

Abstract
The nucleoprotein (NP) gene of the 1918 pandemic influenza A virus has been amplified and sequenced from archival material. The NP gene is known to be involved in many aspects of viral function and to interact with host proteins, thereby playing a role in host specificity. The 1918 NP amino acid sequence differs at only six amino acids from avian consensus sequences, consistent with reassortment from an avian source shortly before 1918. However, the nucleotide sequence of the 1918 NP gene has more than 170 differences from avian strain consensus sequences, suggesting substantial evolutionary distance from known avian strain sequences. Both the gene and protein sequences of the 1918 NP fall within the mammalian clade upon phylogenetic analysis. The evolutionary distance of the 1918 NP sequences from avian and mammalian strain sequences is examined, using several different parameters. The results suggest that the 1918 strain did not retain the previously circulating human NP. Nor is it likely to have obtained its NP by reassortment with an avian strain similar to those now characterized. The results are consistent with the existence of a currently unknown host for influenza, with an NP similar to current avian strain NPs at the amino acid level but with many synonymous nucleotide differences, suggesting evolutionary isolation from the currently characterized avian influenza virus gene pool.
The influenza A virus has a negative-strand RNA genome encoding at least 11 proteins on eight segments. Nucleoprotein (NP), encoded by the 1,565-bp segment 5, is a 498-amino-acid protein that has several different functions throughout the viral life cycle. It acts primarily as a single-strand RNA binding protein and serves as the structural protein in ribonucleoprotein particles (RNPs). In addition, it plays an important role in transcription and in the trafficking of RNPs between the cytoplasm and nucleus (20).
In virions, the viral RNA strands are wrapped around NP monomers and packaged into RNPs, with the three polymerase proteins bound to a short hairpin structure formed by the 5′ and 3′ ends of the RNA segment. NP, along with the three polymerase proteins, is essential for successful transcription and replication (13, 14). NP has been shown to bind PB1, PB2, and M1 directly. Upon infection, RNPs are released into the cytoplasm. The processes of being imported into the nucleus, exported back out to the cytoplasm, and then prevented from re-entering the nucleus also all appear to depend on the interaction of NP with host proteins. NP has been shown to interact with the cellular proteins alpha-importin, filamentous actin, and the nuclear export receptor CRM1 (reviewed in reference 20).
Structure and function studies have identified specific amino acids and regions involved in NP functions. Several highly conserved arginine and tryptophan residues have been shown to contribute to the ability of NP to bind RNA (7). NP has three regions that serve as nuclear localization signals (4) and another that contributes to cytoplasmic accumulation (6). NP is highly conserved, with a maximum amino acid difference of 11% among strains (20), probably because it must bind to multiple proteins, both viral and cellular. Numerous studies suggest that NP is a major determinant of host specificity (27, 28). One study found that a human-avian strain reassortant with NP as the only protein of avian strain origin was just as attenuated in the squirrel monkey respiratory tract as a reassortant containing all six avian strain core protein-encoding segments (30). The ability of these reassortants to replicate in eggs and in primary chicken kidney cultures suggests that their attenuation in the squirrel monkey is due to the inability of avian strain NP to interact with primate host cell factors. There is also evidence of selective immune pressure on the mammalian strain NP due to recognition of several epitopes on the NP protein by host T cells (2, 3, 5, 21, 33).
Phylogenetic analyses of NP sequences from many strains result in trees with two main branches, one consisting of mammalian-adapted strains and one of avian-adapted strains (11, 12, 29). The NP gene segment was not replaced in the pandemics of 1957 and 1968, so it is likely that the sequences in the mammalian clade are descended from the 1918 NP segment. The mammalian branches, unlike the avian branch, show a slow but steady accumulation of changes over time. Extrapolation of the rate of change along the human branch back to a putative common ancestor suggests that this NP entered the mammalian lineage sometime after 1900 (12, 29). One study suggests a much older common ancestor (11).
The 1918 influenza pandemic differed from the subsequent influenza pandemics of 1957 and 1968 in its virulence and in the age distribution of its victims. Analyses of the hemagglutinin (HA), NA, NS, and MA segments (1, 23-25) suggest that while these gene sequences were more closely related to those of avian strains than to those of any subsequent mammalian strains, they may not have emerged directly from avian sources immediately before the pandemic. Given the importance of NP in host adaptation and the phylogenetic evidence that the NP segment might have been circulating in humans for many decades before 1918, it was of interest to sequence the NP segment of the 1918 strain and carry out extensive phylogenetic analyses in hopes of determining its origin.
MATERIALS AND METHODS
Isolation of A/Brevig Mission/1/18 RNA and amplification of the NP segment.
RNA was isolated by methods previously described in other publications (15, 22). The 1,497-nucleotide open reading frame of gene segment 5 of strain A/Brevig Mission/1/18 (Brevig18) was amplified in 18 overlapping fragments, varying in length from 119 to 139 nucleotides. Each fragment was reverse transcribed, amplified, and sequenced at least twice. Reverse transcription-PCR, isolation of products, and sequencing have been described previously (23, 25). A list of primers used is available upon request.
Viral strains.
NP segment sequences used in this analysis were obtained from the Influenza Sequence Databank (ISD) (17). See Table 1 for a list of strains and abbreviations used.
TABLE 1.
Strains used in phylogenetic analyses
| Abbreviation | Strain name | GenBank no. |
|---|---|---|
| Shanghai90 | A/Shanghai/6/90 (H3N2) | L07357 |
| SwDandong83 | A/Swine/Dandong/9/83 (H3N2) | M63770 |
| Memphis78 | A/Memphis/18/78 (H3N2) | L07351 |
| Udorn72 | A/Udorn/307/72 (H3N2) | D00051 |
| Victoria68 | A/Victoria/5/68 (H2N2) | M63753 |
| SwHongKong76 | A/Swine/HongKong/6/76 (H3N2) | M22571 |
| Beijing68 | A/Beijing/1/68 (H3N2) | L07340 |
| HongKong68 | A/HongKong/1/68 (H3N2) | X15890 |
| Singapore57 | A/Singapore/1/57 (H2N2) | M63752 |
| AnnArbor60 | A/AnnArbor/6/60 (H2N2) | M23976 |
| Loygang57 | A/Loygang/4/57 (H1N1) | M76604 |
| England55 | A/England/19/55 (H1N1) | M63751 |
| Brazil78 | A/Brazil/11/78 (H1N1) | D00599 |
| Fort Warren50 | A/Fort Warren/1/50 (H1N1) | D00601 |
| Fm47 | A/Fort Monmouth/1/47 (H1N1) | U02086 |
| Hickox40 | A/Hickox/40 (H1N1) | M63749 |
| WS33 | A/Wilson-Smith/33 (H1N1) | M30746 |
| PR34 | A/Puerto Rico/8/34 (H1N1) | NC_002019 |
| Brevig18 | A/Brevig Mission/1/18 (H1N1) | AY744935 |
| SwineIowa46 | A/Swine/Iowa/46 (H1N1) | M63759 |
| Sw4149 | A/Swine/41/49 (H1N1) | M63760 |
| SwJamesburg42 | A/Swine/Jamesburg/42 (H1N1) | M63758 |
| SwIowa30 | A/Swine/Iowa/15/30 (H1N1) | M30747 |
| SwOhio35 | A/Swine/Ohio/23/35 (H1N1) | M63756 |
| Sw2937 | A/Swine/29/37 (H1N1) | M63757 |
| SwineMay54 | A/Swine/May/54 (H1N1) | M63761 |
| Sw Wisconsin57 | A/Swine/Wisconsin/1/57 (H1N1) | M63762 |
| SwWisconsin61 | A/Swine/Wisconsin/1/61 (H1N1) | M63763 |
| TyNC88 | A/Turkey/North Carolina/1790/88 | M76609 |
| SwNeb98 | A/Swine/Nebraska/209/98 (H3N2) | AF251407 |
| SwTennessee77 | A/Swine/Tennessee/24/77 (H1N1) | M30748 |
| SwHongKong82 | A/Swine/Hong Kong/127/82 (H3N2) | M22570 |
| EqPrag56 | A/Equine/Prague/56 (H7N7) | M22572 |
| EqMiami63 | A/Equine/Miami/1/63 (H3N8) | M22575 |
| EqKy86 | A/Equine/Kentucky/2/86 (H3N8) | M30751 |
| GullMD77 | A/Gull/Maryland/704/77 (H13N6) | M27521 |
| GullAstrakhan84 | A/Gull/Astrakhan/227/84 (H13N6) | M30753 |
| DkPA69 | A/Duck/Pennsylvania/1/69 (H6N1) | M63775 |
| MallNY78 | A/Mallard/New York//6750/78(H2N2) | D00050 |
| DkMM74 | A/Duck/Memphis/928/74 (H3N8) | M63776 |
| DkMT53 | A/Duck/Manitoba/1/53 (H10N7) | M63773 |
| TyOnt66 | A/Turkey/Ontario/7732/66 (H5N9) | M63774 |
| ChPA83 | A/Chicken/Pensylvania/1/83 (H5N2) | M30768 |
| TyMn80 | A/Turkey/Minnesota/833/80 (H4N2) | M30769 |
| GsGuangdong96 | A/Goose/Guangdong/1/96 (H5N1) | AF144303 |
| FPV34 | A/FPV/Rostock/34 (H7N1) | M22576 |
| ChGerm49 | A/Chicken/Germany/N/49 (H10N7) | M24453 |
| SwNeth85 | A/Swine/Netherlands/12/85 (H1N1) | M30749 |
| SwGerm81 | A/Swine/Germany/2/81 (H1N1) | M22579 |
| DkBav77 | A/Duck/Bavaria/2/77 (H1N1) | M22574 |
| TernSA61 | A/Tern/South Africa/61 (H5N3) | M30767 |
| DkHK75 | A/Duck/Hong Kong/7/75 (H3N2) | M22673 |
Phylogenetic analyses.
Phylogenetic analyses of NP were carried out using neighbor joining (NJ) (16). In most cases the proportion of differences was used as the distance measure. Character evolution was analyzed using the MacClade program (18), following a parsimony analysis using PAUP, with ACTRAN as the optimization method (32). Phylogenetically important positions (PIPs) and phylogenetically important regions (PIRs) were determined as described previously (8, 10). Only PIPs that reduced the number of steps in the tree by three or more were counted.
We calculated the probability of two PIPs being adjacent to one another randomly on the NP protein as follows. There are 40 PIPs scattered among the 498 amino acids making up the protein. Thus, the probability that any given amino acid is a PIP is 40/498. The probability that the adjacent, N-terminal amino acid is also a PIP is 39/497. The same probability holds if the adjacent amino acid is on the C-terminal side. Thus, the probability that either adjacent amino acid is a PIP is 2 × 39/497. We treated the initial change and the adjacent change as two independent events, even though they probably are not, since a change in one amino acid may foster or suppress a change in the adjacent amino acid. Nevertheless, assuming this boundary condition (independent events), the total probability that two adjacent amino acids are PIPs is 2 × 39/497 × 40/498 = ∼0.013.
Nucleotide sequence accession number. The nucleotide gene segment sequence of Brevig18 has been deposited in both the ISD and GenBank and given GenBank accession number AY744935.
RESULTS
Sequence of the NP gene.
The open reading frame sequence of the NP gene segment of Brevig18 (corresponding to nucleotides 46 to 1542, as aligned to A/WS/33 [GenBank accession number M30746]), containing 1,497 nucleotides, and a theoretical translation are shown in Fig. 1.
FIG. 1.

Sequence of the NP gene segment coding region of the 1918 strain. The coding sequence begins at nucleotide 46. The NP theoretical translation is shown above the sequence. The PIRs are boxed; within the PIRs, the PIPs are marked with asterisks. The numbering of the gene segment is aligned to that of A/WS/33 (GenBank accession number M30746).
Functional areas of the NP gene.
The NP protein has been found to have roles throughout the influenza virus replication cycle. However, no specific motifs or amino acids have been associated with increased virulence or replication efficiency. By contrast, a decrease of NP function has been associated with the loss of certain regions or changes in some amino acid residues (7, 34). These sites are virtually universally conserved among NP sequences, and those of the 1918 strain match the conserved sequence at these residues. At least seven different sites, containing 74 amino acids, have been proposed to be T-cell recognition sites (3, 7, 33, 34). The 1918 strain matches the avian consensus at all 74 residues, whereas six of these amino acids are different in later human strains.
Phylogenetic analyses of the NP gene. (i) PAUP and NJ analyses.
A phylogenetic analysis of 52 NP genes was conducted, using NJ as the tree-building algorithm and the proportion of differences as the distance measure (16). Only the coding portion of the gene was analyzed. The tree defines two large clades (Fig. 2): a mammalian clade with human and classical swine subclades and an avian clade containing equine, gull, and waterfowl subclades. The waterfowl subclade is further divided into North American and Eurasian branches. The sequence of the 1918 NP gene is within and near the root of the swine viruses. Separate analyses of synonymous and nonsynonymous substitutions also placed the 1918 virus NP gene in the mammalian clade. When synonymous substitutions were analyzed, the 1918 virus gene was placed within and near the root of swine viruses, as seen in Fig. 2. When nonsynonymous viruses were analyzed, the 1918 virus gene was placed within and near the root of the human viruses. Both analyses yielded trees with high bootstrap values: 100 for the synonymous tree and 91 for the nonsynonymous tree (not shown).
FIG. 2.
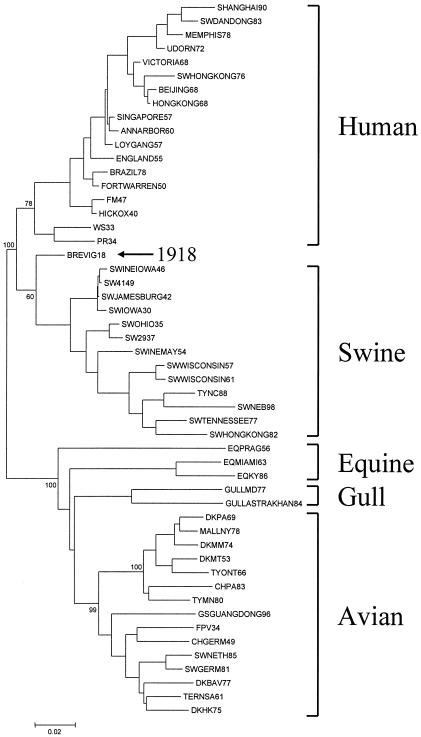
Total nucleotide phylogenetic tree of influenza virus NP gene sequences. The tree was constructed using NJ, with the proportion of nucleotide differences as the distance measure. Bootstrap values are given for selected nodes of the tree. The position of the 1918 strain sequence is given by the arrow, and a distance bar is shown below the tree.
A parsimony analysis (PAUP) using the same data set and midpoint rooting produced two trees of 2,236 steps. Both trees placed the 1918 viral NP nucleotide sequence within and near the root of the human lineage. If the 1918 strain sequence were moved to near the root of the swine strain lineage (18), the tree length would increase 12 steps. Moving the 1918 strain sequence to the North American or Eurasian avian strain branches of the tree increased the tree length by 120 or 122 steps, respectively.
The 1918 strain viral protein sequence is also placed within and near the root of the human strain viral NPs by the NJ method. As was found with the other gene segments of the 1918 virus (1, 23-25), the 1918 strain NP is grouped phylogenetically with subsequent mammalian viruses but retains many sequence features found in avian viruses. A BLAST search using amino acid sequences demonstrates that the protein of the 1918 strain is as similar, or more similar, to avian strain NP proteins as to mammalian strain NP proteins. For example, there are eight amino acid differences between the 1918 strain and DkBav77, but there are 11 differences between the 1918 strain and SwIowa30 and 16 differences between the 1918 strain and WS/33. A BLAST search using nucleotide sequences, however, demonstrates the reverse: the 1918 strain is most similar to mammalian strain sequences. For example, there are 67 differences between the 1918 strain and SwIowa30 (95.5% identity) and 65 differences between the 1918 strain and WS33 (95.6% identity), but there are 193 differences between the 1918 strain and DkBav77 (87.1% identity). It is somewhat paradoxical that phylogenetic analyses and nucleotide comparisons suggest that the NP of the 1918 strain is more closely related to mammalian strain sequences than to avian strain ones, when the NP protein of the 1918 strain differs from its nearest avian strain relative at only eight amino acids. The placement of the 1918 strain NP in the mammalian clade suggests that the small number of amino acid differences between the 1918 strain NP and those of avian strains is conserved throughout the mammalian clade and thus is sufficient to define a separate clade. If the 1918 strain NP had been retained from the previously circulating human strain, its position outside the avian clade would be expected. However, the small number of differences from the avian strain might suggest a recent avian origin. We looked at several other measures of sequence relatedness to try to resolve this paradox.
(ii) Regression analyses.
Previous phylogenetic analyses of NP have come to different conclusions about the date of entry of the NP gene into the mammalian clade. One group estimated an entry date in the mid-1800s (11), while analyses performed by others suggest a date just prior to the 1918 pandemic (12, 29).
A regression analysis using one of the PAUP trees suggested a change of approximately three steps per year for mammalian viral isolates (Fig. 3). Both human and swine strain sequences appear to evolve at the same rate when analyzed by this method. The regression line extrapolates to around 1915 (Fig. 3) for the initial circulation of the mammalian strain progenitor, but the lack of pre- and post-1918 sequences precludes a more detailed analysis. The data point for the 1918 strain falls just above the regression line, which may simply be the result of random scatter, since the 1954 time point falls below the regression line to an equal extent. To quantify the rate of change more precisely, we used MEGA software to determine the exact number of nucleotide changes between the swine sequences used in Fig. 3. All pairwise combinations were examined, and the numbers of nucleotide changes per year were calculated. The average number of nucleotide changes per year calculated by this method was 3.3, a number identical to that found by others (12).
FIG. 3.
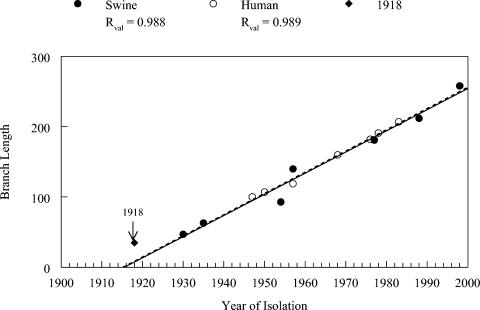
Changes in NP gene sequences over time. The number of nucleotide changes from a hypothetical mammalian strain ancestor were plotted versus the year of viral isolation for swine strain (closed circles) and human strain (open circles) lineages. The position of the 1918 strain sequence is shown by the arrow.
(iii) Phylogenetically important regions of the protein.
Previously published studies have defined PIRs as clusters of amino acids that shorten phylogenetic trees in comparison to randomly generated trees (8, 10). These regions potentially reflect important functional and evolutionary differences among groups of sequences. To identify PIRs of the NP protein, we performed a parsimony analysis of the proteins encoded by the sequences used in Fig. 2, using PAUP. We then imported one of the PAUP trees into MacClade and rearranged the tree to look like the NJ tree of Fig. 2. An examination of all 498 amino acid positions resulted in the identification of six PIRs, ranging in size from 2 to 7 amino acids in length (Fig. 1; also Materials and Methods). For example, the first PIR in Fig. 1 encompasses amino acid positions 31 to 34, with positions 31, 33, and 34 containing significant phylogenetic information, while position 32 has no phylogenetic information. We calculate the probability of two phylogenetically important positions being adjacent to one another randomly at ∼ 0.013 (see Materials and Methods). Thus, while some PIRs (primarily the short, 2-amino-acid variety) may be spurious and due to happenstance, we feel that most are probably due to selection and may identify host-specific functional regions of the protein.
PIRs of the HA and NA proteins of influenza virus primarily identify antigenic amino acid residues, although some also identify receptor binding and glycosylation residues (8, 10). PIRs A, B, and E of NP contain amino acids known to be involved in NP function and/or immunogenicity. The other three PIRs, C, D, and F, have not been associated with any specific function of the NP protein as far as we have been able to determine, although C and D are immediately adjacent to T-cell recognition regions.
PIR A is within a putative nuclear export signal (19). At two of the sites in this PIR (positions 31 and 34), the 1918 strain sequence matches the avian consensus strain, while those of later human strains have changed. At the third site (position 33), the 1918 strain and all subsequent human strains differ from the avian consensus strain. PIR B is within a cytotoxic T-cell recognition region (33) and a nuclear localization region. The PIPs within PIR B appear to define the equine clade. Sequences of the 1918 strain match the human, swine, and avian strain sequences at these sites. In both PIRs C and D, which are adjacent to identified T-cell recognition regions, the 1918 strain matches the avian consensus strain at all PIPs, while at four sites (positions 351 and 353 in PIR C and 372 and 375 in PIR D) later human strains have acquired a different amino acid, possibly consistent with positive selection pressure due to T-cell recognition. PIR E is also within a known cytotoxic T-lymphocyte site (3). At all four PIPs, the 1918 strain matches the avian consensus strain, while later human strains have changed, again consistent with positive selection pressure at these sites. At PIR F, the 1918 strain matches the avian consensus strain at one PIP (position 455) and the Eurasian avian consensus strain at a second PIP (position 450). Later human strains differ from the 1918 strain at both of these PIPs. Again, the pattern is consistent with positive selection pressure on these sites in the human strain lineage.
(iv) Amino acids that distinguish the 1918 strain NP from avian strain NPs.
The HA1 domain of the 1918 strain HA differs from the avian consensus strain sequence at 25 amino acids. One of these, E190D, is part of the HA receptor binding site and was also found to have changed when an avian H1N1 virus emerged in European pigs, suggesting an important role for this amino acid in mammalian strain adaptation (23, 31). The 1918 strain NP was examined to determine whether there were any similarly informative amino acids. There are six amino acid positions that distinguish the 1918 strain NP protein from its avian strain counterparts (Table 2). Three of the positions, 16, 283, and 313, have substitutions that the 1918 strain viral protein shares with other viruses in the human clade. One of the substitutions, at position 283, is especially intriguing, since the leucine found in the avian, equine, and swine viruses is replaced with proline in Brevig and in human viruses. This replacement could result in a drastic, localized conformational alteration in the protein. At the other three positions, 33, 100, and 136, the 1918 strain sequence is conserved in both human and swine viruses, separating them from their avian and equine counterparts. Only position 33 is in a PIR as described above. A seventh position, 473, has S in the 1918 strain and in several human and swine isolates. Most human and swine isolates and all equine and avian isolates have N at this position, except FPV34, which also has S.
TABLE 2.
Amino acids that distinguish the 1918 strain from the avian clade
| Amino acid position | Amino acid in indicated strain:
|
||||
|---|---|---|---|---|---|
| 1918 | Human | Swine | Equine | Avian | |
| 16 | D | D | G | G | G |
| 33 | I | I | I | V | V |
| 100 | I | V | Ia | R | R |
| 136 | M | M | M/I | L | L |
| 283 | P | P | L | L | L |
| 313 | Y | Y | F | F | F |
A few strains have V at this position.
We compared the amino acid changes that differentiated the 1918 virus from its avian virus relatives and asked whether any of these changes had also taken place during the introduction of an avian virus into European swine in the late 1970s. We found no amino acid replacements in the 1918 strain and avian-like swine viruses at identical amino acid positions.
(v) Ratio of synonymous to nonsynonymous changes.
We compared the synonymous/nonsynonymous (S/N) ratios within different lineages in the NP phylogenetic tree. As expected, within the avian clade, where influenza virus is thought to be endemic, the S/N ratio averages 15.2. This finding suggests that in avian strains the NP is so well adapted that most amino acid changes would be deleterious or neutral, so that silent nucleotide changes predominate. Within the mammalian clade, the S/N ratio averages 3.9, suggestive of an actively evolving protein in which many amino acid changes provide a selective advantage. When the sequence from the 1918 strain was compared to those of older mammalian isolates, the S/N ratios varied from 2.7 (1918 strain versus WS33) to 5.1 (1918 strain versus SwIowa30). The S/N ratios are similar when more-recent human and swine isolates are compared with the 1918 virus. For example, the ratio is 3.0 for a more-recent human isolate (1918 strain versus AnnArbor60) and 4.7 for a more-recent swine isolate (1918 strain versus SwWisc61). These numbers differ substantially from those comparing the 1918 strain and avian strains. Comparing the sequence of the 1918 strain to those of 10 avian species chosen from both the Eurasian and North American avian subclades gave an average S/N ratio of 13.9, with a range of 10.4 to 18.0. These data reinforce the results of the BLAST searches; compared to avian sequences, the 1918 strain sequence has many changes at the nucleotide level, but only a very few of these result in amino acid changes.
(vi) Comparison of consensus sequences.
When the sequence of the 1918 strain is compared to any individual avian strain NP sequence, it is found to be closely related at the protein level but quite distantly related at the nucleotide level. NP from the 1918 strain differs at 189 nucleotides and 11 amino acids from that of DkMT53, a typical North American bird strain, and 193 nucleotides and 8 amino acids from that of DkBav77, a typical Eurasian bird strain. Avian strains also differ from each other extensively at the nucleotide level, while differing only slightly at the amino acid level. DkBav77 differs from DkMT53 at 130 nucleotides and three amino acids. The sequences clearly fall in two separate clades (Fig. 2) that have probably been distinct for many hundreds, if not thousands, of years (35-37). These numbers are similar to those of the 1918 strain/avian strain comparison ratio made above in that there are many more synonymous substitutions than nonsynonymous ones. A preponderance of synonymous substitutions resulting in no amino acid replacements is expected for a virus well adapted to its host. Perhaps, given that avian influenza viruses have not been exhaustively characterized, there could be a currently uncharacterized avian strain that would share many of the nucleotide differences found in the 1918 strain.
Avian strain sequences vary from each other at many synonymous sites. However, they are all similarly distant from the avian consensus strain sequence. Therefore, if the 1918 virus attained its NP from a bird similar to those currently known, it should also share a similar number of synonymous changes with the avian consensus strain. One would also expect the 1918 strain to be more similar either to the Eurasian consensus strain or the North American consensus strain, as is true of currently sequenced avian strains. In order to determine whether the 1918 strain sequence might resemble an average avian strain sequence more closely than it resembles any individual avian strain sequence, we compared the 1918 strain sequence with consensus sequences derived from the viral isolates shown in Fig. 2. Six consensus sequences were used: those of human, swine (minus the 1918 strain sequence), North American avian, Eurasian avian, mammalian (human and swine combined), and avian (North American avian and Eurasian avian combined) strains. The 1918 virus differs at 173 nucleotides from the avian consensus strain sequence, at 187 nucleotides from the North American avian consensus strain sequence, and at 179 nucleotides from the Eurasian avian consensus strain sequence. Thus, it is only slightly more closely related to the consensus strain sequences than it is to individual strains, and it is no more closely related to one avian clade than to the other. By contrast, individual avian strain sequences are more closely related to the avian consensus strain sequences than they are to each other and much more closely related to the consensus of their own clade than to the consensus of the other clade. Thus, DkMT53 differs from the avian consensus strain sequence at 71 nucleotides, from the North American consensus strain sequence at 40 nucleotides, and from the Eurasian consensus strain sequence at 116 nucleotides. DkBav77 differs from the avian consensus strain sequence at 73 nucleotides, the Eurasian consensus strain sequence at 51 nucleotides, and the North American consensus strain sequence at 137 nucleotides.
It is possible that the 1918 strain sequence might be more similar to avian strains circulating early in the last century than to contemporary strains. However, consistent with substantial evidence that avian strain sequences do not evolve over time as do mammalian strain sequences, sequences obtained from birds collected just prior to 1918 are very similar to contemporary avian strain sequences (9). In a previous study, 151 bases of NP sequence were obtained from two birds, a cinnamon teal collected in Bear River, Utah, in 1916 and a Brant goose, collected in Alaska in 1917. The sequences differed at one nucleotide from each other and were located in the North American avian clade phylogenetically (9, 26). Comparison of the pre-1918 avian strains to the avian consensus sequences show that they have 9/150 (6%) differences from the avian consensus strain, 6/150 (4%) from the North American avian consensus strain, and 12/150 (7.9%) from the Eurasian avian consensus strain. Over these 150 nucleotides, the 1918 strain NP differs from that of the pre-1918 birds at 21 nucleotides (13.9%); 20 of the differences are synonymous and 1 is nonsynonymous. Both the number of differences and the S/N ratio are similar to the relationship of the 1918 strain NP to modern avian strain sequences.
The 1918 strain sequence is more closely related to the mammalian consensus strain sequences than to the avian consensus strain sequences. It differs at 73 nucleotides from the mammalian consensus strain, 104 nucleotides from the human consensus strain, and 83 nucleotides from the swine consensus strain. The observation that the 1918 strain sequence differs at so many nucleotides from the avian consensus strain suggests that it is unlikely that new samples of the currently identified avian clades would be more closely related to the 1918 sequence.
(vii) Comparison of 4-fd sites.
We examined the numbers of changes at sites that are fourfold degenerate (4-fd). These sites can have any of the four bases, with no resulting amino acid replacement. Such sites have been shown to evolve nearly as rapidly as pseudogenes, the most rapidly evolving sequences in genomes. If influenza virus genes have been evolving in birds for long enough to reach evolutionary stasis (35), as is suggested by the high S/N ratios described above, one would predict that at many of the sites where fourfold degeneracy is possible all four bases would be present in the avian clade unless the constraints of RNA secondary structure limit the accumulation of synonymous changes. By contrast, in the mammalian clade, if all subsequent mammalian influenza viruses acquired their NP gene segment from the 1918 virus (i.e., comparatively recently), most of the 4-fd sites would still have the same base. Indeed, of 211 4-fd sites shared by the 1918 strain and SwIowa30 (a closely related mammalian isolate), 85% have the identical base. By contrast, when the 1918 strain is compared with TyMN80 (a closely related avian isolate), of 212 shared 4-fd sites only 58% have the identical base. Among avian isolates, however, where one might expect to find greater heterogeneity at 4-fd sites, the percentages of shared sites are high. For example, 85% of the shared 4-fd sites are identical in a comparison between TyMN80 and DkMT53 (members of the North American avian clade). The number drops to 73% between TyMN80 and DkBav77 (a North American avian strain and a Eurasian avian strain). Since 4-fd sites are expected to become heterogeneous over time (two sequences should show about 25% identity at fourfold sites at equilibrium), these data suggest that nonrandom forces are acting on the avian strain sequences. These could involve periodic homogenization events both within and between avian clades or selection, as discussed above, if a number of 4-fd sites are involved in RNA secondary structure. Nevertheless, the low percentage of shared fourfold sites between sequences of the 1918 strain and avian strain sequences is another indication of substantial evolutionary distance and makes it less likely that the NP gene segment came directly from a currently known avian source just before the pandemic.
DISCUSSION
There are at least three possibilities for the origin of the 1918 NP gene segment. First, it could have been retained from the previously circulating human virus, as was the case with the 1957 and 1968 pandemic strains, whose NP segments are descendants of the 1918 NP. The large number of nucleotide changes from the avian consensus strain and the placement of the 1918 strain sequence in the mammalian clade are consistent with this hypothesis. NJ analyses of nonsynonymous nucleotide sequences or of amino acid sequences place the 1918 strain sequence within and near the root of the human clade. The 1918 strain NP has only a few amino acid differences from most bird strains, but this consistent group of amino acid changes is shared by the 1918 strain NP and its subsequent mammalian strain descendants and is not found in any birds, resulting in the 1918 strain sequence being placed outside the avian clade (Table 2). One or more of these amino acid substitutions may be important for adaptation of the protein to humans and, indeed, positions 33 and 136 were previously identified as being important in defining the human-swine clade versus its avian progenitor (12). However, none of these changes have been found during the recent introduction of an avian H1N1 virus into European swine. Therefore, it would appear that these changes are not the only way an avian strain NP can adapt to function in mammals. The percentage of shared 4-fd sites between 1918 strain and avian strain sequences is quite low, again suggesting substantial evolutionary distance. However, the very small number of amino acid differences from the avian consensus strain argues for recent introduction from birds; 80 years after 1918, the NP gene (for example, A/Nagasaki/93/98) segment had accumulated 34 additional amino acid differences and only about 95 nucleotide differences (a rate of 2.3 amino acid changes per year) from the avian consensus strain. Thus, it seems unlikely that the 1918 strain NP, with only six amino acid differences from the avian consensus strain, could have been in humans for many years before 1918. This conclusion is supported by the regression analysis that suggests that the progenitor of the 1918 virus probably entered the human population around 1915. This hypothesis can be formally tested only if a pre-1918 human influenza virus is obtained.
A second possible origin for the 1918 NP strain segment is direct reassortment from an avian strain virus. The small number of amino acid differences between the 1918 strain and the avian consensus strain supports this hypothesis. Also, in the regions shown to be phylogenetically important (PIRs), the 1918 strain closely resembles the avian consensus strain. While the 1918 strain varies at many nucleotides from the nearest avian strain, avian strains are quite diverse at the nucleotide level. S/N ratios between the 1918 strain and avian strains are similar to the ratios between avian strains, allowing the possibility that avian strains may exist that are more closely related to the 1918 strain. However, the 1918 strain sequence shares far fewer 4-fd sites with avian strains (58%) than do even distantly related avian strains (73%), and comparisons of the 1918 strain sequence to avian consensus strains also underline the great evolutionary distance between the 1918 strain and the known avian clades. So, while the very small number of amino acid differences suggests that adaptation could have been rapid, it is difficult to explain the acquisition of more than 170 silent mutations, even postulating a rapid early mutation rate. Regression analysis shows that the rate of accumulation of nucleotide changes in the human lineage from 1918 to 1998 is around three changes per year, suggesting that the gene would take more than 50 years to accumulate 170 differences from the average avian strain. We know of no plausible mechanism that would favor a preponderance of substitutions at silent sites except for selective removal of nonsilent substitutions. The low ratio of S/N changes in the human lineage since 1918 suggests that many nonsilent substitutions can be tolerated. The great evolutionary distance between the 1918 strain sequence and the avian consensus strains suggests that no avian strain similar to those in the currently identified clades could have provided the 1918 strain with its NP segment. NP sequences from two pre-1918 avian viruses (26) demonstrate that there has been very little change in wild duck influenza sequences over an 80-year period.
A final possibility is that the 1918 strain gene segment was acquired shortly before 1918 from a source not currently represented in the database of influenza virus sequences. The ISD contains more than 300 avian strain NP sequences, of which 175 are full length. A NJ phylogenetic tree (data not shown) places all 175 of them in the same three clades (North American and Eurasian waterfowl and gull) as the 17 avian strain sequences used in this paper's analysis (Fig. 2). The two waterfowl clades are as different from each other as they are from the 1918 strain sequence. The NP gene sequences in the gull clade are even more divergent from those of the other avian clades than is the 1918 strain sequence. There may be unknown influenza virus hosts, for example, other groups of birds, which while similar to currently characterized avian strains at the amino acid level are quite different at the nucleotide level. It is possible that such a host was the source of the 1918 strain NP segment. This hypothesis can be tested only by demonstrating the existence of an additional clade of influenza virus strains similar to the 1918 virus.
Acknowledgments
This study was supported by grants from the National Institutes of Health, the American Registry of Pathology, and the Department of Veterans Affairs and by intramural funds of the Armed Forces Institute of Pathology.
The opinions contained herein are the private views of the authors and are not to be construed as official or as reflecting the views of the Department of the Army or the Department of Defense.
REFERENCES
- 1.Basler, C. F., A. H. Reid, J. K. Dybing, T. A. Janczewski, T. G. Fanning, H. Zheng, M. Salvatore, M. L. Perdue, D. E. Swayne, A. Garcia-Sastre, P. Palese, and J. K. Taubenberger. 2001. Sequence of the 1918 pandemic influenza virus nonstructural gene (NS) segment and characterization of recombinant viruses bearing the 1918 NS genes. Proc. Natl. Acad. Sci. USA 98:2746-2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Belz, G. T., W. Xie, J. D. Altman, and P. C. Doherty. 2000. A previously unrecognized H-2Db-restricted peptide prominent in the primary influenza A virus-specific CD8+ T-cell response is much less apparent following secondary challenge. J. Virol. 74:3486-3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boon, A. C., G. de Mutsert, Y. M. Graus, R. A. Fouchier, K. Sintnicolaas, A. D. Osterhaus, and G. F. Rimmelzwaan. 2002. Sequence variation in a newly identified HLA-B35-restricted epitope in the influenza A virus nucleoprotein associated with escape from cytotoxic T lymphocytes. J. Virol. 76:2567-2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bui, M., J. E. Myers, and G. R. Whittaker. 2002. Nucleo-cytoplasmic localization of influenza virus nucleoprotein depends on cell density and phosphorylation. Virus Res. 84:37-44. [DOI] [PubMed] [Google Scholar]
- 5.Danke, N. A., and W. W. Kwok. 2003. HLA class II-restricted CD4+ T cell responses directed against influenza viral antigens postinfluenza vaccination. J. Immunol. 171:3163-3169. [DOI] [PubMed] [Google Scholar]
- 6.Digard, P., D. Elton, K. Bishop, E. Medcalf, A. Weeds, and B. Pope. 1999. Modulation of nuclear localization of the influenza virus nucleoprotein through interaction with actin filaments. J. Virol. 73:2222-2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Elton, D., L. Medcalf, K. Bishop, D. Harrison, and P. Digard. 1999. Identification of amino acid residues of influenza virus nucleoprotein essential for RNA binding. J. Virol. 73:7357-7367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fanning, T. G., A. H. Reid, and J. K. Taubenberger. 2000. Influenza A virus neuraminidase: regions of the protein potentially involved in virus-host interactions. Virology 276:417-423. [DOI] [PubMed] [Google Scholar]
- 9.Fanning, T. G., R. D. Slemons, A. H. Reid, T. A. Janczewski, J. Dean, and J. K. Taubenberger. 2002. 1917 avian influenza virus sequences suggest that the 1918 pandemic virus did not acquire its hemagglutinin directly from birds. J. Virol. 76:7860-7862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fanning, T. G., and J. K. Taubenberger. 1999. Phylogenetically important regions of the influenza A H1 hemagglutinin protein. Virus Res. 65:33-42. [DOI] [PubMed] [Google Scholar]
- 11.Gammelin, M., A. Altmuller, U. Reinhardt, J. Mandler, V. Harley, P. Hudson, W. Fitch, and C. Scholtissek. 1990. Phylogenetic analysis of nucleoproteins suggests that human influenza A viruses emerged from a 19th-century avian ancestor. Mol. Biol. Evol. 7:194-200. [DOI] [PubMed] [Google Scholar]
- 12.Gorman, O., W. Bean, Y. Kawaoka, I. Donatelli, Y. Guo, and R. Webster. 1991. Evolution of influenza A virus nucleoprotein genes: implications for the origins of H1N1 human and classical swine viruses. J. Virol. 65:3704-3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Honda, A., K. Ueda, K. Nagata, and A. Ishihama. 1988. RNA polymerase of influenza virus: role of NP in RNA chain elongation. J. Biochem. (Tokyo) 104:1021-1026. [DOI] [PubMed] [Google Scholar]
- 14.Huang, T. S., P. Palese, and M. Krystal. 1990. Determination of influenza virus proteins required for genome replication. J. Virol. 64:5669-5673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Krafft, A. E., B. W. Duncan, K. E. Bijwaard, J. K. Taubenberger, and J. H. Lichy. 1997. Optimization of the isolation and amplification of RNA from formalin- fixed, paraffin-embedded tissue: the Armed Forces Institute of Pathology experience and literature review. Mol. Diagn. 2:217-230. [DOI] [PubMed] [Google Scholar]
- 16.Kumar, S., K. Tamura, I. B. Jakobsen, and M. Nei. 2001. MEGA2: molecular evolutionary genetics analysis software, version 2.1. Arizona State University, Tempe, Ariz. [DOI] [PubMed]
- 17.Macken, C., H. Lu, J. Goodman, and L. Boykin. 2001. The value of a database in surveillance and vaccine selection, p. 103-106. In A. D. M. E. Osterhaus, N. Cox, and A. W. Hampson (ed.), Options for the control of influenza IV. Elsevier Science, Amsterdam, The Netherlands.
- 18.Maddison, W. P., and D. R. Maddison. 1992. MacClade: analysis of phylogeny and character evolution, version 3. Sinauer Associates, Sunderland, Mass.
- 19.Neumann, G., M. R. Castrucci, and Y. Kawaoka. 1997. Nuclear import and export of influenza virus nucleoprotein. J. Virol. 71:9690-9700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Portela, A., and P. Digard. 2002. The influenza virus nucleoprotein: a multifunctional RNA-binding protein pivotal to virus replication. J. Gen. Virol. 83:723-734. [DOI] [PubMed] [Google Scholar]
- 21.Potter, P., S. Tourdot, T. Blanchard, G. L. Smith, and K. G. Gould. 2001. Differential processing and presentation of the H-2Db-restricted epitope from two different strains of influenza virus nucleoprotein. J. Gen. Virol. 82:1069-1074. [DOI] [PubMed] [Google Scholar]
- 22.Reid, A. H., R. E. Cunningham, G. Frizzera, and T. J. O'Leary. 1993. bcl-2 rearrangement in Hodgkin's disease. Results of polymerase chain reaction, flow cytometry, and sequencing on formalin-fixed, paraffin-embedded tissue. Am. J. Pathol. 142:395-402. [PMC free article] [PubMed] [Google Scholar]
- 23.Reid, A. H., T. G. Fanning, J. V. Hultin, and J. K. Taubenberger. 1999. Origin and evolution of the 1918 “Spanish” influenza virus hemagglutinin gene. Proc. Natl. Acad. Sci. USA 96:1651-1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reid, A. H., T. G. Fanning, T. A. Janczewski, S. McCall, and J. K. Taubenberger. 2002. Characterization of the 1918 “Spanish” influenza virus matrix gene segment. J. Virol. 76:10717-10723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reid, A. H., T. G. Fanning, T. A. Janczewski, and J. K. Taubenberger. 2000. Characterization of the 1918 “Spanish” influenza virus neuraminidase gene. Proc. Natl. Acad. Sci. USA 97:6785-6790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reid, A. H., T. G. Fanning, R. D. Slemons, T. A. Janczewski, J. Dean, and J. K. Taubenberger. 2003. Relationship of pre-1918 avian influenza HA and NP sequences to subsequent avian influenza strains. Avian Dis. 47:921-925. [DOI] [PubMed] [Google Scholar]
- 27.Scholtissek, C., H. Burger, O. Kistner, and K. F. Shortridge. 1985. The nucleoprotein as a possible major factor in determining host specificity of influenza H3N2 viruses. Virology 147:287-294. [DOI] [PubMed] [Google Scholar]
- 28.Scholtissek, C., I. Koennecke, and R. Rott. 1978. Host range recombinants of fowl plague (influenza A) virus. Virology 91:79-85. [DOI] [PubMed] [Google Scholar]
- 29.Shu, L., W. Bean, and R. Webster. 1993. Analysis of the evolution and variation of the human influenza A virus nucleoprotein gene from 1933 to 1900. J. Virol. 67:2723-2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Snyder, M. H., A. J. Buckler-White, W. T. London, E. L. Tierney, and B. R. Murphy. 1987. The avian influenza virus nucleoprotein gene and a specific constellation of avian and human virus polymerase genes each specify attenuation of avian-human influenza A/Pintail/79 reassortant viruses for monkeys. J. Virol. 61:2857-2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stevens, J., A. L. Corper, C. F. Basler, J. K. Taubenberger, P. Palese, and I. A. Wilson. 2004. Structure of the uncleaved human H1 hemagglutinin from the extinct 1918 influenza virus. Science 303:1866-1870. [DOI] [PubMed] [Google Scholar]
- 32.Swofford, D. L. 1991. PAUP: phylogenetic analysis using parsimony, version 3.1.1. Illinois Natural History Survey, University of Illinois, Champaign, Ill.
- 33.Voeten, J. T., T. M. Bestebroer, N. J. Nieuwkoop, R. A. Fouchier, A. D. Osterhaus, and G. F. Rimmelzwaan. 2000. Antigenic drift in the influenza A virus (H3N2) nucleoprotein and escape from recognition by cytotoxic T lymphocytes. J. Virol. 74:6800-6807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weber, F., G. Kochs, S. Gruber, and O. Haller. 1998. A classical bipartite nuclear localization signal on Thogoto and influenza A virus nucleoproteins. Virology 250:9-18. [DOI] [PubMed] [Google Scholar]
- 35.Webster, R. G., W. J. Bean, O. T. Gorman, T. M. Chambers, and Y. Kawaoka. 1992. Evolution and ecology of influenza A viruses. Microbiol. Rev. 56:152-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Webster, R. G., G. B. Sharp, and E. C. Claas. 1995. Interspecies transmission of influenza viruses. Am. J. Respir. Crit. Care Med. 152:S25-S30. [DOI] [PubMed] [Google Scholar]
- 37.Webster, R. G., S. M. Wright, M. R. Castrucci, W. J. Bean, and Y. Kawaoka. 1993. Influenza—a model of an emerging virus disease. Intervirology 35:16-25. [DOI] [PubMed] [Google Scholar]