

Abstract
Tumor necrosis factor-alpha (TNF-α) has been shown to cause apoptosis of gingival epithelial cells (GECs) in periodontitis. However, the underlying molecular mechanism is still unclear. In this study, we showed that miR-142 expression was significantly elevated in human GECs after exposure to TNF-α. Such induction was in a time- and concentration-dependent manner. Serum miR-142 levels were positively correlated with serum TNF-α levels in patients with chronic periodontitis (r = 0.314, P = 0.0152). Depletion of miR-142 was found to attenuate TNF-α-induced apoptosis, as determined by TUNEL staining and caspase-3 activity assays. In contrast, overexpression of miR-142 significantly reduced viability and induced apoptosis in GECs. Basic leucine zipper transcription factor 2 (BACH2) was identified to be a functional target of miR-142. Overexpression of miR-142 caused a 3-fold reduction of BACH2 protein in primary GECs. Overexpression of BACH2 significantly reversed miR-142- or TNF-α-induced apoptosis of GECs. Similar to the findings with miR-142 mimic, depletion of BACH2 significantly promoted apoptosis in GECs, which was accompanied by decreased expression of Bcl-2 and Bcl-xL and increased expression of Bax and Bim. Overall, miR-142 mediates TNF-α-induced apoptosis in gingival epithelial cells by targeting BACH2 and may represent a potential therapeutic target for periodontitis.
Keywords: Apoptosis, chronic periodontitis, gingival epithelial cells, inflammatory cytokines, microRNA
Introduction
Chronic periodontitis is a common disorder of the oral cavity caused by pathogenic microorganisms [1,2]. This disease is characterized by massive inflammatory cell infiltration into periodontal tissues and overproduction of pro-inflammatory cytokines including tumor necrosis factor-alpha (TNF-α), leading to periodontal tissue destruction and tooth loss. TNF-α is a multifunctional cytokine that plays an important role in periodontitis [3]. It has been documented that TNF-α can increase the permeability of gingival epithelial cells (GECs) and augment the invasion of periodontal pathogens [4]. This cytokine also induces receptor activator of nuclear factor kβ ligand (RANKL) expression in GECs, consequently promoting osteoclast formation and periodontal bone resorption [5]. In addition, TNF-α can exert direct destructive effects on GECs, e.g. triggering apoptotic death [6]. However, the mechanism underlying TNF-α-induced apoptosis in GECs is still unclear.
microRNAs (miRNAs) are endogenous, small noncoding RNAs that participate in a broad range of cellular processes, such as cell proliferation, differentiation, apoptosis, and inflammation [7]. miRNAs can negatively regulate gene expression by binding to the 3’-untranslated region (UTR) of target mRNAs, resulting in mRNA degradation and translational repression. Profiling studies revealed that many miRNAs are differentially expressed in healthy and inflamed gingival tissues from patients with periodontitis [8]. However, only a small subset of miRNAs are functionally validated in the pathobiology of periodontitis. For instance, miRNA-146 was reported to interfere with pro-inflammatory cytokine production in human gingival fibroblasts [9].
miR-142 has been identified to be upregulated in inflamed gingival tissues, relative to healthy gingival tissues [10]. This miRNA can alter cell proliferation, migration, and survival in several types of malignant epithelial cells [11,12]. However, few studies have addressed its role in GECs. In this study, we aimed to determine the expression and biological relevance of miR-142 in GECs in response to TNF-α.
Materials and methods
Patients and serum collection
A total of 31 patients with chronic periodontitis (age: 32-57 years; gender: 14 males and 17 females) were enrolled in this study. The patients with other systemic inflammatory diseases or having received periodontal therapy were excluded. Written informed consent was obtained from each participant. This study was approved by Institutional Ethical Committee and Review Board of Shanghai Jiao Tong University School of Medicine (Shanghai, China).
Blood samples (5 mL) were collected from each participant and allowed to clot at room temperature. Serum was separated by centrifuging at 1,200 g for 10 min. Serum samples were aliquoted and stored at -80°C until analyses.
Cell culture and TNF-α treatment
Primary GECs were collected from healthy gingival tissues after oral surgery as previously described [13]. Briefly, gingival tissues were cut into small pieces and incubated with 0.4% dispase (Sigma-Aldrich, St. Louis, MO, USA). Cells were maintained as monolayers in serum-free keratinocyte growth medium (Invitrogen, Carlsbad, CA, USA) in a humidified incubator containing 5% CO2. Cells at ~80% confluence were treated with different concentrations of TNF-α (PeproTech Inc., Rocky Hill, NJ, USA) for indicated times. Cells were collected for gene expression and apoptosis analysis.
Real-time PCR analysis
Total RNA from serum samples or cells was extracted with TRIzol reagent (Invitrogen). cDNA was obtained with a reverse transcription system kit (Applied Biosystems, Foster, CA, USA), using specific stem-loop primers for miR-142. For real-time PCR, TaqMan MicroRNA Assays (Applied Biosystems) for miR-142 and U6 (an internal control) were used, according to the manufacturer’s protocol. Relative miR-142 expression was calculated after normalization against U6.
Measurement of serum TNF-α levels by enzyme-linked immunosorbent assay (ELISA)
The levels of TNF-α in serum samples from patients with periodontitis were measured using a TNF-α ELISA kit (eBioscience, San Diego, CA, USA) according to the manufacturer’s instructions.
Transfection of RNA molecules
miR-142 mimic, anti-miR-142 inhibitor, and negative control RNAs were purchased from GenePharma (Shanghai, China). BACH2-targeting small interfering RNA (siRNA) and negative control siRNA were obtained from Santa Cruz Biotechnology, Santa Cruz, CA, USA. Primary GECs at ~70% confluence were transfected with 50 nM small RNAs using Lipofectamine 2000 (Invitrogen). A fluorescein-labeled double-stranded RNA (Qiagen, Valencia, CA, USA) was used to assess transfection efficiency. When the transfection efficiency was of > 80%, transfected cells were used for further experiments. At 24 h post-transfection, cells were exposed to TNF-α for 48 h and tested for gene expression and apoptotic response.
Cell viability assay
Cells seeded in 96-well plates (3 × 103 cells per well) were treated with different concentrations of TNF-α for 48 h. Each well was added with the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution (0.5 mg/mL; Sigma-Aldrich) and incubated for 4 h at 37°C. Formazan crystals were dissolved with dimethyl sulfoxide. The absorbance was read at 570 nm.
Apoptosis analysis
Apoptosis was assessed using a terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) assay kit according to the protocol of the manufacturer (Roche Applied Science, Indianapolis, IN, USA). In brief, after treatment with 15 ng/mL TNF-α for 48 h, cells were seeded on 8-well chamber slides and allowed to adhere overnight. Cells were fixed in 4% paraformaldehyde, permeabilized with 0.1% Triton X-100, and incubated with fluorescein isothiocyanate (FITC)-labeled dUTP and terminal deoxynucleotidyltransferase for 1 h in the dark at 37°C. Cells were counterstained with DAPI (Molecular Probes, Eugene, OR, USA) and analyzed using a fluorescence microscope. The percentage of TUNEL-positive cells relative to total cells was calculated.
Caspase-3 activity assay
Caspase-3 activity was analyzed using a colorimetric assay kit (BioVision Inc., Mountain View, CA, USA), according to the instructions of the manufacturer. The absorbance was recorded at 405 nm.
Plasmid construction and transfection
Wild-type BACH2 3’-UTR was amplified by PCR and cloned into the downstream of the firefly luciferase gene in PGL3 vector (Promega, Madison, WI, USA). Mutation of the BACH2 3’-UTR was performed using the QuickChange Site-Directed Mutagenesis Kit (Stratagene, Heidelberg, Germany). The BACH2 expression plasmid (pcDNA3.1-BACH2) was constructed by inserting human BACH2 open reading frame (Origene, Nockville, MD, USA) into pcDNA3.1(+) vector (Invitrogen). All constructs were confirmed by sequencing. To test whether BACH2 overexpression can reverse miR-142- or TNF-α-induced apoptosis, GECs were co-transfected with pcDNA3.1-BACH2 plasmid or vector (1 μg) together with miR-142 mimic (50 nM) or were pre-transfected with pcDNA3.1-BACH2 or vector before TNF-α treatment. Cells were then examined for gene expression and apoptosis.
Luciferase reporter assay
HEK293T cells (1 × 105 cells per well) in 24-well plates were co-transfected with 0.5 μg of 3’-UTR reporter constructs, 20 ng of Renilla luciferase-expressing pRL-TK vector (Promega), together with 50 nM of miR-142 mimic or control miRNA. Luciferase activity assay was carried out 24 h after transfection with the dual luciferase reporter assay system (Promega). The relative luciferase activity was normalized to that of Renilla luciferase.
Western blot analysis
Cells were lysed in radioimmunoprecipitation assay buffer containing protease inhibitors (Sigma-Aldrich). Equal amounts of protein were resolved by SDS-polyacrylamide gel electrophoresis and transferred onto nitrocellulose membranes. Immunoreactions were performed with primary antibodies: anti-BACH2 (1:300 dilution; Santa Cruz Biotechnology), anti-Bcl-2 (1:500 dilution; Cell Signaling Technology, Danvers, MA, USA), anti-Bcl-xL (1:500 dilution; Cell Signaling Technology), anti-Bax (1:500 dilution; Cell Signaling Technology), anti-Bim (1:500 dilution; Abcam, Cambridge, MA, USA), and anti-β-actin antibody (1:2000 dilution; Santa Cruz Biotechnology). After washing, membranes were incubated with horseradish peroxidase-conjugated secondary antibodies (Santa Cruz Biotechnology). The blots were visualized with the chemiluminescent system (Cell Signaling Technology). Densitometry was performed using Quantity One software (Bio-Rad Laboratories, Hercules, CA, USA).
Statistical analysis
Data are expressed as mean ± standard deviation and were analyzed by the Student’s t test or one-way analysis of variance followed by the Tukey-Kramer multiple comparisons test. Correlation analysis was performed using the Spearman method. A P value of < 0.05 was considered statistically significant.
Results
TNF-α stimulates the expression of miR-142 in human GECs
It has been documented that many miRNAs are dysregulated in gingival tissues from patients with periodontitis [8,10]. To search for TNF-α-regulated miRNAs implicated in this disease, we treated primary human GECs with different concentrations of TNF-α and measured the expression changes of candidate miRNAs. As shown in Figure 1A, miR-142 expression was significantly increased in primary GECs after treatment with TNF-α at 5-20 ng/mL, compared to control cells (P < 0.05). A maximal induction of miR-142 was observed at 15 ng/mL. However, the other miRNAs tested, miR-20a, miR-130, miR-203, miR-205, miR-146a, miR-548 did not show significant expression changes after TNF-α treatment (data not shown). Time-course studies demonstrated that TNF-α exposure increased miR-142 levels as early as 8 h after treatment, with a peak level occurring at 20 h (Figure 1B). These results suggest that miR-142 is a TNF-α-responsive miRNA in GECs. In support of this notion, clinical data revealed that serum miR-142 levels were positively correlated with serum TNF-α levels in patients with chronic periodontitis (r = 0.314, P = 0.0152; Figure 1C).
Figure 1.
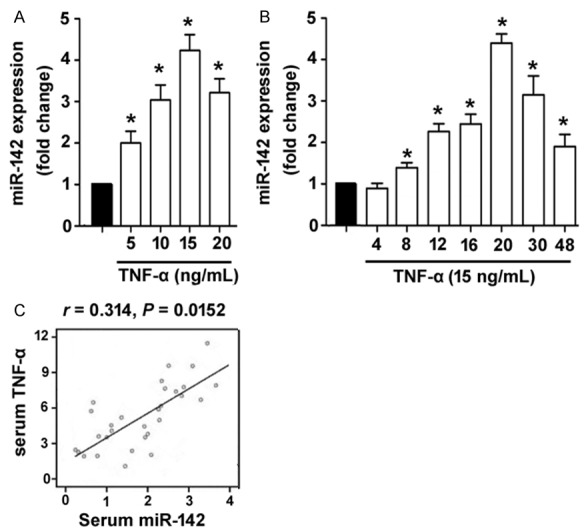
TNF-α stimulates the expression of miR-142 in human GECs. A. Detection of miR-142 abundance in primary GECs exposed to different concentrations of TNF-α for 24 h. B. Time-course studies of miR-142 expression in GECs exposed to 15 ng/mL TNF-α. *P < 0.05 vs. untreated control cells. C. Analysis of the correlation between serum miR-142 and serum TNF-α levels in patients with chronic periodontitis (n = 31).
miR-142 is required for TNF-α-induced apoptosis in human GECs
Next, we tested if miR-142 is involved in TNF-α-induced biological effects in human GECs. MTT assay showed that the viability of GECs was reduced by about 42% after exposure to 15 ng/mL TNF-α for 48 h, which was reversed by inhibition of miR-142 (Figure 2A). Significant apoptosis was detected in GECs treated with 15 ng/mL TNF-α, as determined by TUNEL staining (Figure 2B). Interestingly, TNF-α-induced apoptotic death in GECs was significantly (P < 0.05) prevented by delivery of anti-miR-142 inhibitors (Figure 2B). Measurement of caspase-3 activity, a biomarker of apoptosis, revealed that knockdown of miR-142 led to an ~60% decline in caspase-3 activity in TNF-α-treated GECs (Figure 2C). Additionally, enforced expression of miR-142 recapitulated the suppressive effect of TNF-α on GECs (Figure 2D and 2E). These data collectively indicate that miR-142 participates in TNF-α-induced apoptosis in human GECs.
Figure 2.
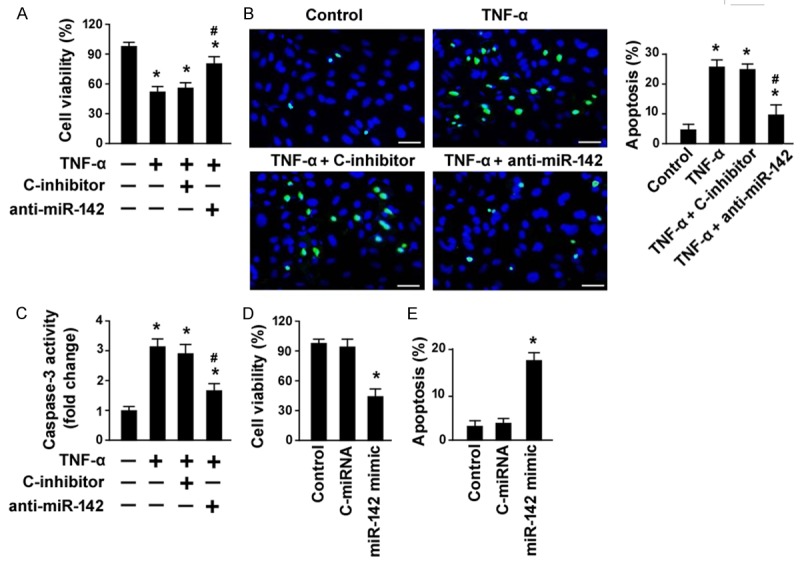
miR-142 is required for TNF-α-induced apoptosis in human GECs. A. MTT assay was performed to detect cell viability in GECs exposed to 15 ng/mL TNF-α for 48 h with or without pre-transfection with anti-miR-142 inhibitor or control inhibitor (C-inhibitor). B. Apoptosis detection by TUNEL staining. Right, quantification of TUNEL-positive cells from three independent experiments. C. Measurement of caspase-3 activity in GECs with indicated treatments. *P < 0.05 vs. untreated control cells; #P < 0.05 vs. C-inhibitor + TNF-α group. D. Analysis of the viability in GECs transfected with control miRNA (C-miRNA) or miR-142 mimic by MTT assay. E. Quantification of apoptosis in GECs transfected with C-miRNA or miR-142 mimic. *P < 0.05 vs. untreated control cells.
miR-142 directly targets BACH2 in human GECs
To determine how miR-142 induces apoptosis in GECs, we performed bioinformatic analysis to search for potential targets of miR-142. As shown in Figure 3A, there is one predicted miR-142 binding site in the 3’-UTR of BACH2 mRNA. To validate whether miR-142 can directly target BACH2 3’-UTR, luciferase reporter assay was carried out using wild-type or mutant BACH2 3’-UTR reporter genes. When the reporters were co-transfected with miR-142 mimic or negative miRNA control into HEK293T cells, miR-142 mimic significantly reduced the luciferase activity of the reporter containing wild-type BACH2 3’-UTR, compared to control miRNA (Figure 3B). However, the activity of the reporter gene with mutant BACH2 3’-UTR was not affected by miR-142 mimic. In agreement with the luciferase reporter assay, overexpression of miR-142 led to 3-fold decrease in BACH2 protein levels in primary GECs, as determined by Western blot analysis (Figure 3C). These results suggest that BACH2 is a direct target gene of miR-142.
Figure 3.
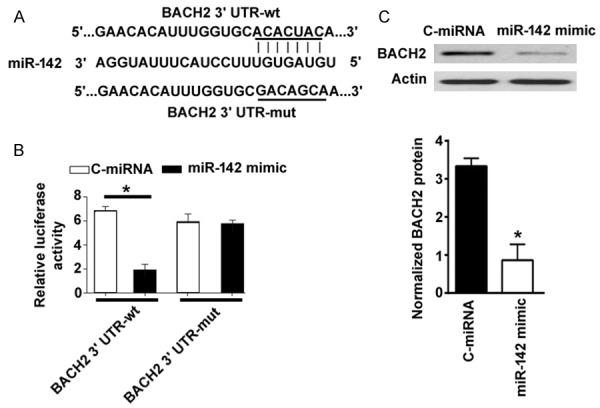
miR-142 directly targets BACH2 in human GECs. A. Prediction of a miR-142 binding site in the 3’-UTR of BACH2 mRNA by TargetScan software (http://www.targetscan.org/vert_71/). B. HEK293T cells were transfected with indicated constructs and tested for luciferase activities. C. Western blot analysis of BACH2 protein in GECs transfected with C-miRNA or miR-142 mimic. *P < 0.05 vs. C-miRNA-transfected cells.
Overexpression of BACH2 reverses miR-142-induced apoptosis in human GECs
Next, we checked whether miR-142 triggers apoptosis in GECs through downregulation of BACH2. To this end, we co-transfected miR-142 mimic with a plasmid expressing BACH2 lacking the 3’-UTR into GECs and measured apoptotic response. Co-transfection with the BACH2-expressing plasmid restored BACH2 levels in GECs (Figure 4A). Enforced expression of BACH2 significantly reduced apoptosis induced by miR-142 mimic (Figure 4B). Thus, miR-142-induced apoptosis in GECs is likely mediated through downregulation of BACH2.
Figure 4.
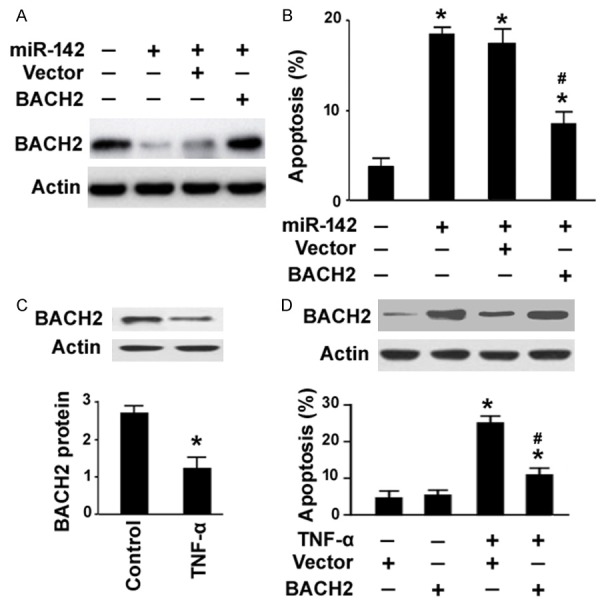
Overexpression of BACH2 confers resistance to miR-142- or TNF-α-induced apoptosis. A. Western blot analysis of BACH2 protein in GECs transfected with indicated constructs. B. Quantification of apoptosis in GECs transfected with indicated constructs for 48 h by TUNEL staining. *P < 0.05 vs. untreated control cells; #P < 0.05 vs. cells co-transfected with miR-142 mimic and vector. C. Western blot analysis of BACH2 protein in GECs treated with or without 15 ng/mL TNF-α. *P < 0.05 vs. untreated control. D. Top, Western blot analysis of BACH2 protein in GECs transfected with vector or BACH2-expressing plasmid before exposure to TNF-α. Bottom, quantification of apoptosis in GECs with the same treatment. *P < 0.05 vs. untreated control cells; #P < 0.05 vs. vector-treated cells exposed to TNF-α.
Overexpression of BACH2 confers resistance to TNF-α-induced apoptosis
Next, we investigated the effect of overexpression of BACH2 on TNF-α-induced apoptosis in GECs. Western blot analysis revealed that TNF-α treatment resulted in a significant (P < 0.05) inhibition of BACH2 expression in human GECs (Figure 4C). Pre-transfection with the BACH2-expressing plasmid significantly impaired apoptotic response in TNF-α-treated GECs, which was coupled with restoration of BACH2 protein expression (Figure 4D). These data indicate the pro-survival activity of BACH2 in human GECs.
Knockdown of BACH2 triggers apoptosis in human GECs by regulating the Bcl-2 family members
To further validate the role of BACH2 in GECs, we performed loss-of-function experiments. siRNA-mediated knockdown of BACH2 was confirmed by Western blot analysis (Figure 5A). Interestingly, depletion of BACH2 significantly induced apoptosis in human GECs, as compared to delivery of control siRNA (Figure 5B). Moreover, silencing of BACH2 decreased the expression of Bcl-2 and Bcl-xL and increased the expression of Bax and Bim (Figure 5C).
Figure 5.
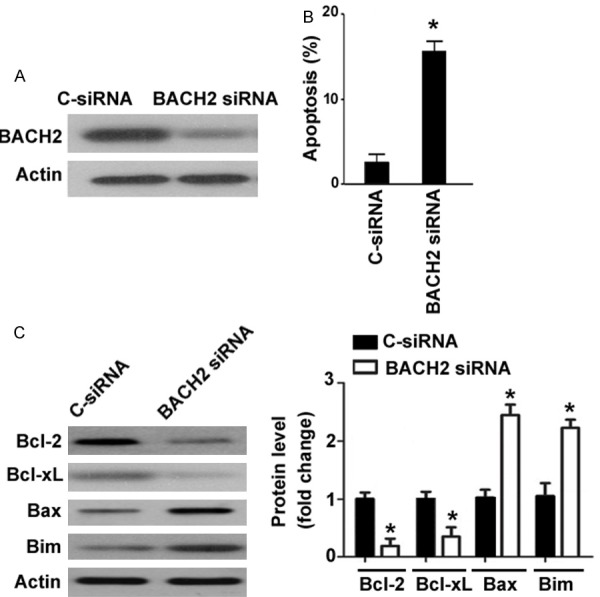
Knockdown of BACH2 triggers apoptosis in human GECs by regulating the Bcl-2 family members. A. Western blot analysis of BACH2 protein in GECs transfected with control siRNA (C-siRNA) or BACH2 siRNA. B. Analysis of apoptosis in GECs transfected with C-siRNA or BACH2 siRNA. C. Western blot analysis of apoptosis-related proteins in GECs transfected with C-siRNA or BACH2 siRNA. *P < 0.05 vs. C-siRNA-transfected cells.
Discussion
miRNA profiling studies have reported that many miRNAs are differentially expressed between inflamed and healthy gingival tissues [8,10]. Functional studies revealed that miR-138 can suppress periodontal progenitor differentiation in an inflammatory microenvironment, thus contributing to bone loss associated with advanced periodontal disease [14]. It has been documented that miR-146a expression was upregulated in patients with periodontitis and that elevated miR-146a was associated with a significant reduction in TNF-α [15]. miR-146a has been shown to play a negative feedback role in the regulation of proinflammatory cytokine secretion in human periodontal ligament cells after lipopolysaccharide stimulation [16]. However, we are still far from understanding of the roles of dysregulated miRNAs in periodontitis. In this study, we showed that miR-142 expression was induced in GECs by TNF-α in a time-and concentration-dependent manner. Moreover, there was a positive correlation between serum miR-142 and TNF-α levels in patients with chronic periodontitis. These results suggest that miR-142 may be involved in TNF-α-related pathogenesis of periodontitis.
miR-142 shows the ability to regulate apoptosis in different types of cells, such as cancer cells [11], tumor-associated macrophages [17], and cardiac myocytes [18]. In this study, we extended the observation to primary GECs and showed that overexpression of miR-142 significantly induced apoptosis in GECs, which phenocopied the effect of TNF-α on GECs. Rescue experiments further demonstrated that inhibition of miR-142 significantly blocked TNF-α-induced apoptotic death in GECs. These results suggest that miR-142 mediates apoptotic response induced by TNF-α in GECs.
Mechanistic studies revealed that overexpression of miR-142 significantly decreased the levels of BACH2 protein in primary GECs. Bioinformatic analysis and luciferase reporter assay showed that miR-142 repressed the expression of BACH2 by binding to the 3’-UTR of BACH2 mRNA. BACH2 has been found to be a risk gene for type 1 diabetes and protects from cytokine-induced β-cell apoptosis [19]. In contrast, BACH2 was reported to cause apoptosis in both NIH3T3 and Raji B-lymphoid cells upon mild oxidative stress [20]. These studies encourage us to hypothesize that BACH2 may be involved in TNF-α/miR-142-induced apoptosis in GECs. In support of this hypothesis, we showed that enforced expression of BACH2 significantly reversed miR-142-induced apoptosis in GECs. Similarly, overexpression of BACH2 conferred resistance to TNF-α-induced apoptosis in GECs. Taken together, miR-142 participates in TNF-α-induced apoptosis in GECs, at least partially, via targeting of BACH2.
BACH2 has been shown to regulate the expression of many genes involved in immune cell development [21,22], alveolar macrophage function [23], and bortezomib cytotoxic response in mantle cell lymphoma (MCL) [24]. In bortezomib sensitive MCL cell lines (Jeko and SP53), BACH2 is implicated in the suppression of pro-survival genes including HO1, Mcl-1, Bcl-2 and Bcl-xL. However, our data demonstrated that BACH2 positively regulated the expression of Bcl-2 and Bcl-xL and negatively regulated the expression of pro-apoptotic genes Bax and Bim in GECs. This discrepancy may reflect that the gene regulatory consequences of BACH2 can be altered by its interaction with other co-transcriptional factors. Indeed, it has been documented that BACH2 competes with the transcription factor BCL6 for binding to the promoters of p53 and other cell cycle checkpoint-control genes in pre-B cells [22]. Nevertheless, the regulation of the Bcl-2 family members provides a possible explanation for BACH2-mediated pro-surivival of GECs.
In conclusion, our data provide evidence that miR-142 acts as a TNF-α-responsive miRNA and contributes to TNF-α-induced apoptosis in human GECs by targeting BACH2. These observations warrant further investigation of the in-vivo roles of miR-142 in an animal model of periodontitis.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81271156).
Disclosure of conflict of interest
None.
References
- 1.Tada H, Matsuyama T, Nishioka T, Hagiwara M, Kiyoura Y, Shimauchi H, Matsushita K. Porphyromonas gingivalis gingipain-dependently enhances IL-33 production in human gingival epithelial cells. PLoS One. 2016;11:e0152794. doi: 10.1371/journal.pone.0152794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Curtis MA, Zenobia C, Darveau RP. The relationship of the oral microbiotia to periodontal health and disease. Cell Host Microbe. 2011;10:302–306. doi: 10.1016/j.chom.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cochran DL. Inflammation and bone loss in periodontal disease. J Periodontol. 2008;79:1569–1576. doi: 10.1902/jop.2008.080233. [DOI] [PubMed] [Google Scholar]
- 4.Miyagawa T, Fujita T, Yumoto H, Yoshimoto T, Kajiya M, Ouhara K, Matsuda S, Shiba H, Matsuo T, Kurihara H. Azithromycin recovers reductions in barrier function in human gingival epithelial cells stimulated with tumor necrosis factor-α. Arch Oral Biol. 2016;62:64–69. doi: 10.1016/j.archoralbio.2015.11.015. [DOI] [PubMed] [Google Scholar]
- 5.Fujihara R, Usui M, Yamamoto G, Nishii K, Tsukamoto Y, Okamatsu Y, Sato T, Asou Y, Nakashima K, Yamamoto M. Tumor necrosis factor-α enhances RANKL expression in gingival epithelial cells via protein kinase A signaling. J Periodontal Res. 2014;49:508–517. doi: 10.1111/jre.12131. [DOI] [PubMed] [Google Scholar]
- 6.Basso FG, Pansani TN, Turrioni AP, Soares DG, de Souza Costa CA, Hebling J. Tumor necrosis factor-alpha and interleukins Il-1β, Il-6, and Il-8 impair in vitro migration and induce apoptosis of gingival fibroblasts and epithelial cells, delaying wound healing. J Periodontol. 2016;87:990–996. doi: 10.1902/jop.2016.150713. [DOI] [PubMed] [Google Scholar]
- 7.Pua HH, Ansel KM. MicroRNA regulation of allergic inflammation and asthma. Curr Opin Immunol. 2015;36:101–8. doi: 10.1016/j.coi.2015.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee YH, Na HS, Jeong SY, Jeong SH, Park HR, Chung J. Comparison of inflammatory microRNA expression in healthy and periodontitis tissues. Biocell. 2011;35:43–49. [PubMed] [Google Scholar]
- 9.Xie YF, Shu R, Jiang SY, Liu DL, Ni J, Zhang XL. MicroRNA-146 inhibits pro-inflammatory cytokine secretion through IL-1 receptor-associated kinase 1 in human gingival fibroblasts. J Inflamm (Lond) 2013;10:20. doi: 10.1186/1476-9255-10-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xie YF, Shu R, Jiang SY, Liu DL, Zhang XL. Comparison of microRNA profiles of human periodontal diseased and healthy gingival tissues. Int J Oral Sci. 2011;3:125–134. doi: 10.4248/IJOS11046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xiao P, Liu WL. MiR-142-3p functions as a potential tumor suppressor directly targeting HMGB1 in non-small-cell lung carcinoma. Int J Clin Exp Pathol. 2015;8:10800–10807. [PMC free article] [PubMed] [Google Scholar]
- 12.MacKenzie TN, Mujumdar N, Banerjee S, Sangwan V, Sarver A, Vickers S, Subramanian S, Saluja AK. Triptolide induces the expression of miR-142-3p: a negative regulator of heat shock protein 70 and pancreatic cancer cell proliferation. Mol Cancer Ther. 2013;12:1266–1275. doi: 10.1158/1535-7163.MCT-12-1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yilmaz O, Jungas T, Verbeke P, Ojcius DM. Activation of the phosphatidylinositol 3-kinase/Akt pathway contributes to survival of primary epithelial cells infected with the periodontal pathogen porphyromonas gingivalis. Infect Immun. 2004;72:3743–3751. doi: 10.1128/IAI.72.7.3743-3751.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou X, Luan X, Chen Z, Francis M, Gopinathan G, Li W, Lu X, Li S, Wu C, Diekwisch TG. MicroRNA-138 inhibits periodontal progenitor differentiation under inflammatory conditions. J Dent Res. 2016;95:230–237. doi: 10.1177/0022034515613043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Motedayyen H, Ghotloo S, Saffari M, Sattari M, Amid R. Evaluation of MicroRNA-146a and its targets in gingival tissues of patients with chronic periodontitis. J Periodontol. 2015;86:1380–1385. doi: 10.1902/jop.2015.150319. [DOI] [PubMed] [Google Scholar]
- 16.Jiang SY, Xue D, Xie YF, Zhu DW, Dong YY, Wei CC, Deng JY. The negative feedback regulation of microRNA-146a in human periodontal ligament cells after porphyromonas gingivalis lipopolysaccharide stimulation. Inflamm Res. 2015;64:441–451. doi: 10.1007/s00011-015-0824-y. [DOI] [PubMed] [Google Scholar]
- 17.Xu S, Wei J, Wang F, Kong LY, Ling XY, Nduom E, Gabrusiewicz K, Doucette T, Yang Y, Yaghi NK, Fajt V, Levine JM, Qiao W, Li XG, Lang FF, Rao G, Fuller GN, Calin GA, Heimberger AB. Effect of miR-142-3p on the M2 macrophage and therapeutic efficacy against murine glioblastoma. J Natl Cancer Inst. 2014:106. doi: 10.1093/jnci/dju162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sharma S, Liu J, Wei J, Yuan H, Zhang T, Bishopric NH. Repression of miR-142 by p300 and MAPK is required for survival signalling via gp130 during adaptive hypertrophy. EMBO Mol Med. 2012;4:617–632. doi: 10.1002/emmm.201200234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marroquí L, Santin I, Dos Santos RS, Marselli L, Marchetti P, Eizirik DL. BACH2, a candidate risk gene for type 1 diabetes, regulates apoptosis in pancreatic β-cells via JNK1 modulation and crosstalk with the candidate gene PTPN2. Diabetes. 2014;63:2516–2527. doi: 10.2337/db13-1443. [DOI] [PubMed] [Google Scholar]
- 20.Muto A, Tashiro S, Tsuchiya H, Kume A, Kanno M, Ito E, Yamamoto M, Igarashi K. Activation of Maf/AP-1 repressor Bach2 by oxidative stress promotes apoptosis and its interaction with promyelocytic leukemia nuclear bodies. J Biol Chem. 2002;277:20724–20733. doi: 10.1074/jbc.M112003200. [DOI] [PubMed] [Google Scholar]
- 21.Tanaka H, Muto A, Shima H, Katoh Y, Sax N, Tajima S, Brydun A, Ikura T, Yoshizawa N, Masai H, Hoshikawa Y, Noda T, Nio M, Ochiai K, Igarashi K. Epigenetic regulation of the Blimp-1 gene (Prdm1) in B cells involves Bach2 and histone DEACEtylase 3. J Biol Chem. 2016;291:6316–6330. doi: 10.1074/jbc.M116.713842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Swaminathan S, Huang C, Geng H, Chen Z, Harvey R, Kang H, Ng C, Titz B, Hurtz C, Sadiyah MF, Nowak D, Thoennissen GB, Rand V, Graeber TG, Koeffler HP, Carroll WL, Willman CL, Hall AG, Igarashi K, Melnick A, Müschen M. BACH2 mediates negative selection and p53-dependent tumor suppression at the pre-B cell receptor checkpoint. Nat Med. 2013;19:1014–1022. doi: 10.1038/nm.3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nakamura A, Ebina-Shibuya R, Itoh-Nakadai A, Muto A, Shima H, Saigusa D, Aoki J, Ebina M, Nukiwa T, Igarashi K. Transcription repressor Bach2 is required for pulmonary surfactant homeostasis and alveolar macrophage function. J Exp Med. 2013;210:2191–2204. doi: 10.1084/jem.20130028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen Z, Pittman EF, Romaguera J, Fayad L, Wang M, Neelapu SS, McLaughlin P, Kwak L, McCarty N. Nuclear translocation of B-cell-specific transcription factor, BACH2, modulates ROS mediated cytotoxic responses in mantle cell lymphoma. PLoS One. 2013;8:e69126. doi: 10.1371/journal.pone.0069126. [DOI] [PMC free article] [PubMed] [Google Scholar]