

Abstract
Infection with Japanese encephalitis virus (JEV) causes cerebral inflammation and stimulates inflammatory cytokine expression. Glial cells orchestrate immunocyte recruitment to focal sites of viral infection within the central nervous system (CNS) and synchronize immune cell functions through a regulated network of cytokines and chemokines. Since immune cell infiltration is prominent, we investigated the production of a responding chemoattractant, RANTES (regulated upon activation, normal T-cell expressed and secreted), in response to JEV infection of glial cells. Infection with JEV was found to elicit the production of RANTES from primary neurons/glia, mixed glia, microglia, and astrocytes but not from neuron cultures. The production of RANTES did not seem to be directly responsible for JEV-induced neuronal death but instead contributed to the recruitment of immune cells. RANTES expression required viral replication and the activation of extracellular signal-regulated kinase (ERK) as well as transcription factors, including nuclear factor kappa B (NF-κB) and nuclear factor IL-6 (NF-IL-6). The induction of RANTES expression by JEV infection in glial cells needed the coordinate activation of NF-κB and NF-IL-6. Using enzymatic inhibitors, we demonstrated a strong correlation between the ERK signaling pathway and RANTES expression. However, JEV replication was not dependent on the activation of ERK, NF-κB, and NF-IL-6. Altogether, these results demonstrated that infection of glial cells by JEV provided the early ERK-, NF-κB-, and NF-IL-6-mediated signals that directly activated RANTES expression, which might be involved in the initiation and amplification of inflammatory responses in the CNS.
Chemokines are a family of small (≈8 to 14 kDa), basic, structurally related chemoattractant cytokines that are produced upon activation by a wide spectrum of cell types, including T cells, monocytes, endothelial cells, microglia, and astrocytes. Chemokines have been studied extensively as important regulators of leukocyte trafficking to sites of immune challenge or tissue damage (3, 82, 90). In the brain, chemokines act as chemoattractants for numerous cell types of the central nervous system (CNS) during development and are believed to play a role in neuronal patterning and proliferation (3, 61). The chemokine proteins are classified into subfamilies (CXC, CC, CX3C, and XC) based on the position of the first two of four conserved cysteine residues (90). In general, the members of the CXC family, such as cytokine-induced neutrophil chemoattractant, act primarily on neutrophils, while the majority of CC chemokines, such as monocyte chemoattractant protein-1 (MCP-1), macrophage inflammatory protein 1α (MIP-1α), and regulated-upon-activation, normal T-cell expressed and secreted (RANTES), are monocyte chemoattractants (82).
RANTES is highly chemoattractant for T lymphocytes, monocytes, eosinophils, and basophils (1, 83). Infection with some viruses has been shown to induce RANTES expression in a wide variety of cells (9, 16, 43, 52, 56, 88). Thus, virus-induced RANTES expression could be a major element in the pathogenesis of viral infection. RANTES could be activated by different stimuli through the regulation of transcriptional control (62, 63, 67). The 5′ region of the RANTES gene promoter is divided into five regions (A to E) and plays a distinct role in the induction of gene expression (67). The A/B and E regions contain potential transcription factor binding sites critical to gene expression, NF-κB (nucleotides −58 to −27) and NF-IL-6 (nucleotides −100 to −92), respectively. NF-κB represents a family of dimeric transcription factors that play a central role in the inflammatory responses by regulating gene expression through binding to the cis-acting κB elements (25, 34). Nuclear factor IL-6 (NF-IL-6), a member of the CCAAT/enhancer binding protein (C/EBP) family of transcription factors, belongs to a class of DNA binding proteins called the basic leucine zipper protein family, which includes C/EBPα, C/EBPβ (NF-IL-6), C/EBPγ, and C/EBPδ (NF-IL-6β) (74).
Neurological diseases caused by conventional viruses such as lymphocytic choriomeningitis virus (8, 21), measles virus (89), rubella virus, (89), papovavirus (89), and human immunodeficiency virus type 1 (27, 75) are often characterized by a prolonged asymptomatic period, evidence of immune system recognition, and the presence of inflammatory components among the neuropathological changes (89). The mechanisms by which these viruses cause neurological disease are not fully understood. In many cases, the virus is probably not directly involved in the destruction of brain tissue but may cause damage indirectly by triggering cell-mediated immune responses, such as activating cytotoxic T cells and macrophages. Activated inflammatory cells secrete cytokines, such as interleukin-1 (IL-1) and tumor necrosis factor alpha (TNF-α), which can cause toxic effects in the brain (76). Additionally, other soluble factors, such as neurotoxins (26), quinolinic acid (29), and excitatory neurotransmitters (86), have been implicated as toxin candidates responsible for neurological damage.
Japanese encephalitis virus (JEV), a member of the family Flaviviridae, is an acute zoonotic infection that commonly affects children and is a major cause of acute encephalopathy (10, 58). JEV targets the CNS, clinically manifesting with fever, headache, vomiting, signs of meningeal irritation, and altered consciousness, leading to high mortality and neurological sequelae in some of those who survive (38). After entry into the host, JEV generates a rapid inflammatory response, including peripheral neutrophil leucocytosis or infiltration of neutrophils in extraneural tissue. Clinically, the infection of JEV results in increased levels of cytokines such as macrophage-derived chemotactic factor, TNF-α, and interleukin 8 (IL-8) in the serum and cerebrospinal fluid (35, 78, 85). The increased levels of inflammatory mediators appear to play a protective role or to initiate an irreversible immune response leading to cell death. Accumulating evidence shows that the mortality rate increases with increasing concentrations of cytokines in the serum and cerebrospinal fluid in Japanese encephalitis patients (78).
The host response to infection is central to the effective control and ultimate clearance of invading pathogens or removal of infected cells. The response to cerebral infection with JEV in mice is characterized at the pathological level by significant recruitment and extravasation of immunoinflammatory cells, predominantly macrophages, T cells, and neutrophils, to sites of viral replication in the brain (12, 50, 51). Despite the pathological and socioeconomic importance of JEV, knowledge of the immunological mechanisms occurring during viral infection is limited. The inflammatory response observed within the CNS in viral encephalitis is partly mediated by chemokines, which are released by various cells of the CNS (32). Previously, we had reported that infection of astrocytes with JEV stimulated RANTES expression (16). In this study, we sought to address the underlying mechanisms. We found that microglia and astrocytes were the predominant cells for JEV-induced RANTES expression. Moreover, we found that the signaling pathway of JEV-induced RANTES production was associated with the activation of extracellular signal-regulated kinase (ERK) and increased binding activity of NF-IL-6 and NF-κB.
MATERIALS AND METHODS
Viruses.
The propagation of JEV NT113 (15), isolated from a mosquito, was carried out in C6/36 cells. JEV strains Peking-1 (36), Nakayama (66), and JaGar (47) were provided by M.-D. Kuo (Institute of Preventive Medicine, National Medical Defense Center, Taiwan). C6/36 cells were infected with JEV at a multiplicity of infection of 0.1 with Dulbecco's modified Eagle's medium (DMEM) containing 5% fetal bovine serum (FBS) (Life Technologies) and cultivated for a further 4 to 5 days at 28°C. On the other hand, neural cultures were first adsorbed with JEV (multiplicity of infection = 20) for 1 h at 37°C. After adsorption, the unbound viruses were removed by gentle washing with phosphate-buffered saline (PBS, pH 7.4). Fresh DMEM-2% FBS was added to each plate for further incubation at 37°C. Virus inactivation was done by placing viruses in boiled water for 15 min or exposing viruses to UV light (wavelength, 254 nm) at a distance of 3 cm for 30 min.
Cell cultures.
The animal study protocol was approved by the Animal Experimental Committee of Taichung Veterans General Hospital. Neuron/glia, mixed glia, neuron, microglia, and astrocyte cultures were prepared from the cerebral cortexes of 1-day-old Sprague-Dawley rats (14, 16, 41). In brief, pooled dissected cortexes were digested with 5 ml of papain solution (1.5 mM CaCl2, 0.5 mM EDTA, 0.6 mg of papain per ml, 0.063% DNase I, 5.32 g of Hanks' balanced salt solution per liter, and 0.2 mg of cysteine per ml) at 37°C for 20 min to dissociate the cells. After centrifugation at 200 × g for 5 min, cells were plated on poly-d-lysine-coated (20 μg/ml; molecular weight, 30,000 to 70,000; Sigma Chemical Co.) dishes. One day after seeding, the culture medium was replaced with minimal essential medium (MEM; Life Technologies) supplemented with 10% FBS and 10% horse serum. On the fourth day in vitro, the medium was changed and replaced with fresh serum-containing medium. For cortical neurons, the culture medium was replaced with neurobasal medium supplemented with B27 (Life Technologies). Cytosine arabinoside (10 μM, Sigma Chemical Co.) was added to the medium on the third and fourth days in vitro to inhibit nonneuronal cell division.
These neuron/glia and neuron cultures were used for experiments after 10 to 12 days in vitro. For mixed glia, the cell pellets were resuspended in DMEM/F12 (Life Technologies) supplemented with 10% FBS. The medium was replenished 4 days after plating and changed every 3 days. The resultant mixed glia cultures were used 14 to 16 days after plating. Astrocyte and microglia cultures were separated by shaking mixed glial cultures at a speed of 200 rpm for 24 h. The retained astrocytes and floating microglia were maintained in DMEM/F12 containing 10% FBS. Cell composition was identified and estimated by immunocytochemistry with antibodies against microtubule-associated protein 2 (MAP-2; for neurons; Transduction Laboratories), glial fibrillary acidic protein (GFAP; for astrocytes; Santa Cruz Biotechnology), and ED-1 (for microglia; BioSource). In general, neurons/glia consisted of 50% neurons, 40% astrocytes, and 10% microglia. Mixed glial cultures contained ≈80% astrocytes and ≈20% microglia. The purity of neurons, astrocytes, and microglia was more than 95%.
Immunocytochemical staining.
Cells were washed twice with PBS, fixed with 4% paraformaldehyde for 10 min, and permeabilized with 0.1% Triton X-100 for 15 min. The cells were blocked with 5% milk in PBS for 30 min and then incubated with primary antibody (1:500) overnight at 4°C, followed by washing with PBS. After washing, the cells were incubated with horseradish peroxidase-conjugated secondary antibody (1:500) for 1 h at room temperature. After washing, the color was developed with 3,3′-diaminobenzidine (Sigma Chemical Co.) and observed under a light microscope.
RT-PCR.
For determination of mRNA expression, total RNA extraction and reverse transcription (RT)-PCR were performed as previously reported (16). DNA fragments of specific genes and the internal control were coamplified in a tube containing Taq DNA polymerase (Promega) and 0.8 μM each of the sense and antisense primers. The PCR was performed with a DNA thermal cycler (Perkin Elmer-Cetus) under the following conditions: one cycle of 94°C for 3 min; 28 cycles of 94°C for 50 s, 56°C for 40 s, and 72°C for 45 s; and then one cycle of 72°C for 5 min. The amplified DNA fragments were resolved by 1.5% agarose gel electrophoresis and stained with ethidium bromide.
The oligonucleotides used in this study were as follows: 5′-CACCGTCATCCTCGTTGC-3′ and 5′-CACTTGGCGGTTCTTTCG-3′ for RANTES; 5′-CAGGTCTCTGTCACGCTTCT-3′ and 5′-AGTATTCATGGAAGGGAATAG-3′ for MCP-1; 5′-GCTCTGGAACGAAGTCTTCTCAGC-3′ and 5′-TCCAGCTCAGTGATGTATTCTTGGAC-3′ for MIP-1α; 5′-ATGGTCTCAGCCACCCGCTCG-3′ and 5′-TTACTTGGGGACACCCTTTAGCAT-3′ for cytokine-induced neutrophil chemoattractant; 5′-ACCACAGTCCATGCCATCAC-3′ and 5′-TCCACCACCCTGTTGCTGTA-3′ for glyceraldehydes-3-phosphate dehydrogenase (GAPDH); 5′-TCCTGTGGCATCCACGAAACT-3′ and 5′-GGAGCAATGATCTTGATCTTC-3′ for β-actin; and 5′-GAAGACAGGGGTCATCTAGTGTG-3′ and 5′-AGATCCTGTGTTCTTC-3′ for JEV.
RANTES assay.
RANTES was measured with an enzyme immunoassay kit from R&D, following the procedure provided by the manufacturer.
Western blot analysis.
Cells were washed twice with PBS and harvested in Laemmli sodium dodecyl sulfate (SDS) sample buffer. The protein concentration in the supernatant was determined by the Bradford assay (Bio-Rad, Richmond, Calif.). Protein extracts were separated by SDS-polyacrylamide gel electrophoresis (PAGE) and electrophoretically transferred to polyvinylidene difluoride membranes (Amersham Pharmacia Biotech.). Membranes were first incubated with 5% nonfat milk in PBS for 1 h at room temperature. Membranes were washed with PBS containing 0.1% Tween 20 (PBST) and then incubated for 1 h at room temperature with antibodies, including phosphorylated and nonphosphorylated forms of ERK (1:5,000; Santa Cruz Biotechnology) and JEV NS3 (1:5,000). After washing again with PBST buffer, a 1:10,000 (vol/vol) dilution of horseradish peroxidase-labeled immunoglobulin G was added at room temperature for 1 h. The blots were developed with ECL Western blotting reagents (Amersham Pharmacia Biotech.).
Plasmids, transient transfections, and reporter assay.
The firefly luciferase gene under the control of the human RANTES promoter, including −881 to +54 (pGL-A), −181 to +54 (pGL-B), and −79 to +54 (pGL-C), was provided by H.-S. Liu (National Cheng Kung University, Taiwan) (43). A Renilla luciferase reporter under the control of the herpes simplex virus thymidine kinase promoter, pRL-TK, was used as an internal control to normalize reporter gene activity (19). Rat C6 glioma cells maintained in DMEM-5% FBS at 37°C were transfected with Lipofectamine (Life Technologies) liposomal transfection reagent according to the instructions of the manufacturer. Twenty-four hours later, the transfected cells were infected with JEV at a multiplicity of infection of 5. After 36 h, the cells were lysed, and the luciferase activity was determined. Luciferase activities were determined by a luminometer the a dual-luciferase reporter assay according to the instructions of the manufacturer (Promega Co.).
Preparation of nuclear extracts and electrophoretic mobility shift assay.
Nuclear extracts were prepared as described previously (19). In brief, cells were washed twice with ice-cold PBS and pelleted. The cell pellet was resuspended in hypotonic buffer (10 mM HEPES, pH 8.0; 10 mM KCl; 1.5 mM MgCl2; 5 mM dithiothreitol; 0.5 mM phenylmethylsulfonyl fluoride; 1 mM NaF; 1 mM Na3VO4) and incubated on ice for 15 min. Then the cells were lysed by the addition of 0.5% Nonidet P-40 and vigorous vortexing for 30 s. The nuclei were pelleted by centrifugation at 12,000 × g for 1 min at 4°C and resuspended in extraction buffer (20 mM HEPES, pH 8.0; 420 mM NaCl; 1.5 mM MgCl2; 0.2 mM EDTA; 1 mM dithiothreitol; 10% glycerol; 0.5 mM phenylmethylsulfonyl fluoride; 1 mM NaF; 1 mM Na3VO4). After 15 min on ice, lysates were centrifuged at 12,000 × g for 10 min at 4°C. Supernatants were obtained and stored at −70°C. The protein concentration in the supernatant was determined by the Bradford assay (Bio-Rad, Richmond, Calif.).
The oligonucleotides were synthesized and 5′ labeled with biotin by the manufacturer (Purigo Biotech, Inc.), including NF-κB (5′-AGTTGAGGGGACTTTCCCAGGC), NF-IL-6 (5′-AGCTCGCGTTGTGCAATCTG), and human RANTES promoter A/B site (5′-TTTTGGAAACTCCCCTTAGGGGATGCCCCTCA) and E site (5′-TTCCGTTTTGTGCAATTTCAC) (43, 62, 63). Nuclear extract (10 μg) was used for the electrophoretic mobility shift assay. The binding reaction mixture included 1 μg of poly(dI-dC), 0.1 μg of poly-l-lysine, and 100 fmol of biotin-labeled DNA probe in 20 μl of binding buffer (10 mM HEPES, pH 7.6; 50 mM NaCl; 0.5 mM MgCl2; 0.5 mM EDTA; 1 mM dithiothreitol; 5% glycerol). The DNA-protein complex was analyzed on 6% native polyacrylamide gels and electrically transferred to a nylon membrane. The labeled oligonucleotides were reacted with horseradish peroxidase-labeled streptavidin and detected with chemiluminescence reagents.
Chemotaxis assay.
The chemotactic response was evaluated with a modified 24-well Transwell (Costar), as previously described (65). One hundred microliters of RAW 264.7 cell suspension was added to the upper well of the chamber. The lower well received 250 μl of fresh medium and 250 μl of conditioned medium from mixed glia. The upper and lower wells were separated by polycarbonate filters with a 3-μm pore size, and the chamber was incubated for 8 h at 37°C. After incubation, nonmigrating cells were scraped off the upper surfaces of the filters. Migrating cells attached to the lower surfaces were fixed in ethanol for 10 min and stained with crystal violet. Labeled cells were counted microscopically at 400× within a total area of 2 mm2.
Statistical analysis.
The data are expressed as mean values ± standard error of the mean. Statistical analysis was carried out by one-way analysis of variance followed by Dunnett's test to assess statistical significance between treated and untreated groups through all experiments. A P of <0.05 was considered statistically significant.
RESULTS
JEV is a potent inducer of RANTES expression in neural cells.
To define the mechanisms involved in the initiation of a proinflammatory milieu in the CNS leading to eventual encephalitis, the ability of JEV to induce RANTES chemokine expression in primary neuron/glia cultures was assessed. Neuron/glia cultures consisted of neurons (≈50%), astrocytes (≈40%), and microglia (≈10%) 12 days after preparation (Fig. 1A). Mock-infected cultures (48 h) demonstrated minimal RANTES production. In contrast, supernatants from JEV-infected cultures showed a significant increase in RANTES production in a time-dependent manner (P < 0.01 versus the mock-infected control) (Fig. 1C). To address the regulation of JEV-induced RANTES secretion, we determined RANTES expression at mRNA levels in JEV-infected neuron/glia cultures by RT-PCR. GAPDH gene expression was used as an internal control. Figure 1B shows that RANTES mRNA expression was evidently induced at 24 h postinfection and reached a very high level at 48 h postinfection. Thus, the increase in RANTES protein from JEV-infected culture supernatants was coupled with an increase in cellular RANTES mRNA synthesis.
FIG. 1.
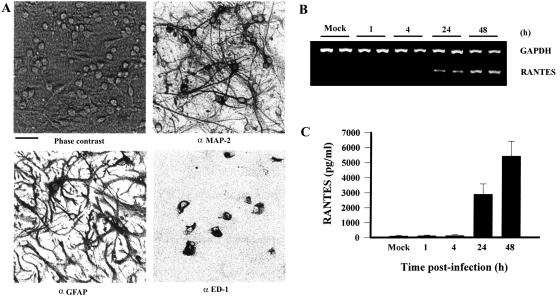
Induction of RANTES expression in JEV-infected neuron/glia cultures. After 12 days in vitro, neuron/glia cultures were subjected to immunocytochemistry with antibodies against MAP-2, GFAP, and ED-1. Neuron/glia cultures before harvest were also photographed under phase contrast. Scale bar, 50 μm (A). Neuron/glia cultures were mock-infected or infected with JEV (NT113) over time. Total cellular RNAs were harvested and subjected to RT-PCR for the detection of RANTES and GAPDH (B). Culture supernatants were collected, and the concentration of RANTES was determined by enzyme-linked immunosorbent assay (n = 4) (C).
The loss of neuron-specific MAP-2 immunoreactivity and the integrity of neuronal processes are early markers of neuronal damage (14, 17). The integrity of neuronal processes was lost gradually and was apparent 48 h after JEV infection in neuron/glia cultures (Fig. 2A). However, microglia and astrocytes within neuron/glia cultures were not significantly affected by JEV infection (data not shown). JEV RNA measurable by RT-PCR was initially detected after 24 h of JEV infection in neuron/glia cultures, and the signal increased up to 48 h postinfection (Fig. 2C). The induction of RANTES expression was parallel to virus-induced neuronal loss and the kinetics of viral replication. This indicates an association between JEV-upregulated RANTES expression and virus infection.
FIG. 2.
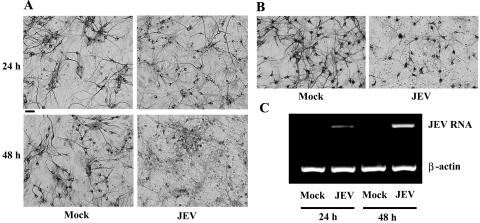
Neuronal toxicity of JEV. Neuron/glia cultures were mock infected or infected with JEV (NT113) over time. The integrity of neurons was determined by immunocytochemistry with antibody against MAP-2 (A), and the levels of JEV RNA and β-actin mRNA were determined by RT-PCR (C). The integrity of neurons 24 h after mock or JEV (NT113) infection was also assessed in neuron-enriched cultures by immunocytochemistry with antibody against MAP-2 (B). Scale bar, 50 μm.
To determine the specific CNS cells which contribute to overall RANTES induction after JEV infection, mixed glia, neurons, astrocytes, and microglia isolated from neonatal rats were also infected with JEV in vitro (Fig. 3A). RANTES, as in neuron/glia cultures, was also expressed in mixed glia, microglia, and astrocytes (P < 0.01 versus the mock-infected control). The induction of RANTES gene expres-sion was most prominent in mixed glia. The level of RANTES expression in microglia and astrocytes was relatively equal to but generally much lower than that in neuron/glia cultures and mixed glia. Although JEV infection damaged neurons (Fig. 2B), their infection failed to induce RANTES production in neuron-enriched cultures (Fig. 3A). These results indicate that, with the exception of neurons, neurons/glia, mixed glia, microglia, and astrocytes are all capable of significantly expressing RANTES as a response to JEV infection, although the level of RANTES induced may vary depending on the cell population. Moreover, we found that supernatants from JEV-infected microglia further stimulated RANTES production by astrocytes (P < 0.01) (Fig. 3B), suggesting that soluble factors from microglia contributed to the increased RANTES production in the presence of both astrocytes and microglia after JEV infection.
FIG. 3.
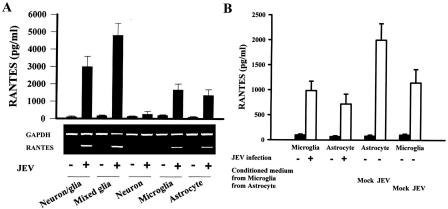
Cell specificity of RANTES production. Neuron/glia, mixed glia, neuron, microglia, and astrocyte cultures were mock infected or infected with JEV (NT113) for 24 h. The production of RANTES and expression of RANTES mRNA were determined by enzyme-linked immunosorbent assay (n = 4) and RT-PCR, respectively (A). The supernatants of mock- and JEV-infected microglia and astrocytes (24 h) were collected and mixed with an equal volume of fresh medium. Microglia-derived conditioned medium was added to new astrocyte cultures and vice versa. The level of RANTES in supernatants before and 24 h after addition was determined by enzyme-linked immunosorbent assay (n = 3) (B).
That JEV replicates in neurons is a well-established fact. Previously, we demonstrated that glial cells could be infected by JEV, as evidenced by immunocytochemistry, RT-PCR, and functional changes (16, 41). Measurable JEV RNA was detected in neuron/glia cultures after infection (Fig. 2C). In this study, the infectivity of JEV for microglia (≈80% of infectivity) and astrocytes (20 to 30% of infectivity) was assessed by immunocytochemical staining with antibody against nonstructural protein 3 (NS3) in each enriched cultures (Fig. 4). Furthermore, immunoreactivity with an antibody against NS3 was strongly localized at microglia in JEV-infected mixed glia. However, immunoreactivity with the antibody against RANTES was localized at both microglia and astrocytes (Fig. 5). These in vitro data further demonstrate that JEV was able to infect glial cells and stimulate RANTES expression from microglia and astrocytes.
FIG. 4.

Glial infectivity of JEV. Microglia and astrocyte cultures were infected with JEV (NT113) for 24 h. The morphological characteristics of microglia and astrocytes were assessed by immunocytochemistry with antibodies against ED-1 (microglia) and GFAP (astrocytes). The infected cells in each culture were assessed by immunocytochemistry with antibody against JEV NS3. Scale bar, 50 μm.
FIG. 5.
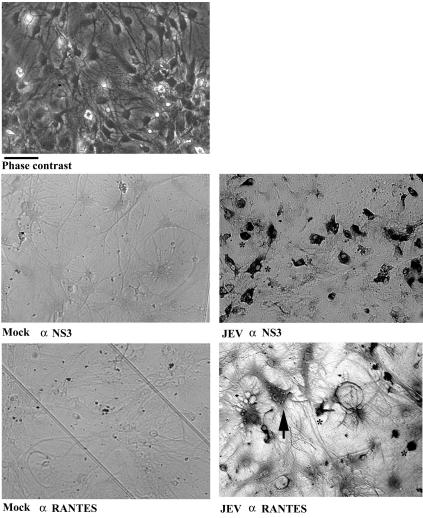
Vial protein and RANTES immunoreactivity in mixed glia. Mixed glia were mock infected or infected with JEV (NT113) for 24 h, and the immunoreactivity of viral protein and RANTES was examined by immunocytochemistry with antibodies against JEV NS3 and RANTES. JEV-infected cultures were photographed before harvest under phase contrast. The immunoreactive cells marked by arrows and asterisks are representatives of astrocytes and microglia, respectively. by their morphological characteristics. Scale bar, 50 μm.
To further determine if replication-competent virus is essential for the induction of RANTES, mixed glia cultures were stimulated with boiling-inactivated and UV-inactivated JEV. Both treatments dramatically decreased the ability of JEV to induce RANTES protein secretion (P < 0.01) and mRNA expression (Fig. 6A), suggesting that viral replication and/or productive infection is likely required for such upregulation of RANTES gene expression.
FIG. 6.
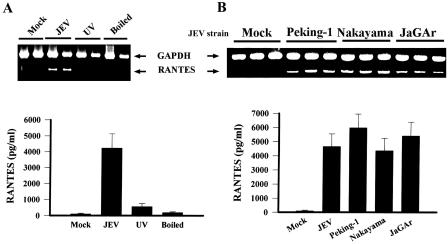
Production of RANTES depends on viral replication. Mixed glia were mock infected or infected with JEV (NT113), UV-inactivated JEV (NT113), or boiling-inactivated JEV (NT113) for 24 h. The production of RANTES and expression of RANTES mRNA were determined by enzyme-linked immunosorbent assay (n = 3) and RT-PCR, respectively (A). Mixed glia were mock infected or infected with other available JEV strains (Peking-1, Nakayama, and JaGar) for 24 h. The production of RANTES and expression of RANTES mRNA were determined by enzyme-linked immunosorbent assay (n = 3) and RT-PCR, respectively (B).
To determine whether other JEV strains share the ability to induce similar RANTES gene expression, three well-characterized JEV strains were analyzed. JEV strains Peking-1 (36), Nakayama (66), and JaGAr (47) all stimulated significant RANTES protein secretion and mRNA expression from mixed glia cultures after 24 h of infection (Fig. 6B).
Induction of RANTES production by JEV infection is inhibited by MEK inhibitor and antioxidant.
Many stimuli have been shown to elicit specific biological responses through activation of the mitogen-activated protein kinase (MAPK) signaling cascade and oxidative stress. The MAPK signaling cascade and oxidative stress also play important roles in the induction of proinflammatory molecules such as RANTES after viral infection (9, 43, 70). Activation of MAPK and the generation of free radicals were demonstrated in JEV-infected cells (18, 40, 77).
To correlate their activation and RANTES gene expression in mixed glia induced by JEV, U0126, an enzyme inhibitor of MEK/ERK (23), and the antioxidant N-acetyl-l-cysteine (NAC) (80) were used. Addition of U0126 significantly blocked JEV-induced RANTES production (P < 0.01) (Fig. 7A). MEK lies immediately upstream of ERK in the signaling cascade and is responsible for its phosphorylation and activation (23). To further characterize the involvement of the MEK/ERK signaling pathway in JEV-induced RANTES production, the level of ERK phosphorylation was assessed. A hyperphosphorylated form of ERK could be detected after 8 h of JEV infection. However, no modification in the protein level of ERK was detected (Fig. 7B). The phosphorylation of ERK was inhibited by treatment of mixed glia with U0126 (Fig. 7B). No decreased expression of JEV viral protein was detected after inhibitor treatments (Fig. 7B), indicating that the ERK signaling pathway plays an important role in JEV-induced RANTES production. This assumption was further supported by the finding that NAC also reduced JEV-induced RANTES production (P < 0.01) and ERK phosphorylation without decreasing viral amplification (Fig. 7). These findings imply that, indeed, inducible RANTES secretion after JEV infection is regulated through MEK/ERK activation.
FIG. 7.
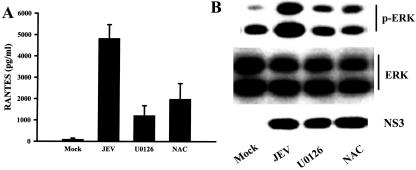
Requirement of ERK activation for RANTES expression. After mock or JEV (NT113) infection, mixed glia were cultivated in medium alone or in the presence of U0126 (10 μM) or NAC (5 mM). The level of RANTES in the supernatants (24 h postinfection) was determined by enzyme-linked immunosorbent assay (n = 4) (A). The levels of phosphorylated ERK (8 h postinfection), total ERK (8 h postinfection), and JEV NS3 protein (24 h postinfection) were determined by Western blot analysis. One of three separate experiments is shown (B).
Activation of RANTES reporter genes in JEV-infected cells.
Due to the low transfection rate in primary cells, the reporter assay was done in rat C6 glioma cells, which also responded to JEV infection by releasing RANTES (data not shown).Theluciferase reporter constructs included pGL-A (−881 to +54), pGL-B (−181 to +54), and pGL-C (−79 to +54) (43). The luciferase activity of RANTES reporter genes pGL-A (2.4-fold above that in mock-infected cells, P < 0.05) and pGL-B (4.1-fold above that in mock-infected cells P < 0.01) in the JEV-infected cells was higher than that of the mock-infected cells. In contrast, the luciferase activity in the reporter of pGL-C was hardly detected (Fig. 8).
FIG. 8.
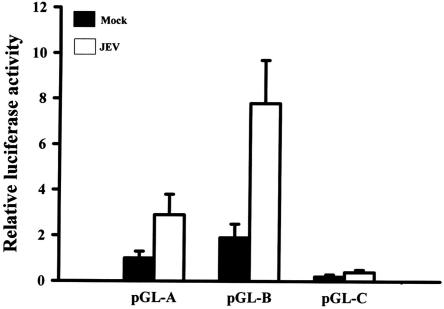
Activation of RANTES promoter activity in JEV-infected cells. C6 glioma cells were transiently cotransfected with the three RANTES promoter reporters (pGL-A, pGL-B, and pGL-C) and the Renilla luciferase reporter gene. Twenty-four hours later, cells were mock infected or infected with JEV (NT113) for another 36 h. The cells were then lysed, and the lysates were harvested. Firefly and Renilla luciferase activities were detected by the dual-luciferase reporter assay system. Firefly luciferase activity was normalized to Renilla luciferase activity. The mock-infected pGL-A group was the control (n = 3).
Formation of NF-IL-6-protein complex increases in JEV-infected cells.
We further identified the essential region in the RANTES promoter for the induction of RANTES expression by JEV infection. Since the luciferase activity induced by JEV infection was prominent in the pGL-B reporter, the region between nucleotides −181 and −79 of the RANTES promoter was first characterized. The RANTES promoter E site region contains a potential binding site for NF-IL-6 (−100 to −92) (62, 63). To determine whether activation of RANTES promoter was caused by binding of factors to this promoter region, nuclear extracts from mixed glia infected with either the control or JEV were mixed with biotin-labeled oligonucleotides of the RANTES promoter E region (nucleotides −109 to −89) and NF-IL-6 consensus binding site for the electrophoretic mobility shift assay. Intense bands representing a DNA-protein complex were observed in JEV-infected cells with both oligonucleotides, E and NF-IL-6 (Fig. 9). The DNA-protein complex formed with E was NF-IL-6 specific, because a 50-fold excess of the unlabeled E and NF-IL-6 oligonucleotides competed efficiently with complex formation, whereas oligonucleotides containing the A/B or NF-κB binding site failed to compete (Fig. 10). Furthermore, formation of the binding complex with E or NF-IL-6 decreased in the presence of U0126 and NAC (Fig. 11). These findings indicate that a close relationship exists between NF-IL-6 and RANTES gene expression after JEV infection.
FIG. 9.
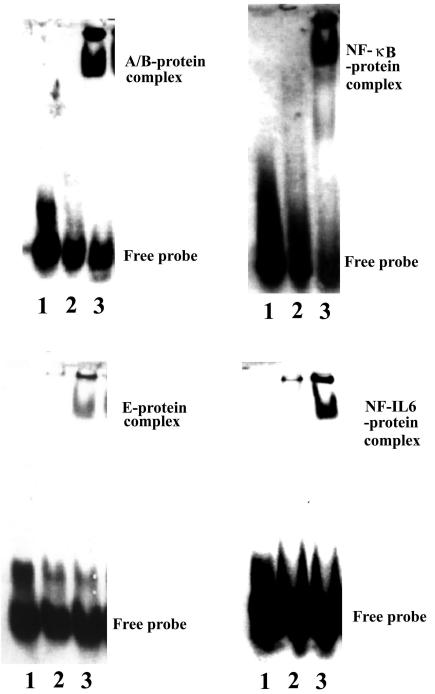
Induction of transcription factors in JEV-infected cells. Mixed glia were mock infected or infected with JEV (NT113) for 8 h. Nuclear extracts were prepared and subjected to the electrophoretic mobility shift assay with the A/B, E, NF-κB, and NF-IL-6 oligonucleotides. Lane 1, free probe only; lane 2, mock infection; lane 3, JEV infection. Similar results were obtained from two independent experiments.
FIG. 10.
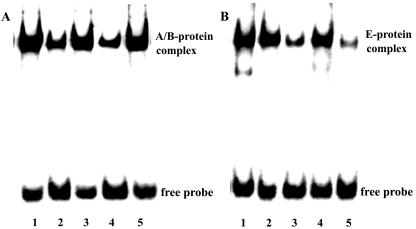
Specificity of NF-κB and NF-IL-6 binding. Mixed glia were infected with JEV (NT113) for 8 h. The isolated nuclear extracts were subjected to the electrophoretic mobility shift assay with the biotin-labeled A/B (A) or E (B) oligonucleotide. The characteristics of A/B-protein and E-protein complex were assessed by the competition assay (lanes 2 to 5) with unlabeled oligonucleotides (50-fold). Lane 1, labeled A/B or E control; lane 2, unlabeled A/B; lane 3, unlabeled E; lane 4, unlabeled NF-κB; lane 5, unlabeled NF-IL-6. Similar results were obtained from two independent experiments.
FIG. 11.
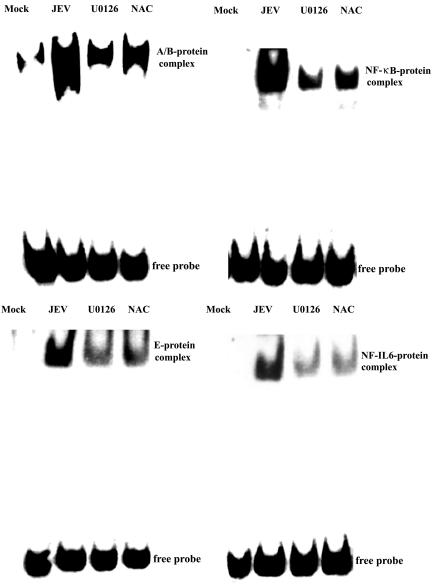
Induction of transcription factors depends on activation of ERK. After mock or JEV (NT113) infection, mixed glia were cultivated for a further 8 h in medium alone or in the presence of U0126 (10 μM) or NAC (5 mM). The isolated nuclear extracts were subjected to the electrophoretic mobility shift assay with the A/B, E, NF-κB, and NF-IL-6 oligonucleotides. Similar results were obtained from three independent experiments.
Formation of NF-κB-protein complex in JEV-infected cells.
NF-κB represents a family of dimeric transcription factors that play a central role in inflammatory responses by regulating gene expression (25). The RANTES promoter A/B site contains sequence homologous to the NF-κB binding site (nucleotides −58 to −27) (62, 63). We next examined whether JEV infection would increase the binding activity for the RANTES promoter A/B site (nucleotides −58 to −27) and NF-κB consensus binding site. JEV infection increased the formation of the RANTES promoter A/B site-protein complex and NF-κB-protein complex (Fig. 9), suggesting the involvement of the A/B site and NF-κB during JEV infection. The A/B site-protein complex was efficiently suppressed by adding the unlabeled A/B and NF-κB but not the E and NF-IL-6 oligonucleotides (Fig. 10). Similar to E and NF-IL-6, U0126 and NAC reduced the formation of each DNA-protein complex (Fig. 11). Since the luciferase activity of the pGL-C (nucleotides −79 to + 54) reporter was hardly detected (Fig. 8), the EMSA results suggested that the formation of A/B site-protein complex or NF-κB-protein complex alone was not enough to trigger RANTES gene expression.
Chemotaxis of RANTES.
RANTES is highly chemoattracted to T lymphocytes, monocytes, eosinophils, and basophils (1). Therefore, the chemotactic activity of the supernatants of mixed glia infected with JEV was evaluated on the mouse monocyte/macrophage cell line RAW264.7. As shown in Fig. 12A, supernatants infected with JEV exhibited a significantly enhanced capacity to stimulate RAW264.7 cell migration compared with the mock-infected control. During this period, JEV infection itself did not affect the migration of RAW264.7 (data not shown). The chemotactic activity of supernatants from JEV-infected cells was completely lost when the supernatants were boiled before the chemotaxis assay and reduced when RANTES neutralizing antibodies were added to the supernatants before the chemotaxis assay. The data indicate that JEV-infected mixed glia release functional RANTES to attract immune cells.
FIG. 12.

Chemotaxis of RANTES. Mixed glia were mock infected or infected with JEV (NT113) for 24 h. Supernatants isolated from JEV-infected cells were modified by treatments before chemotactic assay, including inactivation by boiling for 15 min and neutralization with RANTES and MCP-1 neutralizing antibodies (50 μg/ml) for 30 min. Chemotactic activity against RAW264.7 monocytes/macrophages was measured by Transwell assay as described in Materials and Methods. Mock infection was the control. *, P < 0.05, and **, P < 0.01 versus JEV; and ##, P < 0.01 versus the mock-infected control, n = 4 (A). Mixed glia were infected with the mock-infected control or JEV (NT113) for 24 h. Total RNAs were isolated and subjected to RT-PCR for the detection of MCP-1, Mip-1α, cytokine-induced neutrophil chemoattractant (CINC), and GAPDH mRNAs. One of three independent experiments is shown (B).
Chemokines are a group of chemotactic cytokines with numerous members. Due to the partial inhibition of RANTES antibody on chemotaxis, we speculated that other members of the chemokine group could also be produced by mixed glia after infection with JEV. As shown in representative experiments in Fig. 12B, after a 24-h infection, elevated mRNA levels of cytokine-induced neutrophil chemoattractant, MIP-1α, and MCP-1 mRNA were detected in JEV-infected mixed glia. In addition to RANTES neutralizing antibody, MCP-1 neutralizing antibody also reduced the chemotactic activity. Furthermore, the combination of both neutralizing antibodies effectively decreased the chemotactic activity (Fig. 12A). The data indicate that infection with JEV can trigger mixed glia to release several kinds of chemokines.
DISCUSSION
Glia are a major source of cytokine production and are thus critical effectors of CNS inflammation. They orchestrate immunocyte recruitment to focal areas of viral infection within the brain and synchronize immune cell functions through a regulated network of cytokines and chemokines (30, 37, 60). Activated glia are mainly scavenger cells but also perform various other functions in tissue repair and neural regeneration (24, 37, 59, 71). However, elevated inflammatory responses may be either beneficial in the healing phases by actively monitoring and controlling the extracellular environment and removing dead or damaged cells or harmful by causing the bystanders damage (7, 11, 20, 22, 39, 53, 57).
The mechanisms that control inflammatory responses within the central nervous system after viral infection are only beginning to be understood. In the present study, the susceptibility of neural cells, including neurons/glia, mixed glia, astrocytes, and microglia, to JEV infection was demonstrated by RT-PCR, Western blot analysis, and immunocytochemistry (Fig. 2C, 4, 5, and 7). We showed that neurons/glia, mixed glia, astrocytes, and microglia but not neurons produced RANTES in response to JEV infection (Fig. 1 to 3 and 5). JEV-induced RANTES production was largely mediated by the ERK (Fig. 7) and NF-IL-6 (Fig. 9 to 11) signaling pathways and inhibited by the action of NAC (Fig. 7). The released RANTES did not seem to affect neuronal viability in our assay conditions but stimulated monocyte/macrophage migration (Fig. 12).
That JEV replicates in neurons is a well-established fact. In a fatal human case, JEV antigens were localized mainly in neurons but not in the neighboring glial cells, suggesting that the principal target cells for JEV in the CNS are neurons (33). However, the infectivity of JEV for glial cells remains uncertain. Primary neurons were easily infected by JEV (≈90% infectivity) in vitro (16). Previously, we demonstrated that glial cells could be infected by JEV as evidenced by immunocytochemistry, RT-PCR, and functional changes (16, 41). In this study, JEV RNA was detected in neuron/glia cultures after infection (Fig. 2C). The generation of viral protein NS3 was measured by Western blot analysis in mixed glia after infection (Fig. 7). Immunocytochemical staining with antibody against JEV NS3 revealed the amplification of JEV in microglia (≈80% infectivity) and astrocytes (20 to 30% infectivity) in each enriched cultures (Fig. 4). In contrast, the immunoreactivity of JEV NS3 was mainly localized to microglia in JEV-infected mixed glia (Fig. 5). These findings elicit the susceptibility of each neural cell to JEV infection with variable infectivity. The preference of microglia for JEV infection in mixed glia requires further characterization. However, these in vitro data demonstrate that JEV was able to infect glial cells such as microglia and astrocytes.
Several lines of evidence suggest that JEV infection causes a breakdown of the blood-brain barrier and generates a rapid inflammatory response, including peripheral neutrophil leucocytosis and infiltration of neutrophils into extraneural tissue (12, 50, 51). Researchers have demonstrated the antiviral effect of nitric oxide against JEV (44, 81). Accumulating evidence has shown that the mortality rate increases with increasing concentrations of cytokines, including macrophage-derived chemotactic factor, TNF-α, and IL-8 in the serum and cerebrospinal fluid in Japanese encephalitis patients (35, 78, 85). However, these studies did not directly address the cellular origin of their expression or the potential mechanisms.
Despite the higher infectivity and cytotoxicity (Fig. 2), neurons were unable to release RANTES after JEV infection (Fig. 3). In this study, we have provided clear evidence that central astrocytes and microglia are capable of producing RANTES after JEV infection (Fig. 3 and 5). The prominent effector cell for JEV-induced RANTES generation was the coculture of both astrocytes and microglia (Fig. 3). To evaluate the inductive characteristics, we found that conditioned culture medium from JEV-infected microglia further stimulated RANTES production by astrocytes. In contrast, astrocyte-conditioned medium did not stimulate further expression of RANTES from microglia (Fig. 3). Furthermore, JEV NS3 was detected strongly in microglia in mixed glia after infection. However, the immunoreactivity of RANTES was localized at microglia and astrocytes in mixed glia after infection (Fig. 5). These findings suggest that soluble factors released from microglia after JEV infection might play a role in stimulating RANTES production from astrocytes. This assumption might explain the synergistic production of RANTES by mixed glia after JEV infection. The potential candidates released by microglia to stimulate RANTES production from astrocytes have not yet been fully identified. TNF-α and IL-1β have been shown to trigger astrocytes to upregulate chemokine expression (31, 73). Therefore, the TNF-α and IL-1β released after JEV infection could function to stimulate astrocytes to release RANTES.
The activation of glia by viral infection could lead to the restriction of viral spreading, whereas the consequence is frequently CNS dysfunction and/or destruction (6, 13, 28, 55, 68, 69). Elevated concentrations of chemokines may be partially responsible for neuronal death in various diseases, including viral infection (2). In this in vitro study, we found that infection of neuron/glia cultures with JEV triggered RANTES expression in a time-dependent manner (Fig. 1). The neurotoxic action of JEV (Fig. 2) was accompanied by increased production of RANTES (Fig. 1) and viral amplification (Fig. 2) in neuron/glia cultures after JEV infection, suggesting a potential link between RANTES and JEV-induced neuronal damage. However, the evidence of their association appears to be limited because RANTES neutralizing antibody was unable to inhibit JEV-induced neuronal damage (data not shown), and JEV directly caused neuronal death in neuron cultures without the production of RANTES (Fig. 1 to 3).
The chemokines expressed within tissues are probably the major mediators of the accumulation of several inflammatory cells at sites of inflammation (3, 82, 90). Besides chemotactic activity, chemokines also have mitogenic and activating properties for particular cell types, which make them likely amplifiers of the immune response in many pathological conditions (46, 61, 84). Infection of some viruses has been shown to induce RANTES expression in a wide variety of cells, which mostly contributes to the development of inflammatory processes (9, 16, 43, 52, 56, 88). Lokensgard et al. (45) reported that astrocytes produced chemokines that induced chemotaxis of microglia and other cells of the monocyte/macrophage lineage, in response to human cytomegalovirus infection. In this study, we found that infection of glial cells with JEV produced RANTES and other chemokines with chemoattracting activity (Fig. 12). Therefore, RANTES expressed on glial cells after JEV infection might function as an important cellular signal to attract inflammatory cell infiltration and amplify immune responses.
Many stimuli have been shown to elicit specific biological responses through activation of the MAPK signaling cascade and oxidative stress. It has been reported that the MAPK signaling cascade and oxidative stress also play important roles in the induction of proinflammatory cytokine expression and viral replication after viral infection (6, 9, 43, 70). Evidence has demonstrated that JEV infection stimulated MAPK activation and generated free radicals in infected cells (18, 40, 77, 87). U0126 and NAC reduced both JEV-induced RANTES production and ERK phosphorylation (Fig. 7). Moreover, U0126 and NAC further decreased the DNA binding activity of NF-IL-6 and NF-κB after JEV infection (Fig. 11). These findings indicate that MAPK and free radicals are involved in JEV-induced RANTES expression and that MAPK lies upstream of the activation of NF-κB. In contrast, we found that treatment of infected cells with U0126 or NAC was unable to reduce viral protein synthesis (Fig. 7). Consistent with previous reports, MAPK inhibitors and antioxidants could not inhibit JEV replication (18, 40, 77). Therefore, we suggest that MAPK inhibitors and antioxidants suppressed the signaling pathways leading to RANTES expression rather than inhibited JEV replication. Because of the rare and inconsistent detection of p38 protein kinase and c-Jun N-terminal kinase in JEV-infected glial cells, the contribution of these MAPKs to JEV-induced RANTES expression was not addressed in this study.
In this study, we showed that JEV-mediated upregulation of RANTES production occurred through transcriptional activation because of the induction of mRNA expression (Fig. 1) and promoter activity (Fig. 8). Our data demonstrated that the RANTES promoter region from nucleotides −181 to +54 played an essential role in the activation of RANTES gene expression after JEV infection (Fig. 8). This region contained putative NF-κB and NF-IL-6 binding sites, termed the A/B and E regions, respectively (67). Our data clearly showed a relationship between NF-IL-6 and JEV-induced RANTES expression. The formation of E-protein and NF-IL-6-protein complexes was activated in JEV-infected cells, and the activation was antagonized by nonlabeled NF-IL-6 (Fig. 9 and 10).
The importance of the E region and NF-IL-6 in JEV-induced RANTES expression was also supported by the following two findings. RANTES production (Fig. 7) and formation of the NF-IL-6-protein complex (Fig. 11) were suppressed simultaneously by U0126 and NAC. Furthermore, induction of RANTES promoter activity was lost after deletion of the region containing the NF-IL-6 binding sequence (Fig. 8). It has been reported that NF-κB binds to the A/B region of the RANTES promoter and is involved in activation of the RANTES promoter (62, 63, 67, 79). Our data showed that JEV infection activated the specific NF-κB-protein binding activity (Fig. 9 and 10). Similar to that of NF-IL-6, inactivation of NF-κB-protein binding activity was strongly associated with the reduction in RANTES production (Fig. 7 and 11). However, JEV failed to stimulate RANTES promoter activity when it contained only the A/B region (Fig. 8).
It has become increasingly evident that the combinatory effects of transcription factors are very important in gene expression (19, 49). For example, NF-IL-6 together with NF-κB could induce IL-8 expression in Rous sarcoma virus-infected epithelial cells (49) and inducible nitric oxide synthase expression in endotoxin- or cytokine-stimulated microglia (19). Furthermore, evidence has been provided for a function and physical interaction between NF-IL-6 and NF-κB through cooperative binding of the two transcription factors or specific interactions between these proteins and the basal transcriptional machinery (5, 54, 64). Similarly, we found that NF-IL-6 and NF-κB were activated to bind the RANTES promoter within E and A/B sites in JEV-infected glial cells (Fig. 9 and 10). The binding of NF-κB to the A/B site alone was not sufficient to induce RANTES expression. Therefore, we suggest that induction of RANTES expression by JEV infection in glial cells needs the coordinate activation of at least two factors, including NF-IL-6 and NF-κB.
It is conceivable that the activation signal may be induced via viral receptor engagement or viral replication leading to gene expression. UV- and heat-inactivated JEV failed to activate ERK, NF-IL-6, and NF-κB (data not shown) and induce RANTES expression (Fig. 6), suggesting a requirement for viral amplification for the expression of RANTES. Both NF-κB and NF-IL-6 were activated in many different viral infections and played crucial roles in viral replication and triggering of apoptotic cell death (9, 16, 25, 42, 43, 48, 49, 52, 56, 79, 88). However, we found that their inactivation by both U0126 and NAC did not appear to affect viral replication (Fig. 7). Therefore, the their activation was not required for JEV replication but instead was essential for JEV-induced inflammatory gene expression. This concept was supported by previous studies which demonstrated that several approaches to blocking NF-κB activation failed to influence JEV production (18, 40, 77).
In conclusion, the results of this study and of other reports provide evidence that glial cells might be involved in JEV-induced inflammatory responses by releasing chemokines. Both astrocytes and microglia respond to JEV infection by releasing RANTES through a process related to viral replication. Stimulation of MAPK signaling pathways after JEV infection leads to upregulation of RANTES expression in glial cells through activating NF-κB and NF-IL-6 to bind to the promoter. These two transcription factors cooperate to effect RANTES expression. Our study provides the signaling pathway for RANTES expression in JEV-infected glial cells, which will shed light on the regulation of brain inflammation in viral infection.
Acknowledgments
This work was supported by grants TCVGH-937312C from Taichung Veterans General Hospital and NSC-88-2314-B-075A-020 from the National Science Council, Taiwan, Republic of China.
REFERENCES
- 1.Alam, R., S. Stafford, P. Forsythe, R. Harrison, D. Faubion, M. A. Lett-Brown, and J. A. Grant. 1993. RANTES is a chemotactic and activating factor for human eosinophils. J. Immunol. 150:3442-3448. [PubMed] [Google Scholar]
- 2.Asensio, V. C., and I. L. Campbell. 1997. Chemokine gene expression in the brains of mice with lymphocytic choriomeningitis. J. Virol. 71:7832-7840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Asensio, V. C., and I. L. Campbell. 1999. Chemokines in the CNS: plurifunctional mediators of diverse states. Trends Neurosci. 22:504-512. [DOI] [PubMed] [Google Scholar]
- 4.Bakhiet, M., A. Gad, S. Stromblad, W. A. Kuziel, A. Seiger, and J. Anderson. 2001. RANTES promotes growth and survival of human first trimester forebrain astrocytes. Nat. Cell Biol. 3:150-157. [DOI] [PubMed] [Google Scholar]
- 5.Baldwin, Jr. A. S. 1996. The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu. Rev. Immunol. 14:649-683. [DOI] [PubMed] [Google Scholar]
- 6.Barber, S. A., L. Bruett, B. R. Douglass, D. S. Herbst, M. C. Zink, and J. E. Clements. 2002. Visna virus-induced activation of mitogen-activated protein kinase is required for virus replication and correlates with virus-induced neuropathology. J. Virol. 76:817-828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brosnan, C. F., L. Battistini, C. S. Raine, D. W. Dickson, A. Casadevall, and S. C. Lee. 1994. Reactive nitrogen intermediates in human neuropathology: an overview. Dev. Neurosci. 16:152-161. [DOI] [PubMed] [Google Scholar]
- 8.Byrne, J. A., and M. B. A. Dolstone. 1984. Biology of cloned cytotoxic T lymphocytes specific for lymphocytic choriomeningitis virus: clearance of virus in vivo. J. Virol. 51:682-686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Casola, A., N. Burger, T. Liu, M. Jamaluddin, A. R. Brasier, and R. P. Garofalo. 2001. Oxidant tone regulates RANTES gene expression in airway epithelial cells infected with respiratory syncytial virus. J. Biol. Chem. 276:19715-19722. [DOI] [PubMed] [Google Scholar]
- 10.Chambers, T. J., C. S. Hahn, R. Galler, and C. M. Rice. 1990. Flavivirus genome organization, expression, and replication. Annu. Rev. Microbiol. 44:649-688. [DOI] [PubMed] [Google Scholar]
- 11.Chao, C. C., S. Hu, T. W. Moliter, E. G. Shaskan, and P. K. Peterson. 1992. Activated microglia mediate neuronal cell injury via a nitric oxide mechanism. J. Immunol. 149:2736-2741. [PubMed] [Google Scholar]
- 12.Chaturvedi, U. C., A. Mathur, P. Tandon, S. M. Natu, S. Rajvanshi, and H. O. Tandon. 1979. Variable effect on peripheral blood leucocytes during JE virus infection of man. Clin. Exp. Immunol. 38:492-498. [PMC free article] [PubMed] [Google Scholar]
- 13.Cheeran, M. C., S. Hu, S. L. Yager, G. Gekker, P. K. Peterson, and J. R. Lokensgard. 2001. Cytomegalovirus induces cytokine and chemokine production differentially in microglia and astrocytes: antiviral implications. J. Neurovirol. 7:135-147. [DOI] [PubMed] [Google Scholar]
- 14.Chen, C. J., and S. L. Liao. 2003. Zinc toxicity on neonatal cortical neurons: involvement of glutathione depletion. J. Neurochem. 85:443-453. [DOI] [PubMed] [Google Scholar]
- 15.Chen, C. J., M. D. Kuo, L. J. Chien, S. L. Hsu, Y. M. Wang, and J. H. Lin. 1997. RNA-protein interactions: involvement of NS3, NS5, and 3′ noncoding regions of Japanese encephalitis virus genomic RNA. J. Virol. 71:3466-3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen, C. J., S. L. Liao, M. D. Kuo, and Y. M. Wang. 2000. Astrocytic alteration induced by Japanese encephalitis virus infection. Neuroreport 11:1933-1937. [DOI] [PubMed] [Google Scholar]
- 17.Chen, C. J., S. L. Liao, W. Y. Chen, J. S. Hong, and J. S. Kuo. 2001. Cerebral ischemia/reperfusion injury in rat brain: effects of naloxone. Neuroreport 12:1245-1249. [DOI] [PubMed] [Google Scholar]
- 18.Chen, C. J., S. L. Raung, M. D. Kuo, and Y. M. Wang. 2002. Suppression of Japanese encephalitis virus infection by non-steroidal anti-inflammatory drugs. J. Gen. Virol. 83:1897-1905. [DOI] [PubMed] [Google Scholar]
- 19.Chen, C. J., S. L. Raung, S. L. Liao, and S. Y. Chen. 2004. Inhibition of inducible nitric oxide synthase expression by baicalein in endotoxin/cytokine-stimulated microglia. Biochem. Pharmacol. 67:957-965. [DOI] [PubMed] [Google Scholar]
- 20.Dickson, D. W., S. C. Lee, L. A. Mattiace, S. H. Yen, and C. Brosnan. 1993. Microglia and cytokines in neurological disease, with special reference to AIDS and Alzheimer's disease. Glia 7:75-83. [DOI] [PubMed] [Google Scholar]
- 21.Doherty, P. C., M. B. C. Dunlop, C. R. Parish, and R. M. Zinkernagel. 1976. Inflammatory process in murine lymphocytic choriomeningitis is maximal in H-2K or H-2D compatible interactions. J. Immunol. 117:187-190. [PubMed] [Google Scholar]
- 22.Espey, M. G., O. N. Chernyshev, Jr., J. F. Reinhard, M. A. Namboodiri, and C. A. Colton. 1997. Activated human microglia produce the excitotoxin quinolinic acid. NeuroReport 8:431-434. [DOI] [PubMed] [Google Scholar]
- 23.Favata, M. F., K. Y. Horiuchi, E. J. Manos, A. J. Daulerio, D. A. Stradley, W. S. Feeser, D. E. Van-Dyk, W. J. Pitts, R. A. Earl, F. Hobbs, R. A. Copeland, R. L. Magolda, P. A. Scherle, and J. M. Trzaskos. 1998. Identification of a novel inhibitor of mitogen-activated protein kinase. J. Biol. Chem. 273:18623-18632. [DOI] [PubMed] [Google Scholar]
- 24.Gehrmann, J., Y. Matsumoto, and G. W. Kreutzberg. 1995. Microglia: intrinsic immunoeffector cell of the brain. Brain Res. Rev. 20:269-287. [DOI] [PubMed] [Google Scholar]
- 25.Ghosh, S., M. J. May, and E. B. Kopp. 1998. NF-κB and Rel proteins: evolutionarily conserved mediators of immune responses. Annu. Rev. Immunol. 16:225-260. [DOI] [PubMed] [Google Scholar]
- 26.Giulian, D., K. Vaca, and C. A. Noonan. 1990. Secretion of neurotoxin by mononuclear phagocytes infected with HIV-1. Science 250:1593-1596. [DOI] [PubMed] [Google Scholar]
- 27.Grant, I., and J. H. Atkinson. 1990. Neurogenic and psychogenic behavior correlates of HIV infection, p. 291-304. In B. H. Waksman (ed.), Immunological mechanisms in neurologic and psychiatric disease. Raven Press, New York, N.Y.
- 28.Hein, A., J. P. Martin, F. Koehren, A. Bingen, and R. Dorries. 2000. In vivo infection of ramified microglia from adult cat central nervous system by feline immunodeficiency virus. Virology 268:420-429. [DOI] [PubMed] [Google Scholar]
- 29.Heyes, M. P., B. J. Brew, A. Martin, and R. W. Price. 1991. Quinolinic acid in cerebrospinal fluid and serum in HIV-1 infection: relationship to clinical and neurological status. Ann. Neurol. 29:202-209. [DOI] [PubMed] [Google Scholar]
- 30.Huang, D., Y. Han, M. R. Rani, A. Glabinski, C. Trebst, T. Sorensen, M. Tani, J. Wang, P. Chien, S. O'Bryan, B. Bielecki, Z. L. Zhou, S. Majumder, and R. M. Ransohoff. 2000. Chemokines and chemokine receptors in inflammation of the nervous system: manifold roles and exquisite regulation. Immunol. Rev. 177:52-67. [DOI] [PubMed] [Google Scholar]
- 31.Hurwitz, A. A., W. D. Lyman, and J. W. Berman. 1995. Tumor necrosis factor alpha and transforming growth factor beta upregulate astrocyte expression of monocyte chemoattractant protein-1. J. Neuroimmunol. 57:193-198. [DOI] [PubMed] [Google Scholar]
- 32.Ishiguro, A., Y. Suzuki, Y. Inaba, K. Fukushima, A. Komiyama, H. P. Koeffler, and T. Shimbo. 1997. The production of IL-8 in cerebrospinal fluid in aseptic meningitis of children. Clin. Exp. Immunol. 109:426-430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnson, R. T., D. S. Burke, M. Elwell, C. J. Leake, A. Nisalak, C. H. Hoke, and W. Lorsomrudee. 1985. Japanese encephalitis: immunocytochemical studies of viral antigen and inflammatory cells in fatal cases. Ann. Neurol. 18:567-573. [DOI] [PubMed] [Google Scholar]
- 34.Karin, M. 1999. The binding of the end: IκB kinase (IKK) and NF-κB activation. J. Biol. Chem. 274:27339-27342. [DOI] [PubMed] [Google Scholar]
- 35.Khanna, N., M. Agnihotri, A. Mathur, and U. C. Chaturvedi. 1991. Neutrophil chemotactic factor produced by Japanese encephalitis virus stimulated macrophages. Clin. Exp. Immunol. 86:299-303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kitaoka, M., and S. Y. Gaidamovich. 1977. Non-infectious antigen for laboratory diagnosis of Japanese encephalitis. Acta Virol. 21:326-330. [PubMed] [Google Scholar]
- 37.Kreutzberg, G. W. 1996. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 19:312-318. [DOI] [PubMed] [Google Scholar]
- 38.Kumar, R., A. Mathur, A. Kumar, G. D. Sethi, S. Sharma, and U. C. Chaturvedi. 1990. Virological investigations of acute encephalopathy in India. Arch. Dis. Child. 65:1227-1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee, S. C., W. Liu, D. W. Dickson, C. F. Brosnan, and J. W. Berman. 1993. Cytokine production by human fetal microglia and astrocytes: differential induction by lipopolysaccharide and IL-1β. J. Immunol. 150:2659-2667. [PubMed] [Google Scholar]
- 40.Liao, C. L., Y. L. Lin, B. C. Wu, C. H. Tsao, M. C. Wang, C. I. Liu, Y. L. Huang, J. H. Chen, J. P. Wang, and L. K. Chen. 2001. Salicylates inhibit flavivirus replication independently of blocking nuclear factor kappa B activation. J. Virol. 75:7828-7839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liao, S. L., S. L. Raung, and C. J. Chen. 2002. Japanese encephalitis virus stimulates superoxide dismutase activity in rat glial cultures. Neurosci. Lett. 324:133-136. [DOI] [PubMed] [Google Scholar]
- 42.Lin, K. I., S. H. Lee, R. Narayanan, J. M. Baraban, J. M. Hardwick, and R. R. Ratan. 1995. Thiol agents and Bcl-2 identify an alphavirus-induced apoptotic pathway that requires activation of the transcription factor NF-κB. J. Cell Biol. 131:1149-1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lin, Y. L., C. C. Liu, J. I. Chuang, H. Y. Lei, T. M. Yeh, Y. S. Lin, Y. H. Huang, and H. S. Liu. 2000. Involvement of oxidative stress, NF-IL-6, and RANTES expression in Dengue-2-virus-infected human liver cells. Virology 276:114-126. [DOI] [PubMed] [Google Scholar]
- 44.Lin, Y. L., Y. L. Huang, S. H. Ma, C. T. Yeh, S. Y. Chiou, L. K. Chen, and C. L. Liao. 1997. Inhibition of Japanese encephalitis virus infection by nitric oxide: antiviral effect of nitric oxide on RNA virus replication. J. Virol. 71:5227-5235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lokensgard, J. R., M. C. Cheeran, G. Gekker, S. Hu, C. C. Chao, and P. K. Peterson. 1999. Human cytomegalovirus replication and modulation of apoptosis in astrocytes. J. Hum. Virol. 2:91-101. [PubMed] [Google Scholar]
- 46.Luster, A. D. 1998. Chemokines-chemotactic cytokines that mediate inflammation. N. Engl. J. Med. 338:436-445. [DOI] [PubMed] [Google Scholar]
- 47.Mangada, M. N., and T. Takegami. 1999. Molecular characterization of the Japanese encephalitis virus representative immunotype strain JaGAr 01. Virus Res. 59:101-112. [DOI] [PubMed] [Google Scholar]
- 48.Marianneau, P., A. Cardona, L. Edelman, V. Deubel, and P. Despres. 1997. Dengue virus replication in human hepatoma cells activates NF-κB, which in turn induces apoptotic cell death. J. Virol. 71:3244-3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mastronarde, J. G., M. M. Monick, N. Mukaida, K. Matsushima, and G. W. Hunninghake. 1996. Induction of interleukin (IL)-8 gene expression by respiratory syncytial virus involves activation of nuclear factor (NF)-κB and NF-IL-6. J. Infect. Dis. 174:262-267. [DOI] [PubMed] [Google Scholar]
- 50.Mathur, A., M. Bharadwaj, R. Kulshreshtha, S. Rawat, A. Jain, and U. C. Chaturvedi. 1988. Immunopathological study of spleen during Japanese encephalitis virus infection in mice. Br. J. Exp. Pathol. 69:423-432. [PMC free article] [PubMed] [Google Scholar]
- 51.Mathur, A., N. Khanna, and U. C. Chaturvedi. 1992. Breakdown of blood-brain barrier by virus-induced cytokine during Japanese encephalitis virus infection. Int. J. Exp. Pathol. 73:603-611. [PMC free article] [PubMed] [Google Scholar]
- 52.Matsukura, S., F. Kokubu, H. Kubo, T. Tomita, H. Tokunaga, M. Kaddkura, T. Yamamoto, Y. Kuroiwa, T. Ohno, H. Suzaki, and M. Adacni. 1998. Expression of RANTES by normal airway epithelial cells after influenza virus A infection. Am. J. Respir. Cell Mol. Biol. 18:255-264. [DOI] [PubMed] [Google Scholar]
- 53.Matsuo, M., Y. Hamasaki, F. Fujiyama, and S. Miyazaki. 1995. Eicosanoids are produced by microglia, not by astrocytes, in rat glial cultures. Brain Res. 685:201-204. [DOI] [PubMed] [Google Scholar]
- 54.Matsusaka, T. K. Fujikawa, Y. Nishio, N. Mukaida, K. Matsushima, T. Kishimoto, and S. Akira. 1993. Transcription factors NF-IL6 and NF-kappa B synergistically activate transcription of the inflammatory cytokines, interleukin 6 and interleukin 8. Proc. Natl. Acad. Sci. USA 90:10193-10197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Meeker, R. B., Y. Azuma, D. C. Bragg, R. V. English, and M. Tompkins. 1999. Microglial proliferation in cortical neural cultures exposed to feline immunodeficiency virus. J. Neuroimmunol. 101:15-26. [DOI] [PubMed] [Google Scholar]
- 56.Melchjorsen, J., and S. R. Paludan. 2003. Induction of RANTES/CCL5 by herpes simplex virus is regulated by nuclear factor kappa B and interferon regulatory factor 3. J. Gen. Virol. 84:2491-2495. [DOI] [PubMed] [Google Scholar]
- 57.Minghetti, L., and G. Levi. 1995. Induction of prostanoid biosynthesis by bacterial lipopolysaccharide and isoproterenol in rat microglial cultures. J. Neurochem. 65:2690-2698. [DOI] [PubMed] [Google Scholar]
- 58.Monath, T. P. 1990. Flaviviruses, p. 763-814. In B. N. Field and D. M. Knipe (ed.), Virology. Raven, New York, N.Y.
- 59.Moore, S., and S. Thanos. 1996. The concept of microglia in relation to central nervous system disease and regeneration. Prog. Neurobiol. 48:441-460. [DOI] [PubMed] [Google Scholar]
- 60.Mucke, L., and M. Eddleston. 1993. Astrocytes in infectious and immune-mediated diseases of the central nervous system. FASEB J. 7:1226-1232. [DOI] [PubMed] [Google Scholar]
- 61.Murdoch, C., and A. Finn. 2000. Chemokine receptors and their role in inflammation and infectious diseases. Blood 95:3032-3043. [PubMed] [Google Scholar]
- 62.Nelson, P. J., B. D. Ortiz, J. M. Pattison, and A. M. Krensky. 1996. Identification of a novel regulatory region critical for expression of the RANTES chemokine in activated T lymphocytes. J. Immunol. 157:1139-1148. [PubMed] [Google Scholar]
- 63.Nelson, P. J., H. T. Kim, W. C. Manning, T. J. Goralski, and A. M. Krensky. 1993. Genomic organization and transcriptional regulation of the RANTES chemokine gene. J. Immunol. 151:2601-2611. [PubMed] [Google Scholar]
- 64.Nerlov, C., and E. B. Ziff. 1995. CCAAT/enhancer binding protein-alpha amino acid motifs with dual TBP and TFIIB binding ability co-operate to activate transcription in both yeast and mammalian cells. EMBO J. 14:4318-4328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ödemis, V., B. Moepps, P. Gierschik, and J. Engele. 2002. Interleukin-6 and cAMP induce stromal cell-derived factor-1 chemotaxis in astroglia by up-regulating CXCR4 cell surface expression: implications for brain inflammation. J. Biol. Chem. 277:39801-39808. [DOI] [PubMed] [Google Scholar]
- 66.Okuno, Y., Y. Okamoto, A. Yamada, K. Baba, and H. Yabuuchi. 1987. Effect of current Japanese encephalitis vaccine on different strains of Japanese encephalitis virus. Vaccine 5:128-132. [DOI] [PubMed] [Google Scholar]
- 67.Ortiz, B. D., P. J. Nelson, and A. M. Krensky. 1997. Awitching gears during T-cell maturation RANTES and late transcription. Immunol. Today 18:468-471. [DOI] [PubMed] [Google Scholar]
- 68.Palma, J. P., and B. S. Kim. 2001. Induction of selective chemokines in glial cells infected with Theiler's virus. J. Neuroimmunol. 117:166-170. [DOI] [PubMed] [Google Scholar]
- 69.Palma, J. P., D. Kwon, N. A. Clipstone, and B. S. Kim. 2003. Infection with Theiler's murine encephalitis virus directly induces proinflammatory cytokines in primary astrocytes via NF-κB activation: potential role for the initiation of demyelinating disease. J. Virol. 77:6322-6331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pazdrak, K., B. Olszewska-Pazdrak, T. Liu, R. Takizawa, A. R. Brasier, R. P. Garofalo, and A. Casola. 2002. MAPK activation is involved in posttranscriptional regulation of RSV-induced RANTES gene expression. Am. J. Physiol. Lung Cell Mol. Physiol. 283:L364-L372. [DOI] [PubMed] [Google Scholar]
- 71.Perry, T., and S. Gordon. 1988. Macrophages and microglia in the nervous system. Trends Neurosci. 11:273-277. [DOI] [PubMed] [Google Scholar]
- 72.Persidsky, Y. 1999. Model systems for studies of leukocyte migration across the blood-brain barrier. J. Neurovirol. 5:579-590. [DOI] [PubMed] [Google Scholar]
- 73.Peterson, P. K., S. Hu, J. Salak-Johnson, T. W. Molitor, and C. C. Chao. 1997. Differential production of and migratory response to beta chemokines by human microglia and astrocytes. J. Infect. Dis. 175:478-481. [DOI] [PubMed] [Google Scholar]
- 74.Poli, V., F. P. Mancini, and R. Cortese. 1990. IL-6-DBP, a nuclear protein involved in interleukin-6 signal transduction, defines a new family of leucine zipper proteins related C/EBP. Cell 63:643-653. [DOI] [PubMed] [Google Scholar]
- 75.Price, R. W., B. J. Brew, and M. Rosenblum. 1990. The AIDS dementia complex and HIV-1 brain infection: a pathogenetic model of virus-immune interaction, p. 269-290. In B. H. Waksman (ed.), Immunological mechanisms in neurologic and psychiatric disease. Raven Press, New York, N.Y. [PubMed]
- 76.Quagliarello, V. J., B. Wispelwey, W. J. Long, and W. M. Scheid. 1991. Recombinant human interleukin-1 induces meningitis and blood-brain barrier injury in the rat. J. Clin. Investig. 87:1360-1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Raung, S. L., M. D. Kuo, Y. M. Wang, and C. J. Chen. 2001. Role of reactive oxygen intermediates in Japanese encephalitis virus infection in murine neuroblastoma cells. Neurosci. Lett. 315:9-12. [DOI] [PubMed] [Google Scholar]
- 78.Ravi, V., S. Parida, A. Desai, A. Chandramuki, M. Gourie-Devi, and G. E. Grau. 1997. Correlation of tumor necrosis factor levels in the serum and cerebrospinal fluid with clinical outcome in Japanese encephalitis patients. J. Med. Virol. 51:132-136. [PubMed] [Google Scholar]
- 79.Ray, P., L. Yang, D. H. Zhang, S. K. Ghosh, and A. Ray. 1997. Selective up-regulation of cytokine-induced RANTES gene expression in lung epithelial cells by overexpression of IκBR. J. Biol. Chem. 272:20191-20197. [DOI] [PubMed] [Google Scholar]
- 80.Sato, M., T. Miyazaki, T. Nagaya, Y. Murata, N. Ida, K. Maeda, and H. Seo. 1996. Antioxidants inhibit tumor necrosis factor-alpha mediated stimulation of interleukin-8, monocyte chemoattractant protein-1, and collagenase expression in cultured human synovial cells. J. Rheumatol. 23:432-438. [PubMed] [Google Scholar]
- 81.Saxena, S. K., A. Mathur, and R. C. Srivastava. 2001. Induction of nitric oxide synthase during Japanese encephalitis virus infection: evidence of protective role. Arch. Biochem. Biophys. 391:1-7. [DOI] [PubMed] [Google Scholar]
- 82.Schall, T. J., and K. B. Bacon. 1994. Chemokines, leukocyte trafficking, and inflammation. Curr. Opin. Immunol. 6:865-873. [DOI] [PubMed] [Google Scholar]
- 83.Schall, T. J., J. B. Jongstra, J. Dyer, J. Jorgensen, C. Clayberger, M. M. Davis, and A. M. Krensky. 1988. A human T cell-specific molecule is a member of a new gene family. J. Immunol. 141:1018-1025. [PubMed] [Google Scholar]
- 84.Segerer, S., P. J. Nelson, and D. Schlondorff. 2000. Chemokines, chemokine receptors, and renal disease: from basic science to pathophysiologic and therapeutic studies. J. Am. Soc. Nephrol. 11:152-176. [DOI] [PubMed] [Google Scholar]
- 85.Singh, A., R. Kulshreshtha, and A. Mathur. 2000. Secretion of the chemokine interleukin-8 during Japanese encephalitis virus infection. J. Med. Virol. 49:607-612. [DOI] [PubMed] [Google Scholar]
- 86.Stone, T. W., and J. H. Connick. 1985. Quinolinic acid and other kynurenines in the central nervous system. Neuroscience 15:597-617. [DOI] [PubMed] [Google Scholar]
- 87.Su, H. L., C. L. Liao, and Y. L. Lin. 2002. Japanese encephalitis virus infection initiates endoplasmic reticulum stress and an unfolded protein response. J. Virol. 76:4162-4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Waterman, P. M., M. Kitabwalla, I. Tikhonov, and C. D. Pauza. 2003. Simian/human immunodeficiency virus (89.6) expressing the chemokine genes MIP-1alpha, RANTES, or lymphotactin. Viral Immunol. 16:35-44. [DOI] [PubMed] [Google Scholar]
- 89.Wolinsky, J. S. 1990. Subacute sclerosing panencephalitis progressive rubella panencephalitis, and multifocal leukoencephalopathy, p. 259-268. In B. H. Waksman (ed.), Immunological mechanisms in neurologic and psychiatric disease. Raven Press, New York, N.Y. [PubMed]
- 90.Zlotnik, A., and O. Yoshie. 2000. Chemokines: a new classification system and their role in immunity. Immunity 12:121-127. [DOI] [PubMed] [Google Scholar]