):e1000079. doi: 10.1371/journal.pgen.1000079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Parrizas M, Maestro MA, Boj SF, Paniagua A, Casamitjana R, Gomis R, et al. Hepatic nuclear factor 1-alpha directs nucleosomal hyperacetylation to its tissue-specific transcriptional targets. Mol Cell Biol. 2001;21(9):3234–43. doi: 10.1128/MCB.21.9.3234-3243.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Johnson JS, Evans-Molina C. Translational implications of the beta-cell epigenome in diabetes mellitus. Transl Res. 2015;165(1):91–101. doi: 10.1016/j.trsl.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sandovici I, Hammerle CM, Ozanne SE, Constancia M. Developmental and environmental epigenetic programming of the endocrine pancreas: consequences for type 2 diabetes. Cell Mol Life Sci. 2013;70(9):1575–95. doi: 10.1007/s00018-013-1297-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Andrali SS, Sampley ML, Vanderford NL, Ozcan S. Glucose regulation of insulin gene expression in pancreatic beta-cells. Biochemical Journal. 2008;415:1–10. doi: 10.1042/BJ20081029. [DOI] [PubMed] [Google Scholar]
- 70.Deering TG, Ogihara T, Trace AP, Maier B, Mirmira RG. Methyltransferase Set7/9 maintains transcription and euchromatin structure at islet-enriched genes. Diabetes. 2009;58(1):185–93. doi: 10.2337/db08-1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Evans-Molina C, Robbins RD, Kono T, Tersey SA, Vestermark GL, Nunemaker CS, et al. Peroxisome Proliferator-Activated Receptor gamma Activation Restores Islet Function in Diabetic Mice through Reduction of Endoplasmic Reticulum Stress and Maintenance of Euchromatin Structure. Molecular and cellular biology. 2009;29(8):2053–2067. doi: 10.1128/MCB.01179-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Maganti AV, Maier B, Tersey SA, Sampley ML, Mosley AL, Oezcan S, et al. Transcriptional Activity of the Islet beta Cell Factor Pdx1 Is Augmented by Lysine Methylation Catalyzed by the Methyltransferase Set7/9. Journal of Biological Chemistry. 2015;290(15):9812–9822. doi: 10.1074/jbc.M114.616219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bacos K, Gillberg L, Volkov P, Olsson AH, Hansen T, Pedersen O, et al. Blood-based biomarkers of age-associated epigenetic changes in human islets associate with insulin secretion and diabetes. Nat Commun. 2016;7:11089. doi: 10.1038/ncomms11089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Arda HE, Li L, Tsai J, Torre EA, Rosli Y, Peiris H, et al. Age-Dependent Pancreatic Gene Regulation Reveals Mechanisms Governing Human beta Cell Function. Cell Metab. 2016;23(5):909–20. doi: 10.1016/j.cmet.2016.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bernstein DL, Kameswaran V, Le Lay JE, Sheaffer KL, Kaestner KH. The BisPCR(2) method for targeted bisulfite sequencing. Epigenetics Chromatin. 2015;8(27) doi: 10.1186/s13072-015-0020-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yang BT, Dayeh TA, Kirkpatrick CL, Taneera J, Kumar R, Groop L, et al. Insulin promoter DNA methylation correlates negatively with insulin gene expression and positively with HbA(1c) levels in human pancreatic islets. Diabetologia. 2011;54(2):360–367. doi: 10.1007/s00125-010-1967-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Akirav EM, Lebastchi J, Galvan EM, Henegariu O, Akirav M, Ablamunits V, et al. Detection of beta cell death in diabetes using differentially methylated circulating DNA. Proc Natl Acad Sci U S A. 2011;108(47):19018–23. doi: 10.1073/pnas.1111008108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fisher MM, Watkins RA, Blum J, Evans-Molina C, Chalasani N, DiMeglio LA, et al. Elevations in Circulating Methylated and Unmethylated Preproinsulin DNA in New-Onset Type 1 Diabetes. Diabetes. 2015;64(11):3867–72. doi: 10.2337/db15-0430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Herold KC, Usmani-Brown S, Ghazi T, Lebastchi J, Beam CA, Bellin MD, et al. beta cell death and dysfunction during type 1 diabetes development in at-risk individuals. J Clin Invest. 2015;125(3):1163–73. doi: 10.1172/JCI78142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Husseiny MI, Kaye A, Zebadua E, Kandeel F, Ferreri K. Tissue-specific methylation of human insulin gene and PCR assay for monitoring beta cell death. PLoS One. 2014;9(4):e94591. doi: 10.1371/journal.pone.0094591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lebastchi J, Deng S, Lebastchi AH, Beshar I, Gitelman S, Willi S, et al. Immune therapy and beta-cell death in type 1 diabetes. Diabetes. 2013;62(5):1676–80. doi: 10.2337/db12-1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lehmann-Werman R, Neiman D, Zemmour H, Moss J, Magenheim J, Vaknin-Dembinsky A, et al. Identification of tissue-specific cell death using methylation patterns of circulating DNA. Proc Natl Acad Sci U S A. 2016;113(13):E1826–34. doi: 10.1073/pnas.1519286113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rieck S, Kaestner KH. Expansion of beta-cell mass in response to pregnancy. Trends in Endocrinology and Metabolism. 2010;21(3):151–158. doi: 10.1016/j.tem.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tarabra E, Pelengaris S, Khan M. A Simple Matter of Life and Death-The Trials of Postnatal Beta-Cell Mass Regulation. International Journal of Endocrinology. 2012:516718–516718. doi: 10.1155/2012/516718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Krishnamurthy J, Ramsey MR, Ligon KL, Torrice C, Koh A, Bonner-Weir S, et al. p16(INK4a) induces an age-dependent decline in islet regenerative potential. Nature. 2006;443(7110):453–457. doi: 10.1038/nature05092. [DOI] [PubMed] [Google Scholar]
- 86.Chen H, Gu X, Su Ih, Bottino R, Contreras JL, Tarakhovsky A, et al. Polycomb protein Ezh2 regulates pancreatic beta-cell Ink4a/Arf expression and regeneration in diabetes mellitus. Genes & development. 2009;23(8):975–985. doi: 10.1101/gad.1742509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhou JX, Dhawan S, Fu H, Snyder E, Bottino R, Kundu S, et al. Combined modulation of polycomb and trithorax genes rejuvenates beta cell replication. J Clin Invest. 2013;123(11):4849–58. doi: 10.1172/JCI69468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bernstein DL, Le Lay JE, Ruano EG, Kaestner KH. TALE-mediated epigenetic suppression of CDKN2A increases replication in human. Journal of Clinical Investigation. 2015;125(5):1998–2006. doi: 10.1172/JCI77321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hall E, Dayeh T, Kirkpatrick CL, Wollheim CB, Dekker Nitert M, Ling C. DNA methylation of the glucagon-like peptide 1 receptor (GLP1R) in human pancreatic islets. BMC Med Genet. 2013;14:76. doi: 10.1186/1471-2350-14-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Liang H, Ward WF. PGC-1alpha: a key regulator of energy metabolism. Adv Physiol Educ. 2006;30(4):145–51. doi: 10.1152/advan.00052.2006. [DOI] [PubMed] [Google Scholar]
- 91.Volkmar M, Dedeurwaerder S, Cunha DA, Ndlovu MN, Defrance M, Deplus R, et al. DNA methylation profiling identifies epigenetic dysregulation in pancreatic islets from type 2 diabetic patients. EMBO J. 2012;31(6):1405–26. doi: 10.1038/emboj.2011.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bhandare R, Schug J, Le Lay J, Fox A, Smirnova O, Liu C, et al. Genome-wide analysis of histone modifications in human pancreatic islets. Genome Res. 2010;20(4):428–33. doi: 10.1101/gr.102038.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pasquali L, Gaulton KJ, Rodriguez-Segui SA, Mularoni L, Miguel-Escalada I, Akerman I, et al. Pancreatic islet enhancer clusters enriched in type 2 diabetes risk-associated variants. Nat Genet. 2014;46(2):136–43. doi: 10.1038/ng.2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dayeh TA, Olsson AH, Volkov P, Almgren P, Ronn T, Ling C. Identification of CpG-SNPs associated with type 2 diabetes and differential DNA methylation in human pancreatic islets. Diabetologia. 2013;56(5):1036–46. doi: 10.1007/s00125-012-2815-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Donohoe DR, Bultman SJ. Metaboloepigenetics: interrelationships between energy metabolism and epigenetic control of gene expression. J Cell Physiol. 2012;227(9):3169–77. doi: 10.1002/jcp.24054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Xie R, Carrano AC, Sander M. A systems view of epigenetic networks regulating pancreas development and beta-cell function. Wiley Interdiscip Rev Syst Biol Med. 2015;7(1):1–11. doi: 10.1002/wsbm.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Milagro FI, Campion J, Garcia-Diaz DF, Goyenechea E, Paternain L, Martinez JA. High fat diet-induced obesity modifies the methylation pattern of leptin promoter in rats. J Physiol Biochem. 2009;65(1):1–9. doi: 10.1007/BF03165964. [DOI] [PubMed] [Google Scholar]
- 98.Shen W, Wang C, Xia L, Fan C, Dong H, Deckelbaum RJ, et al. Epigenetic modification of the leptin promoter in diet-induced obese mice and the effects of N-3 polyunsaturated fatty acids. Sci Rep. 2014;4:5282. doi: 10.1038/srep05282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Multhaup ML, Seldin MM, Jaffe AE, Lei X, Kirchner H, Mondal P, et al. Mouse-human experimental epigenetic analysis unmasks dietary targets and genetic liability for diabetic phenotypes. Cell Metab. 2015;21(1):138–49. doi: 10.1016/j.cmet.2014.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Smith GC, Konycheva G, Dziadek MA, Ravelich SR, Patel S, Reddy S, et al. Pre- and postnatal methyl deficiency in the rat differentially alters glucose homeostasis. J Nutrigenet Nutrigenomics. 2011;4(4):175–91. doi: 10.1159/000330227. [DOI] [PubMed] [Google Scholar]
- 101.Lindholm ME, Marabita F, Gomez-Cabrero D, Rundqvist H, Ekstrom TJ, Tegner J, et al. An integrative analysis reveals coordinated reprogramming of the epigenome and the transcriptome in human skeletal muscle after training. Epigenetics. 2014;9(12):1557–69. doi: 10.4161/15592294.2014.982445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ronn T, Volkov P, Davegardh C, Dayeh T, Hall E, Olsson AH, et al. A six months exercise intervention influences the genome-wide DNA methylation pattern in human adipose tissue. PLoS Genet. 2013;9(6):e1003572. doi: 10.1371/journal.pgen.1003572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Balkau B, Mhamdi L, Oppert JM, Nolan J, Golay A, Porcellati F, et al. Physical activity and insulin sensitivity: the RISC study. Diabetes. 2008;57(10):2613–8. doi: 10.2337/db07-1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Martinez JA, Milagro FI, Claycombe KJ, Schalinske KL. Epigenetics in adipose tissue, obesity, weight loss, and diabetes. Adv Nutr. 2014;5(1):71–81. doi: 10.3945/an.113.004705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A. 2005;102(30):10604–9. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ling C, Poulsen P, Simonsson S, Ronn T, Holmkvist J, Almgren P, et al. Genetic and epigenetic factors are associated with expression of respiratory chain component NDUFB6 in human skeletal muscle. J Clin Invest. 2007;117(11):3427–35. doi: 10.1172/JCI30938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ronn T, Poulsen P, Hansson O, Holmkvist J, Almgren P, Nilsson P, et al. Age influences DNA methylation and gene expression of COX7A1 in human skeletal muscle. Diabetologia. 2008;51(7):1159–68. doi: 10.1007/s00125-008-1018-8. [DOI] [PubMed] [Google Scholar]
- 108.Ravelli AC, van der Meulen JH, Michels RP, Osmond C, Barker DJ, Hales CN, et al. Glucose tolerance in adults after prenatal exposure to famine. Lancet. 1998;351(9097):173–7. doi: 10.1016/s0140-6736(97)07244-9. [DOI] [PubMed] [Google Scholar]
- 109.Constancia M, Hemberger M, Hughes J, Dean W, Ferguson-Smith A, Fundele R, et al. Placental-specific IGF-II is a major modulator of placental and fetal growth. Nature. 2002;417(6892):945–8. doi: 10.1038/nature00819. [DOI] [PubMed] [Google Scholar]
- 110.Petry CJ, Dorling MW, Pawlak DB, Ozanne SE, Hales CN. Diabetes in old male offspring of rat dams fed a reduced protein diet. Int J Exp Diabetes Res. 2001;2(2):139–43. doi: 10.1155/EDR.2001.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sandovici I, Smith NH, Nitert MD, Ackers-Johnson M, Uribe-Lewis S, Ito Y, et al. Maternal diet and aging alter the epigenetic control of a promoter-enhancer interaction at the Hnf4a gene in rat pancreatic islets. Proc Natl Acad Sci U S A. 2011;108(13):5449–54. doi: 10.1073/pnas.1019007108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Pinney SE, Jaeckle Santos LJ, Han Y, Stoffers DA, Simmons RA. Exendin-4 increases histone acetylase activity and reverses epigenetic modifications that silence Pdx1 in the intrauterine growth retarded rat. Diabetologia. 2011;54(10):2606–14. doi: 10.1007/s00125-011-2250-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Thompson RF, Fazzari MJ, Niu H, Barzilai N, Simmons RA, Greally JM. Experimental intrauterine growth restriction induces alterations in DNA methylation and gene expression in pancreatic islets of rats. J Biol Chem. 2010;285(20):15111–8. doi: 10.1074/jbc.M109.095133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Park JH, Stoffers DA, Nicholls RD, Simmons RA. Development of type 2 diabetes following intrauterine growth retardation in rats is associated with progressive epigenetic silencing of Pdx1. J Clin Invest. 2008;118(6):2316–24. doi: 10.1172/JCI33655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Alonso-Magdalena P, Vieira E, Soriano S, Menes L, Burks D, Quesada I, et al. Bisphenol A exposure during pregnancy disrupts glucose homeostasis in mothers and adult male offspring. Environ Health Perspect. 2010;118(9):1243–50. doi: 10.1289/ehp.1001993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chang H, Wang D, Xia W, Pan X, Huo W, Xu S, et al. Epigenetic disruption and glucose homeostasis changes following low-dose maternal bisphenol A exposure. Toxicology Research. 2016;5:1400–1409. doi: 10.1039/c6tx00047a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Li G, Chang H, Xia W, Mao Z, Li Y, Xu S. F0 maternal BPA exposure induced glucose intolerance of F2 generation through DNA methylation change in Gck. Toxicol Lett. 2014;228(3):192–9. doi: 10.1016/j.toxlet.2014.04.012. [DOI] [PubMed] [Google Scholar]
- 118.Radford EJ, Ito M, Shi H, Corish JA, Yamazawa K, Isganaitis E, et al. In utero effects. In utero undernourishment perturbs the adult sperm methylome and intergenerational metabolism. Science. 2014;345(6198):1255903. doi: 10.1126/science.1255903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ng SF, Lin RC, Laybutt DR, Barres R, Owens JA, Morris MJ. Chronic high-fat diet in fathers programs beta-cell dysfunction in female rat offspring. Nature. 2010;467(7318):963–6. doi: 10.1038/nature09491. [DOI] [PubMed] [Google Scholar]
- 120.de Castro Barbosa T, Ingerslev LR, Alm PS, Versteyhe S, Massart J, Rasmussen M, et al. High-fat diet reprograms the epigenome of rat spermatozoa and transgenerationally affects metabolism of the offspring. Mol Metab. 2016;5(3):184–97. doi: 10.1016/j.molmet.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Donkin I, Versteyhe S, Ingerslev LR, Qian K, Mechta M, Nordkap L, et al. Obesity and Bariatric Surgery Drive Epigenetic Variation of Spermatozoa in Humans. Cell Metab. 2016;23(2):369–78. doi: 10.1016/j.cmet.2015.11.004. [DOI] [PubMed] [Google Scholar]
- 122.Chou DH, Holson EB, Wagner FF, Tang AJ, Maglathlin RL, Lewis TA, et al. Inhibition of histone deacetylase 3 protects beta cells from cytokine-induced apoptosis. Chem Biol. 2012;19(6):669–73. doi: 10.1016/j.chembiol.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]