

Abstract
The oncogenic potential of latent Epstein-Barr virus (EBV) can be regulated by epigenetic factors controlling LMP1 and EBNA2 gene transcription. The EBV latency control region (LCR) constitutes ∼12 kb of viral sequence spanning the divergent promoters of LMP1 and EBNA2 and encompasses the EBV latent replication origin OriP and RNA polymerase III-transcribed EBV-encoded RNA genes. We have used the chromatin immunoprecipitation assay to examine the chromatin architecture of the LCR in different types of EBV latency programs. We have found that histone H3 K4 methylation (H3mK4) was enriched throughout a large domain that extended from internal repeat 1 (IR1) to the terminal repeat in type III latency where EBNA2 and LMP1 genes are expressed. In type I latency where EBNA2 and LMP1 genes are transcriptionally silent, the H3mK4 domain contracts and does not enter the EBNA2 or LMP1 promoters. In contrast, histone H3 K9 methylation (H3mK9), associated with silent heterochromatin, was enriched in the EBNA2 and LMP1 upstream control regions in type I but not type III cells. MTA [5′-deoxy-5′(methylthio)adenosine], a pharmacological inhibitor of protein methylation, globally reduced histone H3mK4 and inhibited EBNA2 transcription in type III cells. 5′-Azacytidine, an inhibitor of DNA methylation that derepresses EBNA2 transcription in type I latency, caused H3mK4 expansion and a corresponding loss of H3mK9 at IR1. The chromatin boundary protein and transcription repressor CCCTC-binding factor was enriched at the EBNA2 transcription control region in type I but not type III cells. We also present evidence that OriP binding factors EBNA1 and ORC2 can interact with sequences outside of OriP including a region within IR1 that may influence EBNA2 transcription status. These results indicate that types I and III latency programs have distinct histone methylation patterns in the LCR and suggest that chromatin architecture coordinates gene expression of LMP1 and EBNA2.
Epstein-Barr virus (EBV) is a human gammaherpesvirus that is causally associated with endemic forms of Burkitt's lymphoma (BL), nasopharyngeal carcinoma, and lymphoproliferative disease in immunosuppressed individuals (reviewed in reference 29 and 49). EBV efficiently immortalizes primary B lymphocytes in cell culture and can establish latent infections in human B lymphocytes and epithelial cells in vivo. During latent infection the viral genome persists as a chromatin-associated multicopy episome that expresses a limited set of viral genes. Distinct latency transcription patterns have been correlated with different cell types and viral pathogenesis, but it remains unclear what regulates these different gene expression patterns (53, 54, 68).
EBNA2 and LMP1 are essential for EBV-induced cellular proliferation and immortalization of primary B lymphocytes (9, 25). However, their expression can be down-regulated during latency. At least four different gene expression patterns have been characterized for various forms of EBV latency found in different tumors or host cell types (5, 6, 53). For example, EBV immortalized lymphoblastoid cell lines (LCLs) express the full panoply of viral oncogenes in a transcription program referred to as type III latency (54). In contrast, viral latency in healthy donors or in most BL-derived cell lines expresses a more restricted transcription program referred to as type I, where EBNA2 and LMP1 transcription are repressed. EBNA2 is a transcriptional regulatory protein that binds to the LMP1 and EBNA2 promoters and enforces the type III latency program (1, 17, 20, 22, 71, 78). The regulation of EBNA2 expression is, therefore, critical in determining the latency program and the proliferative capacity of EBV-infected cells.
The transcriptional control of EBNA2 is complex (22, 41, 42, 69, 70, 75, 76). During the establishment of EBV latency, EBNA2 transcription initiates at a promoter in internal repeat 1 (IR1) referred to as Wp. Once EBNA2 protein levels reach a threshold, the protein activates a second promoter upstream of Wp, referred to as Cp. EBNA2 stimulates the Cp and LMP1 promoters through a cellular DNA binding protein called CSL (CBF1 or RBP-Jk) (20, 22, 35, 77). CSL can also interact with cellular coactivators, like intracellular Notch, and corepressors, like CIR, that may regulate EBV latency transcripts in the absence of EBNA2 (11, 23, 24, 77).
Epigenetic events, like DNA methylation and histone modifications, constitute another level of regulatory mechanisms for EBNA2 and LMP1 expression (3, 15, 16, 34, 39, 40, 51, 68, 70). DNA methylation of proximal promoter elements correlates with the transcription repression of EBNA2 and LMP1 transcription in type I latency (3, 16, 58, 66). Inhibitors of DNA methylation relieve this repression and induce a switch from type I to type III latency patterns of EBV gene expression (3, 26). Histone modifications are also known to be important for EBNA2 regulation since transcription activation correlates with promoter-specific histone hyperacetylation and histone acetyltransferase association (2, 72). Moreover, the CSL corepressors CIR and EBNA3A/C are known to associate with histone deacetylases during transcription repression of Cp and LMP1 promoters (24, 31).
The genomic organization of the LMP1 and EBNA2 genes suggests that the common upstream region containing OriP and EBV-encoded RNAs (EBERs) may be important in the coordinate regulation of these latency transcripts. Genetic evidence suggests that EBNA1 interaction with OriP can regulate the DNA methylation status and transcription activity of EBNA2 and LMP1 (18, 42, 46, 47, 65). OriP is known to interact with cellular replication and licensing factors that include components of the origin recognition complex (ORC) and minichromosome maintenance proteins (12, 14, 50, 59). The ORC has been implicated in the organization of chromatin structure and transcription repression in lower eukaryotes (55). It is not known whether OriP or ORC contributes to the establishment of chromatin structure that influences viral gene expression of LMP1 or EBNA2 during EBV latency.
Chromatin organization is another epigenetic regulatory mechanism that maintains stable gene expression patterns. The histone code hypothesis argues that posttranslational modifications of histone tails recruit factors that alter chromatin structure and determine biological activity of DNA loci (27, 64). Histone acetylation typically occurs in the proximal promoter regions of active genes (21, 63). Histone methylation correlates with larger domains of gene activity or inactivity (48, 61). Methylation of histone H3 on lysine 4 (H3mK4) correlates with euchromatin and higher gene activity (8, 33). In contrast, methylation of histone H3 on lysine 9 (H3mK9) correlates with transcriptional repression and colocalizes to regions of pericentromeric heterochromatin typically found in regions of repetitive DNA (7, 33, 44, 45). The establishment of active or inactive genetic domains by the spreading of histone modifications has been described at the mammalian β-globin locus control region and yeast mating type silencing elements (10, 33, 48). Insulator and boundary elements limit this spreading of histone modification and thereby establish discrete chromatin neighborhoods (10, 19, 32). Several insulator binding proteins have been found to repress gene expression by inhibiting enhancer communication with promoter elements (10, 43). CCCTC-binding factor (CTCF) is a ubiquitous GC-rich DNA binding protein that can block enhancer-promoter interactions. CTCF binding is highly sensitive to DNA methylation and is thought to play a critical role in maintaining heritable states found in imprinted genes (30).
In this work, we investigate chromatin modifications at the EBV latency control region (LCR) in various latency types. We investigate whether the region between LMP1 and EBNA2 is organized by histone modifications and chromatin structure that may dictate transcription status. We also explore the potential links between OriP binding factors with the EBNA2 transcription control regions. Finally, we present evidence that insulator protein CTCF contributes to the organization of chromatin in type I latency. Our findings suggest that histone methylation patterns delineate chromatin domains that correlate with different EBV latency programs.
MATERIALS AND METHODS
Cell lines and antibodies.
MutuI and KemI are type I latency cell lines derived from BL (kind gifts from J. Sample, St. Jude Children's Research Hospital, Memphis, Tenn.). Raji (American Type Culture Collection) is a type III latency cell line derived from BL. LCL3472-EBV (kind gift from D. Herlyn, Wistar Institute, Philadelphia, Pa.) is a type III latency cell line derived from primary lymphoblasts transformed with EBV strain B95-8. All cell lines were maintained in RPMI medium supplemented with 10% fetal bovine serum, glutamine, penicillin, and streptomycin sulfate (Cellgro). MutuI was treated with 5 μM 5-azacytidine (AzaC) (Sigma) for up to 72 h in reverse transcription (RT)-PCR, chromatin immunoprecipitation (ChIP), and methylation-specific PCR experiments, as indicated. LCL3472 cells were treated with 0.3 or 3 mM MTA [5′-deoxy-5′(methylthio)adenosine] (Sigma) for 24 h. All experiments were performed with cells collected at logarithmic growth phase (5 × 105 to 7 × 105 cells/ml). The following rabbit polyclonal antibodies were used: anti-diacetyl H3 (no. 06-599; Upstate), anti-tetraacetyl H4 (no. 06-866; Upstate), anti-acetyl K9 H3 (no. 06-942; Upstate), anti-dimethyl K4 H3 (no. 07-030; Upstate), anti-trimethyl K4 H3 (no. 07-473; Upstate), anti-dimethyl K9 H3 (no. 07-212; Upstate) and anti-CTCF (no. C7948-01; U.S. Biological), anti-Orc2 (no. 559266; Pharmingen), and control rabbit or mouse immunoglobulin G (IgG; Jackson Laboratories). Rabbit polyclonal anti-EBNA1 was raised against a recombinant full-length EBNA1. Primer sequences are available upon request (see Table S1 in the supplemental material).
Nucleosome organization assay.
Briefly, MutuI or LCL3472 cells were harvested and disrupted in a Dounce homogenizer in lysis buffer (0.3 M sucrose, 2 mM magnesium acetate, 3 mM CaCl2, 1% Triton X-100, and 10 mM HEPES [pH 7.9]). The lysate was then spun through a pad (25% glycerol, 5 mM magnesium acetate, 0.1 mM EDTA, and 10 mM HEPES [pH 7.4]) at 1,000 × g for 15 min at 4°C. The nuclei were incubated with micrococcal nuclease (at 15, 30, and 90 U per reaction) at 37°C for 5 min in digestion buffer (25 mM KCl, 4 mM MgCl2, 1 mM CaCl2, 50 mM Tris [pH 7.4], and 12.5% glycerol). The reaction was stopped by an equal volume of stop buffer (2% sodium dodecyl sulfate, 0.2 M NaCl, 10 mM EDTA, 10 mM EGTA, 50 mM Tris [pH 8.0]) and treated with proteinase K (100 μg/ml) for 2 h at 50°C. MNase-resistant DNA was extracted by phenol-chloroform and ethanol precipitation. Recovered DNA was subject to electrophoresis on a 1.6% agarose gel followed by transfer to Zeta-Probe blotting membranes (Bio-Rad) by Southern blot transfer (57). Southern blots were hybridized with a digoxigenin-labeled probe specific for the OriP, Cp, and Wp regions. The membranes were developed by a DIG-detection kit (Roche).
ChIP assays.
The ChIP assay followed the protocol provided by Upstate Biotechnology, Inc., with minor modifications as described previously (13). Formaldehyde cross-linked DNA was sheared by sonication to a mean average length of 500 bp for conventional PCR analysis and to 200 bp for real-time PCR analysis and verified by agarose gel electrophoresis. ChIP DNA was amplified for 22 to 24 cycles, which were determined to be within the linear amplification range. Real-time PCR was performed with Syber Green probe in an ABI Prism 7000 by using 1/100 to 1/2,500 of the ChIP DNA according to manufacturer's specified parameters.
DNA methylation-specific PCR assay.
A DNA methylation-specific PCR was performed essentially as described (67), with some minor modifications. Genomic DNA from 107 MutuI cells, treated or untreated with 5 μM AzaC for up to 72 h and from 107 untreated LCL3472 cells was harvested by using a DNA Isolation Kit (Roche). DNA (10 μg) in 100 μl was then denatured in 0.3 M NaOH at 42°C for 30 min, without using restriction endonuclease. Denatured DNA was mixed directly with 1.02 ml of 40.5% sodium bisulfite and 60 μl of 10 mM hydroquinone, overlaid with 100 μl of mineral oil, and incubated at 55°C for 16 h in the dark. DNA was then purified by using a DNA Purification Kit (QIAGEN), treated with 0.3 M NaOH at 37°C for 15 min, and precipitated with 3 M ammonium acetate and 2.5 volumes of ethanol. Recovered DNA was dissolved in 25 μl of Tris-EDTA buffer (pH 7.5) and stored at −20°C. The methylation status of the C promoter (covering coordinates 10,702 to 11,194) was determined by using methylation-specific PCR. The sequences for the primer pair ub3-ub4 for amplification of the bisulfite-converted unmethylated C promoter and the mb3-mb4 primer pair for the amplification of the bisulfite-converted methylated C promoter were identical to those described previously (67). The following unmethylated DNA-specific primers were used: for Cp ub3 (coordinates 10,702 to 10,723), 5′-CATCCAAAAACCAAACAACTCA; for Cp ub4 (coordinates 11,217 to 11194), 5′-AGTAAGGTGTAATTAATTTTGTTT. The following methylated DNA-specific primers were used: for Cp mb3 (coordinates 10,703 to 10,723), 5′-GTCCGAAAACCGAACGACTCG; for Cp mb4 (coordinates 11,216 to 11,194), 5′-GTAAGGCGTAATTAATTTCGTTC. Bisulfite-treated DNA (2 μl or approximately 40 ng) was amplified by PCR according to the following program: an initial denaturation at 94°C for 10 min, followed by 35 cycles consisting of 94°C for 30 seconds, annealing at a predetermined optimal temperature for each primer pair for 30 sec, and 72°C for 30 sec, with a final extension at 72°C for 3 min. PCR products (15-μl) were analyzed on a 1.8% agarose gel.
Reverse transcriptase PCR assays.
Total RNA was obtained by using QIAGEN′s RNeasy Kit, according to manufacturer's protocol. For cDNA synthesis, 2 μg of total RNA was incubated with 5 μM concentrations of random decamers (Ambion), 150 U of Superscript II reverse transcriptase (Invitrogen), 1.6 U of RNase inhibitor (Ambion), 1 mM deoxynucleoside triphosphate, and 3.3 mM dithiothreitol for 1 h 30 min at 37°C in a 15-μl reaction. After heat inactivation at 65°C for 10 min, the sample was diluted with 85 μl of distilled H2O. PCR was performed by using 1/20 of the reaction mixture for 25 cycles of 45 sec at 94°C, 45 sec at 55°C, and 1 min at 72°C.
RESULTS
Chromatin organization of the EBV latency control region.
The EBV LCR spans a ∼12-kb region between the two major repetitive sequence elements, terminal repeat (TR) and IR1 (Fig. 1A). These repetitive sequences contribute to EBNA2 and LMP1 transcription regulation as does the viral origin of plasmid replication OriP, which is located between these divergent promoters. Previously, it had been shown that EBNA1 and ORC proteins associate with the dyad symmetry (DS) region of OriP (12, 14, 50, 59) in BL cells. ChIP analysis of EBNA1 revealed specific binding centered at DS (site o) with some detection at 500 bp to each side (Fig. 1B). We also found that ORC2 binding extended an additional 1,000 bp rightward (sites n to r). The chromatin organization in this region was also investigated by ChIP assay. Histone H3mK9 was excluded from the actively transcribed EBERs and OriP (sites l to o) but was detected from DS (site p) to IR1 (site t). In contrast, histone H3mK4 spanned all of OriP extending leftward to TR (site f) and rightward to IR1 (site s). H3mK4 was mostly excluded from TR and IR1 sequences and was only weakly detected at genome positions beyond the IR1 and TR boundaries of the EBV LCR. This boundary did not apply to histone H3 and H4 acetylation, which was similar throughout most of the genome regions examined. This analysis suggests that the LCR is organized in chromatin domains reflected in the methylation pattern of histone H3.
FIG. 1.
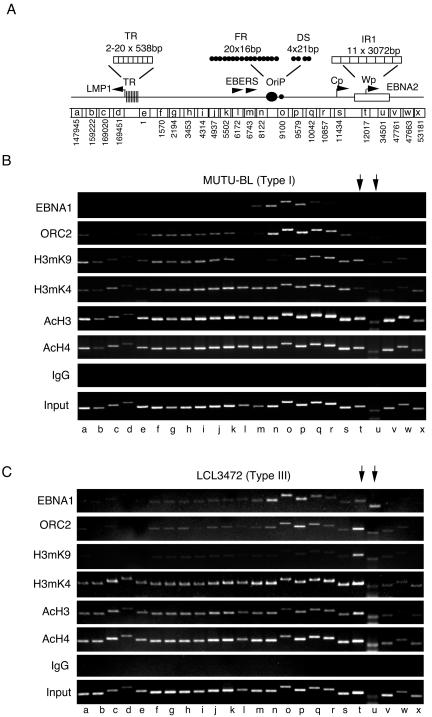
Histone modifications at EBV latency control region. (A) Schematic representation of the EBV latency control regions covering the LMP1 sequences at the left to the EBNA2 transcript region to the right. Sequence numbers below correlate to EBV coordinates (National Center for Biotechnology Information, gi:9625578) and indicate the left-flanking primer used for ChIP amplification. Each ChIP amplicon is ∼400 to 600 bp and is designated by lowercase letters. LMP1, TR, family of repeats (FR), EBERs, OriP, DS, and IR1 are indicated. (B) MutuI BL cell lines were assayed by ChIP with the specific antibodies, control IgG, and input DNA as indicated. EBV genome positions correlate with the schematic in panel A and are indicated by lowercase letters below. (C) LCL3472 cells were assayed in the same manner as described in panel B.
The chromatin organization of the LCR was next examined in an LCL with a type III latency program (Fig. 1C). Similar to results with type I BL cells, EBNA1 and ORC2 in an LCL were enriched at the DS (site o) region with detection at each 500-bp flank. However, in contrast to the results with BL cells, we found that EBNA1 and ORC2 were also detected within the IR1 sequence (sites t and u). Histone H3mK9 was suppressed throughout the LCR with the exception of the IR1 sequence (sites t and u). Histone H3mK4 was enriched through the entire LCR, but unlike type I latency, the pattern extended into IR1 and TR in type III LCL3472 cells (Fig. 1, compare panels B and C at sites t,u and d,e). Histone acetylation was high everywhere with the exception of regions w and x to the right of IR1. These findings indicate that histone modification patterns in the EBV LCR differed between latency types I and III cells.
H3mK4 boundary correlates with EBNA2 transcription.
The H3mK4 pattern was reexamined in four cell lines harboring latent EBV genomes (Fig. 2). Cells with type I latency (MutuI and KemI) did not express any detectable EBNA2 or LMP2 mRNA levels in RT-PCR analysis (Fig. 2A). Cell lines with type III latency (Raji and LCL3472) expressed both EBNA2 and LMP1 transcripts, as was expected (Fig. 2A). These cell lines were then compared by ChIP for H3mK4 across regions g to x (Fig. 2B). Consistent with the results shown in Fig. 1, we found that H3mK4 was enriched in IR1 sequences (sites t and u) in type III cells, Raji and LCL3472, but not detected in type I cells, MutuI and KemI. All regions of the genome were amplified to similar levels from input DNA (Fig. 2C), indicating that these differences do not reflect differences in IR1 copy number within various genomes. These results indicate that an expanded zone of histone H3mK4 correlates with increased transcription activity in type III latency.
FIG. 2.
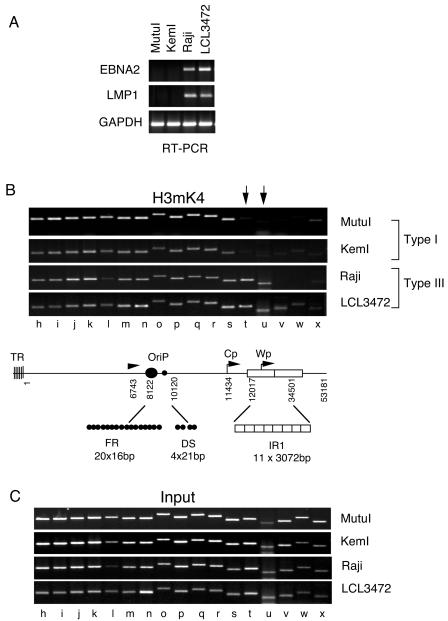
A histone H3mK4 boundary correlates with transcription activity of EBNA2. (A) EBNA2, LMP1, and GAPDH mRNAs were quantitated by RT-PCR analysis of mRNA isolated from MutuI, KemI, Raji, and LCL3472 cell lines. (B) H3mK4 binding to EBV sequence was measured by ChIP assay as described in the legend of Fig. 1B. (C) Input DNA for ChIP analysis shown in panel B above.
Dynamic regulation of the LCR chromatin boundary.
The role of histone H3mK4 on EBV latent gene expression was explored by using pharmacological inhibitors of protein methylation. The S-adenosylmethionine metabolite MTA has been shown to reduce histone H3mK4 in cell culture (37, 62, 74). LCL3472 cells express constitutively high levels of EBNA2 and LMP1 and were shown to have expanded histone H3mK4 at the EBNA2 proximal promoter sequences (Fig. 1 and 2). We now treated LCL3472 cells with 0.3 and 3 mM MTA for 24 h and then assayed for LMP1 and EBNA2 mRNA levels by RT-PCR (Fig. 3A). We found that 3 mM MTA reduced EBNA2 mRNA levels without significantly affecting LMP1 or GAPDH (glyceraldehyde-3-phosphate dehydrogenase) mRNA (Fig. 3A). LCL3472 cells were then examined by ChIP assay with or without 3 mM MTA treatment for 24 h (Fig. 3B). We found that MTA treatment led to a substantial global reduction in histone H3mK4 throughout the EBV LCR (Fig. 3B, top panel). In contrast, MTA treatment had no detectable effect on histone H3 acetylation (Fig. 3B). Interestingly, MTA weakly suppressed EBNA1 binding to OriP but did not alter binding specificity. Although MTA is likely to have pleiotropic effects on treated cells, these results indicate that EBNA2 transcription is highly sensitive to MTA and raise the possibility that histone methylation at the EBNA2 promoter region may be essential for transcription activity in type III LCLs.
FIG. 3.
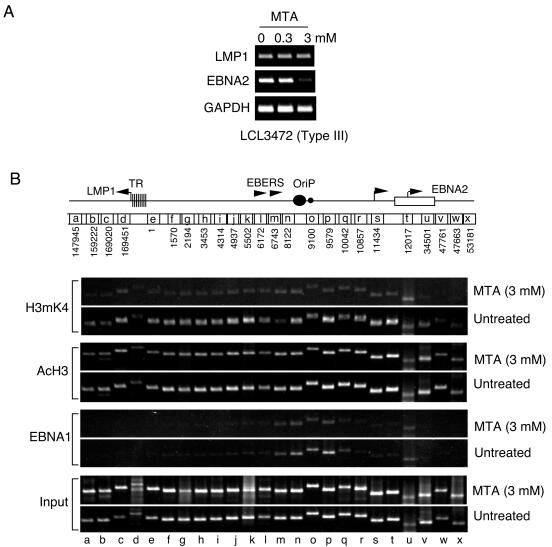
Protein methylase inhibitor MTA blocks EBNA2 transcription. (A) RT-PCR analysis of LMP1, EBNA2, and GAPDH mRNA derived from LCL3472 cells treated with 0, 0.3, or 3 mM MTA as indicated. (B) ChIP assay of LCL34782 cells with or without 3 mM MTA treatment. Antibodies to H3mK4, AcH3, EBNA1, or input are indicated to the right.
Histone modifications have also been linked to DNA methylation patterns (28, 61). DNA methylation patterns at the EBNA2 and LMP1 promoters are known to suppress transcription in type I latency (34). The DNA methylation inhibitor AzaC can activate EBNA2 and LMP1 transcription in type I latency where they are repressed by DNA methylation (26). As expected, treatment of MutuI cells with AzaC induced transcription of LMP1 and EBNA2 after 48 h (Fig. 4A). DNA methylation-specific PCR was used to confirm that AzaC led to the loss of DNA methylation at Cp in MutuI cells (Fig. 4B). In the absence of treatment, most of the Cp DNA was unmethylated in LCL cells (Fig. 4B, lane 2), and methylated in MutuI cells (Fig. 4B, lane 3). Unmethylated Cp DNA was readily detected in MutuI cells within the first 24 h of AzaC treatment (Fig. 4B, lane 4). A corresponding decrease in methylated Cp DNA revealed that genomes were progressively demethylated throughout the 72-h treatment interval (Fig. 4B, lane 6). Interestingly, AzaC treatment also increased histone H3mK4 at regions t to x and e to a (indicated by arrows), which overlap some control elements in the EBNA2 and LMP1 promoters known to be affected by DNA methylation (16, 26, 40, 52, 56, 66) (Fig. 4C). The increase in H3mK4 was detectable at 48 h and increased up to 72 h (Fig. 4C). Therefore, demethylation of Cp DNA appears to precede the observable increase in histone H3mK4 by 24 h. These results indicate that inhibition of DNA methylation by AzaC correlates with an expansion of the histone H3mK4 domain encompassing the EBV LCR. These results also indicate that histone modification patterns are dynamic and do not depend strictly on cell type or viral strain.
FIG. 4.
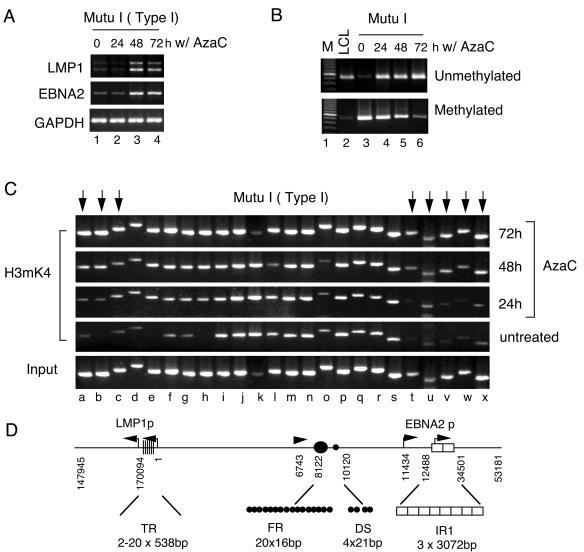
DNA methylation influences histone H3mK4 at LCR boundaries. (A) EBNA2, LMP1, and GAPDH mRNA were quantitated in MutuI cells treated with 5 μM AzaC for 0, 24, 48, or 72 h as indicated. (B) Methylation-specific PCR analysis of Cp in untreated LCL (lane 2) cells or MutuI cells treated with AzaC for 0, 24, 48, and 72 h as indicated (lanes 3 to 6). PCR primers specific for unmethylated DNA or methylated DNAare indicated. (C) MutuI cells treated with AzaC for 0, 24, 48, or 72 h were assayed for H3mK4 binding to EBV regions by ChIP. Input DNA is shown at bottom. EBV DNA regions are indicated by lowercase letters and correspond to amplicons described in the legend of Fig. 1A. (D) Schematic diagram showing LMP1 and EBNA2 promoter control regions.
To further verify these results, we reexamined several key regions of the EBV LCR by using more quantitative real-time PCR analysis of ChIP DNA from MutuI, MutuI+AzaC, and LCL3472 cells (Fig. 5A). The real-time PCR amplicons are ∼100 bp and were used with chromatin fragments sonicated to an average length of ∼200 bp to increase regional specificity within the LCR. We found that EBNA1 bound with high specificity to DS in MutuI, MutuI+AzaC, and LCL3472 cells as expected (Fig. 5B). Significant levels of EBNA1 were also detected at W1 (∼12-fold over IgG) and Cp (∼6-fold over IgG) in LCL3472 but not in MutuI cells with or without AzaC (Fig. 5B). ORC2 had a very similar pattern as EBNA1 and was also enriched at W1 in LCL3472. These results confirm and extend our findings that OriP-associated factors interact within or adjacent to IR1 sequences in type III cells expressing EBNA2.
FIG. 5.
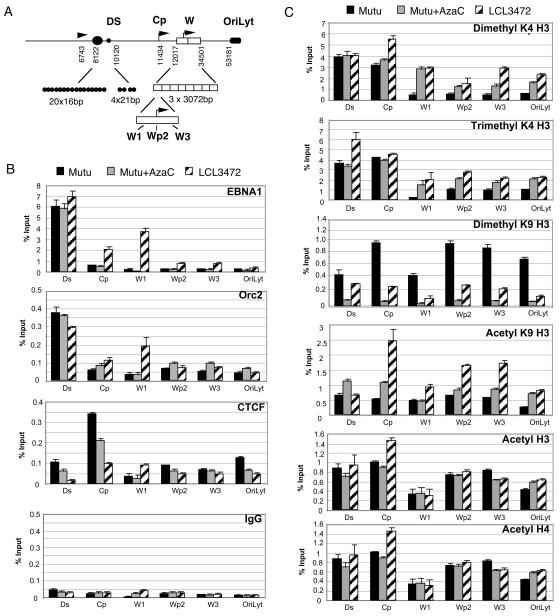
Quantitative differences in histone modification and chromatin binding factors in types I and III latency. (A) Schematic diagram of real-time primer sets covering the DS, Cp, W1, Wp2, W3, and OriLyt regions of the genome. (B). Real-time PCR analysis of ChIP assay with antibodies specific for EBNA1, ORC2, CTCF, or control IgG for MutuI (black bars), MutuI+AzaC (gray bars), or LCL3472 (striped bars) cells. (C) Histone modifications detected by antibodies specific for dimethyl K4 H3, trimethyl K4 H3, dimethyl K9 H3, acetyl K9 H3, acetyl H3, or acetyl H4 were analyzed by real-time PCR as indicated in MutuI, MutuI+5-AzaC, or LCL cells as described in panel B.
Program-specific binding of a chromatin insulator protein.
Chromatin boundary elements limit the spread of histone modifications (48). CTCF is a zinc-finger DNA binding protein found at almost all chromatin boundary elements in higher eukaryotes (10, 43). We found that CTCF was enriched at Cp in MutuI cells but not in LCL3472 cells, and its binding was reduced after AzaC treatment (Fig. 5B). These results indicate that CTCF binds Cp in type I cells repressing EBNA2 transcription. Furthermore, these results suggest that a CTCF-associated chromatin boundary element in Cp regulates the spread of histone modification in the LCR.
AzaC inhibits H3mK9.
Histone modifications in IR1 were also examined by real-time PCR (Fig. 5C). The H3mK4 (dimethyl and trimethyl) pattern confirmed that histone methylation boundary exists in MutuI cells to the right of Cp and left of W1. This boundary was not as apparent in LCL3472 and was intermediate in MutuI cells treated with AzaC. The H3mK9 pattern was highest at Cp and Wp in MutuI cells and generally low in LCL3472. Surprisingly, AzaC treatment eliminated H3mK9 at all sites assayed in MutuI cells suggesting that H3mK9 may be tightly linked to DNA methylation. Acetylation at H3 K9 was enriched at Cp and to a lesser extent at Wp and W3 in LCL3472 cells but not significantly in MutuI cells with or without AzaC treatment. A similar enrichment at Cp was observed with antibodies to hyperacetylated histone H3 and H4, consistent with previous findings (2). These results suggest that histone modifications in the EBNA2 control region vary in response to different transcription regulation patterns and can be affected dramatically by changes in DNA methylation.
Nucleosome phasing at the EBV LCR.
A direct analysis of chromatin structure by micrococcal nuclease (MNase I) digestion pattern was compared for LCL3472 (type III) and MutuI (type I) cells at three sites within the latency control region (Fig. 6). The MNase I patterns at OriP and Cp were significantly weaker in LCL cells relative to MutuI. The MNase I pattern at Wp was readily detected in both cell types, suggesting that this region is organized in nucleosome arrays highly resistant to MNase digestion. The MNase I pattern of total genomic DNA visualized by ethidium bromide staining indicated that input DNA levels were similar in each lane (Fig. 6, right panel). These results indicate that type I and type III EBV LCR have differential sensitivity to MNase I digestion, suggesting that chromatin organization differs in these different latency types.
FIG. 6.
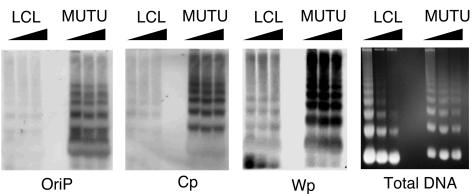
Nucleosome arrays at EBV LCRs. MNase I laddering assay for LCL3472 (LCL) or MutuI (MUTU) cells was assayed by Southern blot hybridization with probes specific for OriP, Cp, or Wp as indicated. Total genomic DNA was visualized by ethidium staining of agarose gels prior to transfer (right panel).
DISCUSSION
Chromatin boundaries define an EBV latency control region.
In this study we examined the histone modification and chromatin organization in the EBV LCR in cell lines with type I or type III latent gene expression patterns. Our studies indicate that histone modification and chromatin organization correlate with different gene expression patterns in these two distinct forms of EBV latency. An examination of histone modification patterns and chromatin binding proteins suggests that EBV is partitioned into discrete chromosomal domains and that the regions between TR and IR1 which regulate LMP1 and EBNA2 transcription constitute an important latency regulatory unit which we refer to here as the EBV LCR. This region encompasses the latency-associated plasmid maintenance element and DNA replication origin, OriP. This region also includes the RNA polymerase III-transcribed EBERs, which are constitutively expressed in all forms of EBV latency. The patterns of histone modification found in these studies suggest that chromatin architecture in the LCR coordinates the gene expression patterns of LMP1 and EBNA2 with other aspects of viral latency, including OriP function.
Correlation of histone H3mK4 with LCR transcription activity.
Histone modifications serve as signals for chromatin binding proteins that influence chromatin structure and regulate gene expression (27, 64). Histone H3mK4 is typically associated with open chromatin that is permissive for transcription activity. In type III latency where EBNA2 and LMP1 transcription is active, histone H3mK4 was enriched throughout the LCR and extended rightward into the W1 region of IR1 and leftward beyond the TR into LMP1 (Fig. 1 and 5). In contrast, in type I latency where EBNA2 and LMP2 transcription is repressed, H3mK4 ends abruptly at the IR1 and TR regions (Fig. 1 and 5). The rightward boundary of histone H3mK4 was examined in four cell types, and the expansion of this modification into IR1 showed a strong correlation with transcription activity of EBNA2 found in latency type III (Fig. 2). The pattern of histone H3mK4 in type I cells strongly suggests that the region between TR and IR1 constitutes a discrete chromatin boundary element that coordinates LMP1 and EBNA2 transcription (Fig. 7).
FIG. 7.
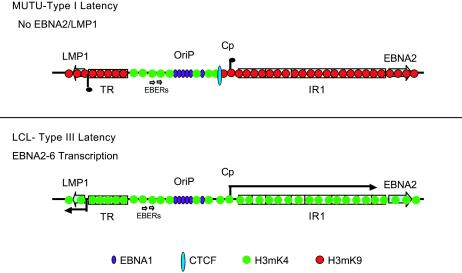
Chromatin structure and long-distance transcription regulation at the EBV LCRs in type III and type I latency. ORC and EBNA1 are shown to interact with Cp and Wp control elements in type III but not type I. H3mK4 is shown to expand from OriP to Wp in type I but is blocked by CTCF at Cp in type I latency. Histone H3mK9 is low in type III but enriched at Cp and Wp in type I latency, corresponding to EBNA2 transcription repression in type I.
OriP/EBERs as a generator of histone H3mK4 open chromatin.
We found that histone H3mK4 was consistently enriched at sequences within and adjacent to OriP, but it is not known what generates this histone modification. Replication activity at OriP and RNA polymerase III transcription of the EBERs are likely to generate an open chromatin structure typically characterized by histone H3mK4 modification. It is also possible that OriP binding factors may have a more direct role in maintaining the H3mK4 signal. ORC components are known to bind OriP and have been implicated in chromatin organization in budding yeast (36) and Drosophila (60). We also found that the OriP binding proteins EBNA1 and ORC2 interacted with sequences adjacent to OriP and with IR1 sequences in type III but not type I latency. This is consistent with genetic evidence linking OriP to activation of EBNA2 transcription control (18, 42, 46, 47, 58, 65). In addition to replication activity and ORC proteins, it is also possible that aberrant chromatin structure at OriP can generate a histone H3mK4 signal. A variant, open chromatin structure at OriP has been detected by using MNase I laddering assays in vivo (73) and by the nucleosome displacement activity of EBNA1 in vitro (4). Based on this, we propose that EBNA1 and ORC interactions with OriP generate a histone H3mK4 signal that spreads in both directions toward the TR and IR1.
Generation of the H3mK9 signal at the EBV internal repeats.
Histone H3mK9 modification is typically associated with transcriptional repression and heterochromatin formation (33). We found that H3mK9 was enriched at the two ends of the LCR in type I but not type III latency, correlating with transcription repression of EBNA2 and LMP1 (Fig. 1). H3mK9 was not detected at OriP and EBERs, suggesting that this central region of the LCR remains free from H3mK9-mediated repression in all forms of EBV latency. The H3mK9 modification is often generated in a repetitive DNA sequence that is predisposed to forming silent heterochromatin (38), and it is possible that EBV TR and IR1 have a heterochromatin organizing function during EBV latency. MNase I studies support a potential role for the repetitive IR1 (Wp) sequence in the formation of facultative heterochromatin in both type I and III latent genomes (Fig. 6). The IR1 MNase laddering pattern was more defined than that found in neighboring regions of the genome, suggesting that densely packed histones occupy this region of the genome. These findings are consistent with a model that H3mK9 signal initiates within the repetitive TR and IR1 sequences and spreads into the LCR until counteracted by H3mK4 patterns generated at OriP and EBERs (Fig. 7). Histone H3mK4 and H3mK9 are thought to generate opposing transcription signals and to be mutually exclusive (48, 61). However, we observed that histone H3mK4 and H3mK9 overlap at some sites in the LCRs of MutuI cells (Fig. 1 and 5). Since EBV exists as a multicopy genome, it is possible that a subpopulation of EBV genomes has H3mK9 while some have H3mK4. Alternatively, it is possible that complex regulatory elements in higher eukaryotes have more intricate patterns of histone tail modifications that include overlapping H3 K4 and K9 methylation.
Boundary elements regulate the expansion of chromatin domains.
Chromatin boundary elements impose epigenetic control on regulatory DNA regions (48). The insulator-like protein CTCF bound Cp in type I but not type III cells, correlating with the H3mK4 boundary and transcriptional repression. CTCF binds to GC-rich elements, and our ChIP data indicate that it binds in the proximity of Cp. The precise binding site for CTCF has not been mapped in Cp, and we have not formally proven that CTCF represses EBNA2 transcription in type I latency. Nevertheless, the correlation between CTCF binding and transcriptional repression at Cp remains intriguing. The reported insulator activities of CTCF suggest that CTCF could inhibit EBNA2 transcription by preventing the expansion of histone H3mK4 initiating at OriP and, consequently, block OriP-dependent enhancer activity at Cp.
The spreading of histone modification signals across large domains has been described for heterochromatin formation in yeast telomeric silencing and Drosophila position effect variegation (48, 55). The contiguous histone methylation patterns observed in the EBV LCR are reminiscent of signal spreading seen in these other model systems of heterochromatic silencing. The limit of H3mK4 and mK9 spreading in type I latency may be critical for the regulation of EBNA2 and LMP1 transcription. Indeed, disruption of histone H3mK4 with MTA led to a dramatic loss of EBNA2 transcription (Fig. 3). However, we cannot rule out that MTA may also inhibit methylation of proteins other than histone H3 that may affect EBNA2 transcription regulation. Despite the potential additional targets of MTA, loss of histone H3mK4 in the LCR correlated with the loss of EBNA2 transcription (Fig. 3). Curiously, LMP1 transcription was not reduced to the same extent as EBNA2 after MTA treatment (Fig. 3A). This observation suggests that EBNA2 regulation is highly sensitive to changes in protein methylation. Consequently, MTA treatment phenocopies type II latency patterns seen in Hodgkin's lymphomas, where LMP1 is expressed in the absence of EBNA2 (53). Future studies may consider whether the alteration of protein methylation contributes to the formation of type II latency.
Coordination of DNA methylation with histone methylation.
DNA methylation has been shown to play a significant role in the epigenetic regulation of EBNA2 and LMP1 transcription (3, 16, 58, 66). AzaC-induced EBNA2 and LMP1 transcription in type I cells correlated with an expansion of the H3mK4 domain into the Cp and Wp regions of IR1. This indicates that inhibition of DNA methylation leads to a rapid change in histone methylation status. Most notably, AzaC treatment caused a dramatic reduction in H3mK9 in IR1 (Fig. 5). Biochemical and genetic links between DNA methylation and histone H3mK9 have been observed in plants and lower eukaryotes but have not been as well characterized in higher eukaryotes (33). Our data strongly suggest that changes in DNA methylation can induce changes in histone methylation at the EBNA2 control regions and suggest that these two methylation processes may be coordinated in mammalian gene expression.
Epigenetic control of EBV gene regulation is likely to be a significant factor in determining the latency program and pathophysiology of EBV infections. Our data indicate that histone H3 methylation on K4 and K9 reflect important changes in the latency program and expression status of EBV latency-associated oncogenes. Our data suggest that histone H3 methylation functions as a biochemical signal that delineates a chromatin domain in the EBV LCR. Our data also indicate that histone methylation is tightly linked to DNA methylation and that these epigenetic signals establish stable regulatory domains that determine gene expression patterns for EBNA2 and LMP1. The links between DNA methylation, histone methylation, and chromatin architecture should be important for further understanding of EBV oncogenic potential and pathogenesis.
Supplementary Material
Acknowledgments
We thank J. Sample and D. Herlyn for cell lines and the Wistar Institute Cancer Core Facilities for fluorescence-activated cell sorter analysis.
This work was funded in part by grants from the NIH (CA93606 and CA05678) to P.M.L. and a Wistar Institute NIH Postdoctoral Training Fellowship to C.M.C.
Footnotes
Supplemental material for this article may be found at http://jvi.asm.org.
REFERENCES
- 1.Abbot, S. D., M. Rowe, K. Cadwallader, A. Ricksten, J. Gordon, F. Wang, L. Rymo, and A. B. Rickinson. 1990. Epstein-Barr virus nuclear antigen 2 induces expression of the virus-encoded latent membrane protein. J. Virol. 64:2126-2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alazard, N., H. Gruffat, E. Hiriart, A. Sergeant, and E. Manet. 2003. Differential hyperacetylation of histones H3 and H4 upon promoter-specific recruitment of EBNA2 in Epstein-Barr virus chromatin. J. Virol. 77:8166-8172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ambinder, R. F., K. D. Robertson, and Q. Tao. 1999. DNA methylation and the Epstein-Barr virus. Semin. Cancer Biol. 9:369-375. [DOI] [PubMed] [Google Scholar]
- 4.Avolio-Hunter, T. M., and L. Frappier. 2003. EBNA1 efficiently assembles on chromatin containing the Epstein-Barr virus latent origin of replication. Virology 315:398-408. [DOI] [PubMed] [Google Scholar]
- 5.Babcock, G. J., L. L. Decker, M. Volk, and D. A. Thorley-Lawson. 1998. EBV persistence in memory B cells in vivo. Immunity 9:395-404. [DOI] [PubMed] [Google Scholar]
- 6.Babcock, G. J., D. Hochberg, and A. D. Thorley-Lawson. 2000. The expression pattern of Epstein-Barr virus latent genes in vivo is dependent upon the differentiation stage of the infected B cell. Immunity 13:497-506. [DOI] [PubMed] [Google Scholar]
- 7.Bannister, A. J., P. Zegerman, J. F. Partridge, E. A. Miska, J. O. Thomas, R. C. Allshire, and T. Kouzarides. 2001. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 410:120-124. [DOI] [PubMed] [Google Scholar]
- 8.Bernstein, B. E., E. L. Humphrey, R. L. Erlich, R. Schneider, P. Bouman, J. S. Liu, T. Kouzarides, and S. L. Schreiber. 2002. Methylation of histone H3 Lys 4 in coding regions of active genes. Proc. Natl. Acad. Sci. USA 99:8695-8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bishop, G. A., and L. K. Busch. 2002. Molecular mechanisms of B-lymphocyte transformation by Epstein-Barr virus. Microbes Infect. 4:853-857. [DOI] [PubMed] [Google Scholar]
- 10.Burgess-Beusse, B., C. Farrell, M. Gaszner, M. Litt, V. Mutskov, F. Recillas-Targa, M. Simpson, A. West, and G. Felsenfeld. 2002. The insulation of genes from external enhancers and silencing chromatin. Proc. Natl. Acad. Sci. USA 99(Suppl. 4):16433-16437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Callahan, J., J. Aster, J. Sklar, E. Kieff, and E. S. Robertson. 2000. Intracellular forms of human NOTCH1 interact at distinctly different levels with RBP-jkappa in human B and T cells. Leukemia 14:84-92. [DOI] [PubMed] [Google Scholar]
- 12.Chaudhuri, B., H. Xu, I. Todorov, A. Dutta, and J. L. Yates. 2001. Human DNA replication initiation factors, ORC and MCM, associate with oriP of Epstein-Barr virus. Proc. Natl. Acad. Sci. USA 98:10085-10089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deng, Z., L. Lezina, C. J. Chen, S. Shtivelband, W. So, and P. M. Lieberman. 2002. Telomeric proteins regulate episomal maintenance of Epstein-Barr virus origin of plasmid replication. Mol. Cell 9:493-503. [DOI] [PubMed] [Google Scholar]
- 14.Dhar, S. K., K. Yoshida, Y. Machida, P. Khaira, B. Chaudhuri, J. A. Wohlschlegel, M. Leffak, J. Yates, and A. Dutta. 2001. Replication from oriP of Epstein-Barr virus requires human ORC and is inhibited by geminin. Cell 106:287-296. [DOI] [PubMed] [Google Scholar]
- 15.Ernberg, I., K. Falk, J. Minarovits, P. Busson, T. Tursz, M. G. Masucci, and G. Klein. 1989. The role of methylation in the phenotype-dependent modulation of Epstein-Barr nuclear antigen 2 and latent membrane protein genes in cells latently infected with Epstein-Barr virus. J. Gen. Virol. 70:2989-3002. [DOI] [PubMed] [Google Scholar]
- 16.Falk, K. I., L. Szekely, A. Aleman, and I. Ernberg. 1998. Specific methylation patterns in two control regions of Epstein-Barr virus latency: the LMP-1-coding upstream regulatory region and an origin of DNA replication (oriP). J. Virol. 72:2969-2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Farrell, C. J., J. M. Lee, E. C. Shin, M. Cebrat, P. A. Cole, and S. D. Hayward. 2004. Inhibition of Epstein-Barr virus-induced growth proliferation by a nuclear antigen EBNA2-TAT peptide. Proc. Natl. Acad. Sci. USA 101:4625-4630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gahn, T. A., and B. Sugden. 1995. An EBNA-1-dependent enhancer acts from a distance of 10 kilobase pairs to increase expression of the Esptein-Barr virus LMP gene. J. Virol. 69:2633-2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gerasimova, T. I., and V. G. Corces. 2001. Chromatin insulators and boundaries: effects on transcription and nuclear organization. Annu. Rev. Genet. 35:193-208. [DOI] [PubMed] [Google Scholar]
- 20.Grossman, S. R., E. Johannsen, X. Tong, R. Yalamanchili, and E. Kieff. 1994. The Epstein-Barr virus nuclear antigen 2 transactivator is directed to response elements by the J kappa recombination signal binding protein. Proc. Natl. Acad. Sci. USA 91:7568-7572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hassan, A. H., K. E. Neely, M. Vignali, J. C. Reese, and J. L. Workman. 2001. Promoter targeting of chromatin-modifying complexes. Front. Biosci. 6:D1054-D1064. [DOI] [PubMed] [Google Scholar]
- 22.Henkel, T., P. D. Ling, S. D. Hayward, and M. G. Peterson. 1994. Mediation of Epstein-Barr virus EBNA2 transactivation by recombination signal-binding protein J kappa. Science 265:92-95. [DOI] [PubMed] [Google Scholar]
- 23.Hsieh, J. J., D. E. Nofziger, G. Weinmaster, and S. D. Hayward. 1997. Epstein-Barr virus immortalization: Notch2 interacts with CBF1 and blocks differentiation. J. Virol. 71:1938-1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hsieh, J. J., S. Zhou, L. Chen, D. B. Young, and S. D. Hayward. 1999. CIR, a corepressor linking the DNA binding factor CBF1 to the histone deacetylase complex. Proc. Natl. Acad. Sci. USA 96:23-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Izumi, K. M. 2001. Identification of EBV transforming genes by recombinant EBV technology. Semin. Cancer Biol. 11:407-414. [DOI] [PubMed] [Google Scholar]
- 26.Jansson, A., M. Masucci, and L. Rymo. 1992. Methylation of discrete sites within the enhancer region regulates the activity of the Epstein-Barr virus BamHI W promoter in Burkitt lymphoma lines. J. Virol. 66:62-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jenuwein, T., and C. D. Allis. 2001. Translating the histone code. Science 293:1074-1080. [DOI] [PubMed] [Google Scholar]
- 28.Jones, P. L., and A. P. Wolffe. 1999. Relationships between chromatin organization and DNA methylation in determining gene expression. Semin. Cancer Biol. 9:339-347. [DOI] [PubMed] [Google Scholar]
- 29.Kieff, E. 1996. Epstein-Barr virus and its replication, p. 2343-2396. In B. N. Fields, D. M. Knipe, and P. M. Howley (ed.), Field's virology, 3rd ed., vol. 2. Lippincott-Raven Publishers, Philadelphia, Pa. [Google Scholar]
- 30.Klenova, E. M., H. C. Morse III, R. Ohlsson, and V. V. Lobanenkov. 2002. The novel BORIS + CTCF gene family is uniquely involved in the epigenetics of normal biology and cancer. Semin. Cancer Biol. 12:399-414. [DOI] [PubMed] [Google Scholar]
- 31.Knight, J. S., K. Lan, C. Subramanian, and E. S. Robertson. 2003. Epstein-Barr virus nuclear antigen 3C recruits histone deacetylase activity and associates with the corepressors mSin3A and NCoR in human B-cell lines. J. Virol. 77:4261-4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuhn, E. J., and P. K. Geyer. 2003. Genomic insulators: connecting properties to mechanism. Curr. Opin. Cell Biol. 15:259-265. [DOI] [PubMed] [Google Scholar]
- 33.Lachner, M., and T. Jenuwein. 2002. The many faces of histone lysine methylation. Curr. Opin. Cell Biol. 14:286-298. [DOI] [PubMed] [Google Scholar]
- 34.Li, H., and J. Minarovits. 2003. Host cell-dependent expression of latent Epstein-Barr virus genomes: regulation by DNA methylation. Adv. Cancer Res. 89:133-156. [DOI] [PubMed] [Google Scholar]
- 35.Ling, P. D., D. R. Rawlins, and S. D. Hayward. 1993. The Epstein-Barr virus immortalizing protein EBNA2 is targeted to DNA by a cellular enhancer binding protein. Proc. Natl. Acad. Sci. USA 90:9237-9241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Loo, S., and J. Rine. 1995. Silencing and heritable domains of gene expression. Annu. Rev. Cell Dev. Biol. 11:519-548. [DOI] [PubMed] [Google Scholar]
- 37.Martens, J. H., M. Verlaan, E. Kalkhoven, and A. Zantema. 2003. Cascade of distinct histone modifications during collagenase gene activation. Mol. Cell. Biol. 23:1808-1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martienssen, R. A. 2003. Maintenance of heterochromatin by RNA interference of tandem repeats. Nat. Genet. 35:213-214. [DOI] [PubMed] [Google Scholar]
- 39.Masucci, M. G., B. Contreras-Salazar, E. Ragnar, K. Falk, J. Minarovits, I. Ernberg, and G. Klein. 1989. 5-Azacytidine up regulates the expression of Epstein-Barr virus nuclear antigen 2 (EBNA-2) through EBNA-6 and latent membrane protein in the Burkitt's lymphoma line rael. J. Virol. 63:3135-3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Minarovits, J., L. F. Hu, S. Minarovits-Kormuta, G. Klein, and I. Ernberg. 1994. Sequence-specific methylation inhibits the activity of the Epstein-Barr virus LMP 1 and BCR2 enhancer-promoter regions. Virology 200:661-667. [DOI] [PubMed] [Google Scholar]
- 41.Nilsson, T., A. Sjoblom, M. G. Masucci, and L. Rymo. 1993. Viral and cellular factors influence the activity of the Epstein-Barr virus BCR2 and BWR1 promoters in cells of different phenotype. Virology 193:774-785. [DOI] [PubMed] [Google Scholar]
- 42.Nilsson, T., H. Zetterberg, Y. C. Wang, and L. Rymo. 2001. Promoter-proximal regulatory elements involved in oriP-EBNA1-independent and -dependent activation of the Epstein-Barr virus C promoter in B-lymphoid cell lines. J. Virol. 75:5796-5811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ohlsson, R., R. Renkawitz, and V. Lobanenkov. 2001. CTCF is a uniquely versatile transcription regulator linked to epigenetics and disease. Trends Genet. 17:520-527. [DOI] [PubMed] [Google Scholar]
- 44.Peters, A. H., S. Kubicek, K. Mechtler, R. J. O'Sullivan, A. A. Derijck, L. Perez-Burgos, A. Kohlmaier, S. Opravil, M. Tachibana, Y. Shinkai, J. H. Martens, and T. Jenuwein. 2003. Partitioning and plasticity of repressive histone methylation states in mammalian chromatin. Mol. Cell 12:1577-1589. [DOI] [PubMed] [Google Scholar]
- 45.Peters, A. H., J. E. Mermoud, D. O'Carroll, M. Pagani, D. Schweizer, N. Brockdorff, and T. Jenuwein. 2002. Histone H3 lysine 9 methylation is an epigenetic imprint of facultative heterochromatin. Nat. Genet. 30:77-80. [DOI] [PubMed] [Google Scholar]
- 46.Puglielli, M. T., M. Woisetschlaeger, and S. H. Speck. 1996. oriP is essential for EBNA gene promoter activity in Epstein-Barr virus immortalized lymphoblastoid cell lines. J. Virol. 70:5758-5768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reisman, D., and B. Sugden. 1986. trans activation of an Epstein-Barr viral transcriptional enhancer by the Epstein-Barr viral nuclear antigen 1. Mol. Cell. Biol. 6:3838-3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Richards, E. J., and S. C. Elgin. 2002. Epigenetic codes for heterochromatin formation and silencing: rounding up the usual suspects. Cell 108:489-500. [DOI] [PubMed] [Google Scholar]
- 49.Rickinson, A. B., and E. Kieff. 1996. Epstein-Barr virus, p. 2397-2446. In B. N. Fields, D. M. Knipe, and P. M. Howley (ed.), Field's virology, 3rd ed., vol. 2. Lippincott-Raven Publishers, Philadelphia, Pa. [Google Scholar]
- 50.Ritzi, M., K. Tillack, J. Gerhardt, E. Ott, S. Humme, E. Kremmer, W. Hammerschmidt, and A. Schepers. 2003. Complex protein-DNA dynamics at the latent origin of DNA replication of Epstein-Barr virus. J. Cell Sci. 116:3971-3984. [DOI] [PubMed] [Google Scholar]
- 51.Robertson, K. D. 2000. The role of DNA methylation in modulating Epstein-Barr virus gene expression. Curr. Top. Microbiol. Immunol. 249:21-34. [DOI] [PubMed] [Google Scholar]
- 52.Robertson, K. D., A. Manns, L. J. Swinnen, J. C. Zong, M. L. Gulley, and R. F. Ambinder. 1996. CpG methylation of the major Epstein-Barr virus latency promoter in Burkitt's lymphoma and Hodgkin's disease. Blood 88:3129-3136. [PubMed] [Google Scholar]
- 53.Rowe, M., A. L. Lear, D. Croom-Carter, A. H. Davies, and A. B. Rickinson. 1992. Three pathways of Epstein-Barr virus gene activation from EBNA1-positive latency in B lymphocytes. J. Virol. 66:122-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rowe, M., D. T. Rowe, C. D. Gregory, e. al., and A. B. Rickinson. 1987. Differences in B-cell growth phenotype reflect novel patterns of Epstein-Barr virus latent gene expression in Burkitt's lymphoma cells. EMBO J. 6:2743-2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rusche, L. N., A. L. Kirchmaier, and J. Rine. 2003. The establishment, inheritance, and function of silenced chromatin in Saccharomyces cerevisiae. Annu. Rev. Biochem. 72:481-516. [DOI] [PubMed] [Google Scholar]
- 56.Salamon, D., M. Takacs, D. Ujvari, J. Uhlig, H. Wolf, J. Minarovits, and H. H. Niller. 2001. Protein-DNA binding and CpG methylation at nucleotide resolution of latency-associated promoters Qp, Cp, and LMP1p of Epstein-Barr virus. J. Virol. 75:2584-2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 58.Schaefer, B. C., J. L. Strominger, and S. H. Speck. 1997. Host-cell-determined methylation of specific Epstein-Barr virus promoters regulates the choice between distinct viral latency programs. Mol. Cell. Biol. 17:364-377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schepers, A., M. Ritzi, K. Bousset, E. Kremmer, J. L. Yates, J. Harwood, J. F. Diffley, and W. Hammerschmidt. 2001. Human origin recognition complex binds to the region of the latent origin of DNA replication of Epstein-Barr virus. EMBO J. 20:4588-4602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shareef, M. M., R. Badugu, and R. Kellum. 2003. HP1/ORC complex and heterochromatin assembly. Genetica 117:127-134. [DOI] [PubMed] [Google Scholar]
- 61.Sims, R. J., K. Nishioka, and D. Reinberg. 2003. Histone lysine methylation: a signature for chromatin function. Trends Genet. 19:629-639. [DOI] [PubMed] [Google Scholar]
- 62.Song, M. R., and A. Ghosh. 2004. FGF2-induced chromatin remodeling regulates CNTF-mediated gene expression and astrocyte differentiation. Nat. Neurosci. 7:229-235. [DOI] [PubMed] [Google Scholar]
- 63.Sterner, D. E., and S. L. Berger. 2000. Acetylation of histones and transcription-related factors. Microbiol. Mol. Biol. Rev. 64:435-459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Strahl, B. D., and C. D. Allis. 2000. The language of covalent histone modifications. Nature 403:41-45. [DOI] [PubMed] [Google Scholar]
- 65.Sugden, B., and N. Warren. 1989. A promoter of Esptein-Barr virus that can function during latent infection can be transactivated by EBNA-1, a viral protein required for viral DNA replication during latent infection. J. Virol. 63:2644-2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Takacs, M., D. Salamon, S. Myohanen, H. Li, J. Segesdi, D. Ujvari, J. Uhlig, H. H. Niller, H. Wolf, G. Berencsi, and J. Minarovits. 2001. Epigenetics of latent Epstein-Barr virus genomes: high resolution methylation analysis of the bidirectional promoter region of latent membrane protein 1 and 2B genes. Biol. Chem. 382:699-705. [DOI] [PubMed] [Google Scholar]
- 67.Tao, Q., L. J. Swinnen, J. Yang, G. Srivastava, K. D. Robertson, and R. F. Ambinder. 1999. Methylation status of the Epstein-Barr virus major latent promoter C in iatrogenic B cell lymphoproliferative disease. Application of PCR-based analysis. Am. J. Pathol. 155:619-625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Thorley-Lawson, D. A., and A. Gross. 2004. Persistence of the Epstein-Barr virus and the origins of associated lymphomas. N. Engl. J. Med. 350:1328-1337. [DOI] [PubMed] [Google Scholar]
- 69.Tierney, R., H. Kirby, J. Nagra, A. Rickinson, and A. Bell. 2000. The Epstein-Barr virus promoter initiating B-cell transformation is activated by RFX proteins and the B-cell-specific activator protein BSAP/Pax5. J. Virol. 74:10458-10467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tierney, R. J., H. E. Kirby, J. K. Nagra, J. Desmond, A. I. Bell, and A. B. Rickinson. 2000. Methylation of transcription factor binding sites in the Epstein-Barr virus latent cycle promoter Wp coincides with promoter down-regulation during virus-induced B-cell transformation. J. Virol. 74:10468-10479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Waltzer, L., F. Logeat, C. Brou, A. Israel, A. Sergeant, and E. Manet. 1994. The human J kappa recombination signal sequence binding protein (RBP-J kappa) targets the Epstein-Barr virus EBNA2 protein to its DNA responsive elements. EMBO J. 13:5633-5638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang, L., S. R. Grossman, and E. Kieff. 2000. Epstein-Barr virus nuclear protein 2 interacts with p300, CBP, and PCAF histone acetyltransferases in activation of the LMP1 promoter. Proc. Natl. Acad. Sci. USA 97:430-435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wensing, B., A. Stuher, P. Jenkins, M. Hollyoake, C. E. Karstegl, and P. J. Farrell. 2001. Variant chromatin structure of the OriP region of Epstein-Barr virus and regulation of EBER1 expression by upstream sequences and OriP. J. Virol. 75:6235-6241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Williams-Ashman, H. G., J. Seidenfeld, and P. Galletti. 1982. Trends in the biochemical pharmacology of 5′-deoxy-5′-methylthioadenosine. Biochem. Pharmacol. 31:277-288. [DOI] [PubMed] [Google Scholar]
- 75.Yoo, L., and S. H. Speck. 2000. Determining the role of the Epstein-Barr virus Cp EBNA2-dependent enhancer during the establishment of latency by using mutant and wild-type viruses recovered from cottontop marmoset lymphoblastoid cell lines. J. Virol. 74:11115-11120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yoo, L. I., J. Woloszynek, S. Templeton, and S. H. Speck. 2002. Deletion of Epstein-Barr virus regulatory sequences upstream of the EBNA gene promoter Wp1 is unfavorable for B-Cell immortalization. J. Virol. 76:11763-11769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zimber-Strobl, U., and L. J. Strobl. 2001. EBNA2 and Notch signalling in Epstein-Barr virus mediated immortalization of B lymphocytes. Semin. Cancer Biol. 11:423-434. [DOI] [PubMed] [Google Scholar]
- 78.Zimber-Strobl, U., L. J. Strobl, C. Meitinger, R. Hinrichs, T. Sakai, T. Furukawa, T. Honjo, and G. W. Bornkamm. 1994. Epstein-Barr virus nuclear antigen 2 exerts its transactivating function through interaction with recombination signal binding protein RBP-J kappa, the homologue of Drosophila Suppressor of Hairless. EMBO J. 13:4973-4982. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.