

Abstract
Imbalance of Aβ production and Aβ removal leads to Aβ accumulation. Aβ degrading enzyme (including neprilysin-NEP, endothelin converting enzyme-ECE) as a therapeutic strategy for lowering brain Aβ deposition has attracted increasing attention. In this study, we investigated alteration of age and region-dependent in APP/PS1 double transgenic mice (3, 6, 9, 12 months) and their age-matched wild type mice including the ability of spatial memory, Aβ deposits, the protein expression, location and activity of NEP and ECE. Our data demonstrated that, as compared with wild type mice, APP/PS1 mice displayed significant cognitive deficit at 9 month revealed by obviously longer in the latency and distance to find the platform and shorter in time spent and swimming distance in the target quadrant. Aβ40 and Aβ42 levels exhibited a significant increase with age in the cerebral cortex and hippocampus of APP/PS1 mice after 6 month, compared with their age-matched wild type mice. And Aβ42 levels were significantly higher than Aβ40 levels in the same age of APP/PS1 mice. Furthermore, NEP protein and activity displayed a marked decrease with age in the cerebral cortex and hippocampus of APP/PS1 mice older than 6 month. Slightly different from NEP, ECE protein was up-regulated with age, while ECE activity showed a significantly decrease with age in cortex and hippocampus of APP/PS1 mice older than 6 month. Double immunofluorescence staining also demonstrated that ECE and NEP highly colocalized in cytoplasmic and membrane, and ECE immunoreactivity tended to increase with age in APP/PS1 mice, especially 12 month APP/PS1 mice. Correlation analysis showed the negative correlation between enzyme (NEP or ECE) activity and Aβ levels in the cerebral cortex and hippocampus of APP/PS1 mice, which was correlated with Aβ accumulation. These results indicate NEP rather than ECE plays more important role in resisting Aβ accumulation. The compensatory upregulation of NEP and ECE could balance Aβ metabolism and protect neuronal functions in infant and juvenile mice. These evidence might provide some clues for the treatment of Alzheimer’s disease.
Keywords: Aβ, neprilysin, endothelin converting enzyme, colocalization, enzyme activity, Alzheimer’s disease
Introduction
Alzheimer’s disease (AD) is the most common neurodegenerative disease, which is characterized by early and progressive cognitive impairment and memory loss [1-5]. According to the statistics, currently over 46.8 million aged people worldwide are suffering from AD [6]. It’s predicted that AD patients will be doubled every 20 years and reach to 131.5 million by 2050 [6]. The main histological hallmarks of AD is senile plaques of beta-amyloid (Aβ), neurofibrillary tangles and cholinergic neuronal loss [7,8]. The pathological mechanisms involving in AD remains unclear and are extensively debated [4,9]. The imbalance between the Aβ production and removal contributes to the abnormal Aβ accumulation, which is triggering neuronal dysfunction and death [4]. Although the abnormal production of Aβ in the brain plays a vital role in the pathogenesis of the familial Alzheimer’s disease, there is little evidence that demonstrated overproduction of Aβ is important for the absence of Aβ deposition in the aging and sporadic AD [10,11].
In the past decade, several proteases have been demonstrated to be capable of degrading Aβ in vitro and in vivo, including neprilysin (NEP), endothelin converting enzymes (ECEs, including ECE-1, ECE-2), insulin degrading enzyme (IDE) [3,4,12,13]. While it has been reported that NEP plays a key role in regulating the steady-state level of Aβ in brain [14].
NEP, also called CD10, EC3.4.24.11, neutral endopeptidase [4,15], is a membrane-bound protein with a 80-110 kDa of molecular weight depending on differences in its glycosylation [16-18], which could cleave several biologically active peptides including bradykinin, endothelins and Aβ [13,19]. As a member of M13 zinc metalloendopeptidase family, NEP is widely expressed in many tissues, especially in the brush-border of intestinal and renal tubular epithelial cells [4,20]. In the nervous system, NEP is localized at pre- and post-synaptic membranes and is ubiquitously expressed in brain regions relevant to amyloid accumulation, such as hippocampus and association cortex which may participates in regulating the neuropeptide signaling [21,22]. Infusion of a specific NEP inhibitor- thiorphan into rat hippocampus, resulted in the elevation of endogenous Aβ levels. On the other hand, upregulating NEP expression led to reduction in Aβ peptide levels, attenuation of amyloid plaques, oxidative stress, inflammation, and decrease of memory impairment in AD transgenic mice [23,24].
Endothelin-converting enzymes ECE-1 and ECE-2 are close homologous of NEP belongs to type II integral membrane zinc metalloendopeptidases. ECEs is widely exist in human brain and has the ability to degrade Aβ in an acidic pH optimum intracellular compartments [25]. In vitro, ECE-1 over-expression in the cells that lacked endogenous expression caused a highly decrease in the accumulation of Aβ. On the contrary, Aβ accumulation was significantly increased when cells that expressed ECE was treated by the metallo-protease inhibitor phosphoramidon [26].
Although both NEP and ECE were capable of degrading Aβ, their expression, changing and interaction in the progression and development of AD has much less been studied. Previously, we found ECE expression increased along with reducing the expression of NEP in Alzheimer’s brain [27]. Meanwhile, the levels and activity of NEP or ECE were significantly increased when SH-SY5Y cells were treated with HNE or Aβ [25,28]. Recently evidence indicated that ECE1 protein expression was elevated in young 5XFAD/NEP+/- as well as hemi- and homozygous NEP knockout mice [29]. These results suggested that certain relationship and interaction might exist between NEP and ECE, and Aβ might be a potential regulatory factor. However, early diagnosis of clinical sporadic AD is pretty difficult and usually found in advanced stages, which makes hard to observe the progression and pathogenesis of AD. Thus we used APP/PS1 transgenic mice, a model of AD, to explore the alteration of NEP and ECE with age, with wild type (WT) mice as control. Herein, we investigated the ability of spatial memory, Aβ40 and Aβ42 levels, and the changes in the expression, colocalization and enzyme activity of NEP and ECE with age in the cerebral cortex and hippocampus of APP/PS1 double transgenic mice and their age-matched WT mice.
Material and methods
Animals
The APPswe/PS1dE9 double transgenic mice, which are on C57BL/6 background, were obtained from Jackson Laboratory. These transgenic mice carry a chimeric human amyloid precursor protein (HuAPP695swe) and human mutant presenilin-1 with the exon-9 deletion (PS1-dE9) under control of the mouse prion protein promoter and produce increasing amounts of amyloid deposits in the brain as well as develop cognitive deficits with age [30]. Experimental mice were generated by crossing male APP/PS1 mice to wild type C57BL/6 female mice, followed by PCR analysis of tail-derived DNA for the presence of the APP, PS1 transgene. The male transgenic APP/PS1 mice aged 3, 6, 9, 12 month and their age-matched male WT mice were used in the present study. Animals were housed at a temperature of 20-25°C, relative humidity of 50-60%, and a 12:12 hour reverse light: dark cycle environment with free access to food and water. All experimental procedures conformed to the guidelines of Care and Use of Laboratory Animals of China for animal experimentation.
Morris water maze
Morris water maze study was performed according to Morris’ protocol [31]. Briefly, each cohort of animals (6 M and 9 M WT mice, n=9, each group; 6 M old APP/PS1 mice, n=6; 9 M old APP/PS1 mice, n=9) was trained to find the submerged platform that was invisible by the addition of white floater in a 1.2 m diameter pool filled with colorless, non-toxic water (23-25°C, 50 cm deep). The experimental conditions and environment during behavioral test remain constant.
Animals were allowed to orient themselves using distal and spatial clues available in the testing room only. Mice were training two trials per day for 7 consecutive days using a platform raised above the surface of the water. If a mouse failed to find the platform within 60 s, the training was terminated. The animal was guided to it by the experimenter and allowed to remain the platform for 20 s. On the eighth day, the platform was removed and a probe trial for memory retention was performed by allowing the mice to swim freely in the pool for 60 s. Mice were monitored by a camera above the pool and all trials were recorded using an AntiLab water maze program for subsequent analysis of time to reach the platform location (latency), swimming speed, path length, and time spent in target quadrant during probe trials. The data were analyzed by two-way ANOVA.
Tissue preparation
WT and APP/PS1 mice were deeply anesthetized with 10% chloral hydrate (5 μl/g, i.p.) and transcardially perfused with 20 ml ice-cold saline solution (pH 7.4) and 10 ml 4% paraformaldehyde (PFA) in phosphate buffered saline (0.1 M sodium phosphate, 0.15 M sodium chloride, PBS, pH 7.4). The brains were removed and fixed in 4% PFA overnight at 4°C. After fixation, the brain tissue was dehydrated in 15% sucrose and 30% sucrose at 4°C, respectively. The tissues were then cut into 20 μm thick sections with a Leica cryostat microtome at -20°C and free-floating sections were collected in PBS. Sections of 20 μm thick were mounted on gelatin-coated slides and stored at -80°C, followed by processing for immunofluorescent staining. In addition, for biochemical analysis, the brains of WT and APP/PS1 mice were quickly removed on ice after decapitation. Cortex was separated, snap-frozen in liquid nitrogen and stored at -80°C.
Measurement of Aβ peptides in brain tissues by ELISA
The cortical Aβ40 and Aβ42 levels of mice in different months of age were measured according to the protocol described previously [32]. Briefly, the cerebral cortical or hippocampal tissue were homogenized in ice-cold guanidine buffer (5.0 M guanidine HCl/50 mM Tris.HCl, pH 8.0) (weight: volume=1:15). The homogenates were aliquoted and stored at -20°C until assay. For the Aβ ELISA assay, the brain homogenates were further diluted at 1:10 with ice-cold reaction buffer (5% BSA, 0.03% Tween-20 in PBS, pH 7.4), centrifuged at 16,000×g for 20 min at 4°C, and the supernatant fractions were collected. The levels of Aβ40 and Aβ42 were determined employing the commercially available human Aβ40 and Aβ42 ELISA kits (Invitrogen) according to the manufacturer’s instructions. Plates were read at 450 nm using Enspire Multimode Reader (Perkin Elmer). Data obtained in brain homogenates were expressed as pictograms of per micrograms of total protein (pg/μg protein). Protein content of brain homogenates was determined by using a Bradford protein assay kit (Beyotime, China) [33,34]. Analyses were always performed in duplicate.
Western blot
Brains (cortex or hippocampus) were isolated at 3, 6, 9, 12 month from APP/PS1 and the age-matched WT mice. Brains tissues were weighted and homogenized in lysis buffer (containing 50 mM pH 7.5 Tris·HCl, 20 mM EDTA, 1% NP-40, 150 mM NaCl, 0.1% SDS, 0.25% sodium deoxycholate) supplemented with 1 x PMSF and protease inhibitor cocktail (Sigma-Aldrich). Samples with equal amount of proteins were loaded to a 10% SDS-PAGE gels and electrophoresed. Separated proteins were transferred to 0.45 μm polyvinylidene difluoride (PVDF) membranes (Millipore). Membranes were blocked in 5% nonfat dry milk in TBST (Tris-buffer saline, 0.05% Tween-20) for 1 hour, then incubated with antibody against NEP (1:1000, Abcam, Cambridge, UK), ECE-1 (1:2500, R&D system, Minneapolis, USA; 1:100, Santa Cruz Technology, Santa Cruz, USA) and GAPDH (1:5000, Kangchen, Shanghai, China) overnight at 4°C. After three washes (8 min each) with TBST, the primary antibody-bound proteins were revealed using HRP-conjugated anti-mouse or anti-goat secondary antibodies (1:5000, Santa Cruz Technology) at RT for 2 hour. The protein signals were detected using the ECL western blotting detection system (Millipore) and BioImaging System (DNR Lumi BIS, Jerusalem, Israel). The bands’ density of NEP, ECE were quantified using Image J software, and normalized to the optical density of GAPDH on the same gel.
Double immunofluorescence staining and imaging analysis
The brain sections (20 μm thickness) attached to gelatin-coated slides were fixed for 20 min with 4% PFA and washed extensively with PBS (pH 7.4). After antigen retrieval by microwaving tissues in 0.01 M citrate buffer (pH 6), sections were incubated for 1 hour in PBS containing 5% BSA and 0.3% Triton X-100 to block non-specific binding, and then incubated with primary anti-NEP (1:25, Abcam, Cambridge, UK) and anti-ECE-1 (1:25, Santa Cruz Technology, Santa Cruz, USA) at 4°C overnight in a humidity chamber. After three 5 min’s washing with PBS, sections were incubated with Alexa Fluor® 488 donkey anti-goat IgG (H+L) (1:500, Invitrogen, California, USA) and Alexa Fluor® 594 donkey anti-mouse IgG (H+L) (1:500, Invitrogen, California, USA) for 2 hour at RT. After rinsing with PBS, the nuclei of sections were counterstained with Hoechst 33342 solution (1 μg/ml, Beyotime, China) for 10 min. The cellular localization of NEP and ECE was observed by laser scanning confocal microscopy (Nikon, Japan) and analyzed by Image-Pro Plus 6.0 to obtain integrated optical density (IOD) and area of interest (AOI). The data was expressed as the average optical density (IOD/AOI). To quantify the colocolization of NEP and ECE positive staining, the analysis of Manders’ overlap coefficient (MOC) and Pearson’s correlation coefficient (PCC) were performed using Image-Pro Plus 6.0.
Enzymatic activity assay
Fluorescence resonance energy transfer assay was performed to determined NEP and ECE activity using black 96-well plates [27]. Mice cerebral cortex or hippocampus from APP/PS1 and age-matched WT mice at 3, 6, 9, 12 month were homogenized in ice cold phosphate-buffered saline (0.1 M PBS, pH 7.4) containing protease inhibitor cocktail (Sigma-Aldrich, 100×) and 1 mM PMSF, and grinded thoroughly with a portable homogenizer. Then samples were diluted to the desired concentrations by addition of 0.1% Triton-X-100 in PBS, and incubated on ice for 30 min. Fluorogenic peptide substrate (10 μM, 50 μl, Mca-RPPGFSAFK-[Dnp]-OH, R&D Systems) dissolved in HEPES buffer (pH 7.4) was added to each well after incubated in the presence or absence of thiorphan (10 μM, NEP specific inhibitor) for 10 min at RT. After another 30 min’s incubate at RT, fluorescent intensity was read with excitation at 320 nm and emission at 405 nm on the Enspire Multimode reader. The protein concentration of each sample was measured using a Bradford protein assay kit (Beyotime, China).
ECE activity was also determined by fluorogenic peptide substrate Mca-RPPGFSAFK (Dnp)-OH with or without ECE inhibitor, phosphormidon (10 μM, Sigma). ECE and NEP activity were both highly pH dependent. There was higher activity in phosphormidon inhibition of ECE but no NEP activity at pH 5.8. Therefore, the inhibition of ECE was represented by the inhibition by phosphoramidon at pH 5.8.
Statistical analysis
Statistical analysis was carried out using SPSS 16.0. All data in the figures were expressed as mean ± standard error (SEM) and analyzed by one-way ANOVA followed by Student’s t test. The data of Morris water maze was analyzed by two-way ANOVA. Linear regression analysis was used to evaluate relations between variables. Statistical significance level was defined as P<0.05.
Results
APP/PS1 mice revealed an age-dependent cognitive deficits
To evaluate spatial memory, we tested learning and retention of a Morris water maze in WT mice and APP/PS1 mice with 6 and 9 months of age. Analysis of latency data across the 7 days of training revealed longer escape latencies and distance in 9 month of APP/PS1 mice (Figure 1A and 1B), an overall cognitive loss was observed in APP/PS1 mice, especially in 9 month’s group, when compared to WT mice (F(3.24)=53.184, P<0.001). Probe trials were performed at the eighth day to test memory retention. As expected, compared to their age-matched WT mice, 9 month of APP/PS1 mice exhibited a significant learning impairment (P<0.01) as indicated by a statistically significant reduction in time spent and swimming distance in the target quadrant (P<0.001) (Figure 1C and 1D). These data demonstrated that 9 month APP/PS1 mice had a significantly cognitive deficits when compared to their age-matched WT mice (Figure 1A-H).
Figure 1.
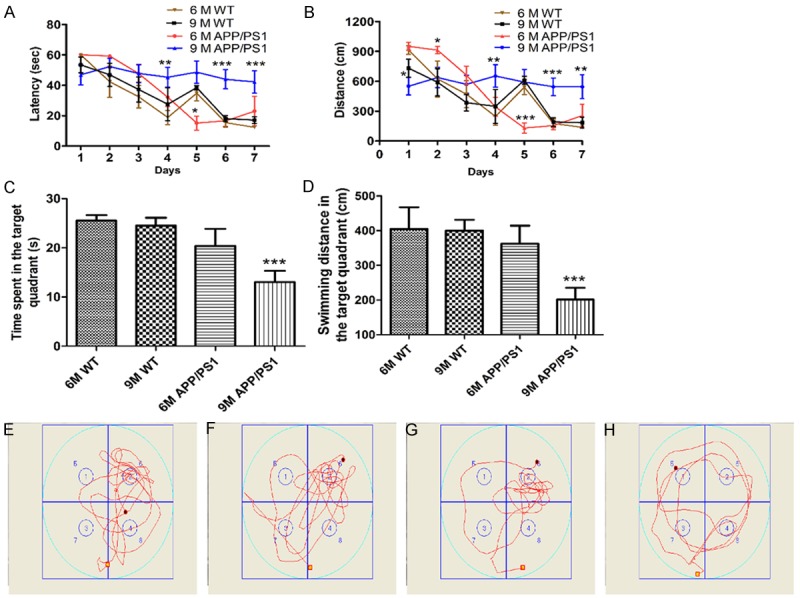
Spatial learning and memory deficits in APP/PS1 mice. A and B: Latency and distance in the target quadrant during training stage; C and D: Time spent and swimming distance in the target quadrant in Probe trial; E-H: The swim-tracking path of WT mice and APP/PS1 mice at 6 and 9 months of age in Probe trial, respectively. 6 M WT mice: n=9; 9 M WT mice: n=9; 6 M APP/PS1 mice: n=6; 9 M APP/PS1 mice: n=9. *P<0.05, **P<0.01, ***P<0.001 as compared with WT mice group.
Aβ levels increased with age in the brains of APP/PS1 mice
We measured and quantified Aβ40 and Aβ42 level in the brain cortex and hippocampus of APP/PS1 mice at 3, 6, 9, 12 month and their age-matched WT mice. In cortex, compared to WT mice, no significantly difference in Aβ40 and Aβ42 levels were detected in 3 month’s APP/PS1 mice, while significant increase revealed when more than 6 month of age in APP/PS1 mice. An up to 40% increase of Aβ40 and 55% of Aβ42 levels were observed, respectively (P<0.001) in 6 month’s APP/PS1 mice, and even higher (~90% and ~130% of Aβ40 and Aβ42) in 9 month’s APP/PS1 mice (P<0.001) (Figure 2A). Meanwhile consistent with cortex, hippocampal levels of Aβ40 and Aβ42 did not increase obviously in 3 month, but dramatically enhanced at 6 month and higher at 9 and 12 month of APP/PS1 mice when compared to WT mice (P<0.01, P<0.001) (Figure 2B). Similar treads in Aβ40 levels both in cortex and hippocampus showed in Figure 2A and 2B (P<0.05, P<0.001).
Figure 2.
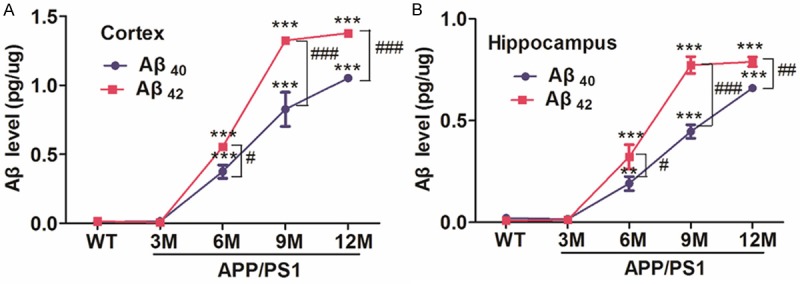
Levels of Aβ40 and Aβ42 in the cerebral cortex and hippocampus from different months of APP/PS1 and their age-mateched WT mice (3, 6, 9, 12 M). A: The levels of Aβ40 and Aβ42 in the cerebral cortex. B: The levels of Aβ40 and Aβ42 in the hippocampus. APP/PS1 mice group: 3 M, n=5; 6 M, n=5; 9 M, n=4; 12 M, n=5; WT group: n=4 each age. **P<0.01, ***P<0.001 compared to WT mice; #P<0.05, ##P<0.01, ###P<0.001 vs their age-matched Aβ40.
Protein levels of NEP and ECE with age in APP/PS1 and WT mice
Evidences suggested that several Aβ degrading enzymes contributed to Aβ degradation and clearance. Cortex and hippocampus were the region susceptible to amyloid accumulation [4,35]. In order to observe changes in NEP and ECE with age, we detected the NEP and ECE protein levels of cortex and hippocampus in APP/PS1 and the age-matched WT mice at 3, 6, 9, 12 month using Western blotting. In the cerebral cortex and hippocampus, no age-related changes in NEP and ECE protein levels were found in WT mice (3, 6, 9, 12 month) (Figure 3A and 3B). In APP/PS1 mice, however, NEP levels showed a significant decrease with age, more obvious down-regulated appeared in hippocampus (P<0.05 at 12 month in cortex). Opposite to NEP, ECE protein significantly increased in the cerebral cortex and hippocampus as shown in Figure 3D and 3F (P<0.05, P<0.01, P<0.001). Notably, compared with their age-matched WT mice, NEP and ECE levels did not significant difference at 3 month APP/PS1 mice, but APP/PS1 mice had significant difference in ECE levels in the cerebral cortex after 6 month (P<0.05, P<0.01, P<0.001) (Figure 3D) and in the hippocampus at 9, 12 month (P<0.001) (Figure 3F), and NEP levels showed a significantly decrease in the hippocampus of APP/PS1 mice after 6 month (P<0.01, P<0.001) (Figure 3E), although there was not reach statistic significant difference in NEP protein levels from APP/PS1 mice cortex (Figure 3C).
Figure 3.
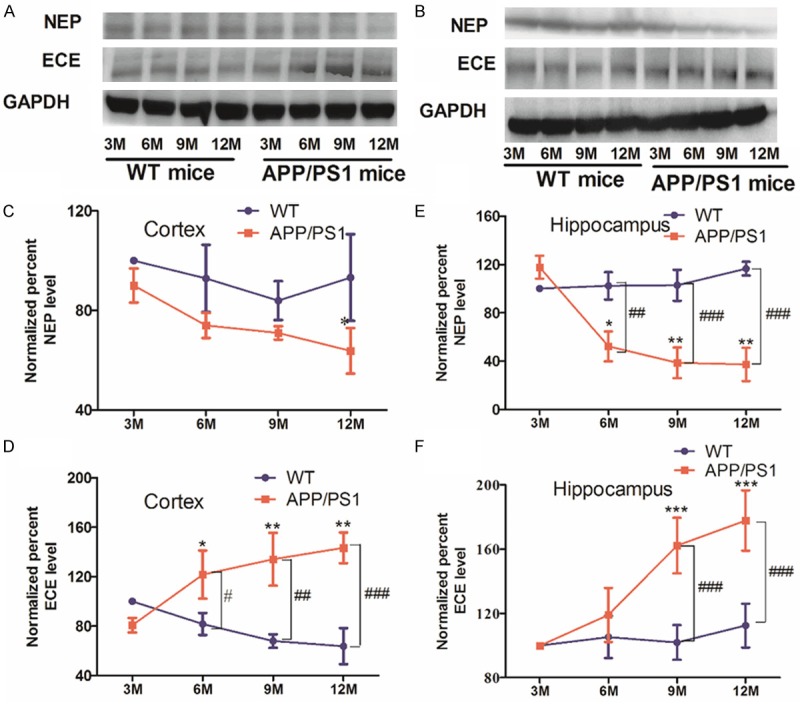
Alterations of NEP and ECE levels in the cerebral cortex or hippocampus of APP/PS1 and WT mice. A and B: Western blot was performed to analysis NEP and ECE protein expression in brain cortex and hippocampus. C-F: NEP and ECE levels were quantified by Image J software for each group. Experiments were repeated three times. *P<0.05, **P<0.01, ***P<0.001 vs 3 M APP/PS1 mice; #P<0.05, ##P<0.01, ###P<0.001 as compared with their age-matched WT mice.
NEP protein highly colocalized with ECE in cytoplasm and membrane
Double immunofluorescence staining of cortical tissue from various age of APP/PS1 and WT mice were performed to further investigate the subcellular location and relation of NEP and ECE during aging with Confocal fluorescence microscopy. Figure 4A-F showed representative confocal fluorescence images of WT and APP/PS1 mice at 3, 6, 9, 12 month. The density of ECE and NEP immunoreactivity were quantified by image analysis of the cerebral cortex (Figure 4b, 4c, 4f, 4g, 4j, 4k, 4n, 4o, 4r, 4s, 4v, 4w). Merged images revealed NEP and ECE highly colocalized in cytoplasmic and membrane (Figure 4d, 4h, 4l, 4p, 4t, 4x). In the cerebral cortex, quantified image analysis (Figure 4G) showed a significant increase of ECE immunoreactivity in APP/PS1 mice at 12 month compared with 3 month of APP/PS1 and WT mice (P<0.05), which was consistent with the above mentioned the results of Western blot. No significant difference was observed in NEP immunoreactivity with age in APP/PS1 and WT mice (Figure 4H). MOC and PCC represented the true degree of colocalization and the correlation of the intensity distribution between two channels (NEP and ECE), respectively. The result showed MOC was up to 0.93 in different month of WT and APP/PS1 mice (Figure 4I), indicated a high colocalization between NEP and ECE. PCC analysis did not show significant difference among different months of WT mice, while a significant difference was found in APP/PS1 mice at 6-12 month compared to 3 month’s APP/PS1 mice (Figure 4J). The PCC values of APP/PS1 mice were obviously lower at 3 month (P<0.01) but significant higher at 12 month (P<0.05) when compared to the same ages of WT mice.
Figure 4.
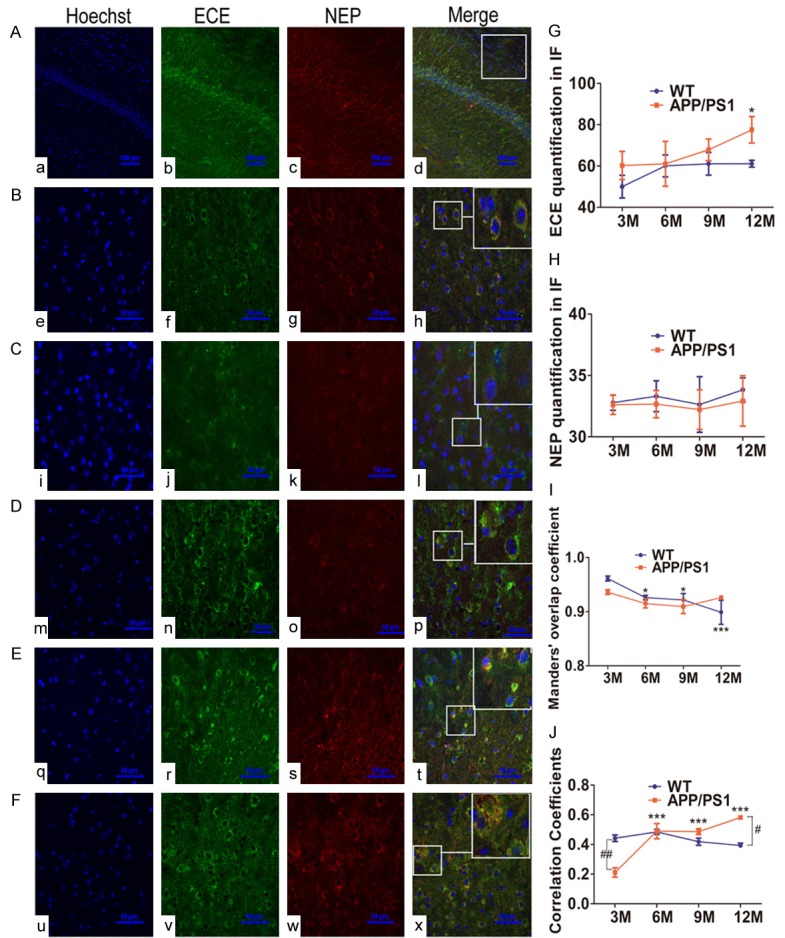
Alterations of NEP and ECE colocalized in brain slices from APP/PS1 and WT mice. A: Images showing ECE and NEP immunoreactivity in 6 M WT mice (200×), staining with anti-ECE (green) (b, f, j, n, r, v) or anti-NEP (red) (c, g, k, o, s, w). The nuclei (a, c, i, m, q, u) were counterstained with Hoechst 33342. Scale bar=100 μm. B-F: Higher magnification views for 6 M WT, 3 M, 6 M, 9 M, 12 M APP/PS1 mice, respectively (600×). Scale bar =50 μm. Colocalization was determined by confocal imaging (d, h, l, p, t, x). G and H: Brain slices from various ages of WT mice and transgenic APP/PS1 mice for relative ECE and NEP average signal density (IOD/AOI) were measured and quantified using Image-Pro Plus 6.0 software, respectively. I and J: MOC and PCC were performed to analyze the colocalization of ECE and NEP. Error bars represent SD, n=3. *P<0.05, ***P<0.001 compared with 3 M WT or APP/PS1 mice. #P<0.05, ##P<0.01 vs the age-matched WT mice.
NEP and ECE activity were decreased with age in APP/PS1 mice
NEP and ECE enzymatic activity were determined in the cerebral cortex and hippocampus with a commercially available fluorogenic substrate. In APP/PS1 mice, the significant decreases of NEP and ECE catalytic activity with age were observed in the cerebral cortex (P<0.05, P<0.01, P<0.001) (Figure 5A and 5B). NEP and ECE activity in hippocampus, differ somewhat from cortex, were increased at 6 month and then sharply decreased after 9 month (P<0.05, P<0.01) (Figure 5C and 5D). Meanwhile, no significant difference with age in NEP and ECE activity was found in WT mice. Compared with their age-matched WT mice, APP/PS1 mice had a significant decrease in NEP activity in the cerebral cortex after 6 month (P<0.05) (Figure 5A), similar to the results of ECE activity in the cerebral cortex at 9 month (Figure 5B). As showed in Figure 5C and 5D, a significant difference in NEP and ECE activity were observed in the hippocampus between 6 month APP/PS1 mice and 6 month WT mice.
Figure 5.
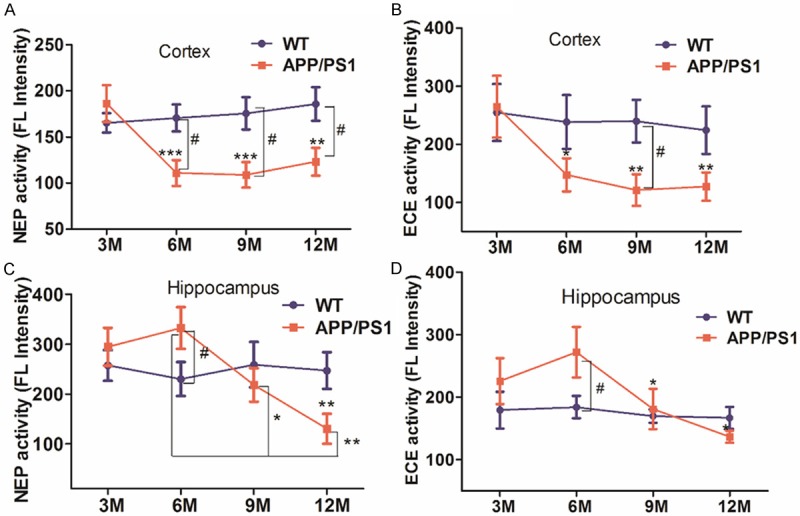
NEP and ECE activity in the cerebral cortex or hippocampus of APP/PS1 and WT mice. A and B: NEP and ECE activity in the cerebral cortex of APP/PS1 and WT mice, respectively. C and D: NEP and ECE activity in the hippocampus of APP/PS1 and WT mice, respectively. 3, 6 M WT and APP/PS1 mice: n=9, per group; 9 M WT and APP/PS1 mice: n=8, per group; 12 M WT and APP/PS1 mice: n=10, per group. *P<0.05, **P<0.01, ***P<0.001 vs 3 M APP/PS1 mice or 6 M APP/PS1 mice; #P<0.05 as compared with their age-matched WT mice.
NEP and ECE activity is inversely correlated with Aβ levels
To evaluate the potential role of NEP and ECE in the clearance of Aβ, we analyzed the correlation between Aβ levels and NEP as well as ECE activity in the cerebral cortex and hippocampus during aging in APP/PS1 mice. A significant negative correlation was found between Aβ40 or Aβ42 levels and NEP or ECE activity in the cerebral cortex of APP/PS1 mice (Figure 6A and 6B). A similar trend of a negative relationship between NEP or ECE activity and Aβ40 or Aβ42 levels in the hippocampus of APP/PS1 mice (Figure 6C and 6D). Among Aβ40 or Aβ42 levels in the cerebral cortex seemed to correlate better than their relative levels in the hippocampus to NEP and ECE activity.
Figure 6.
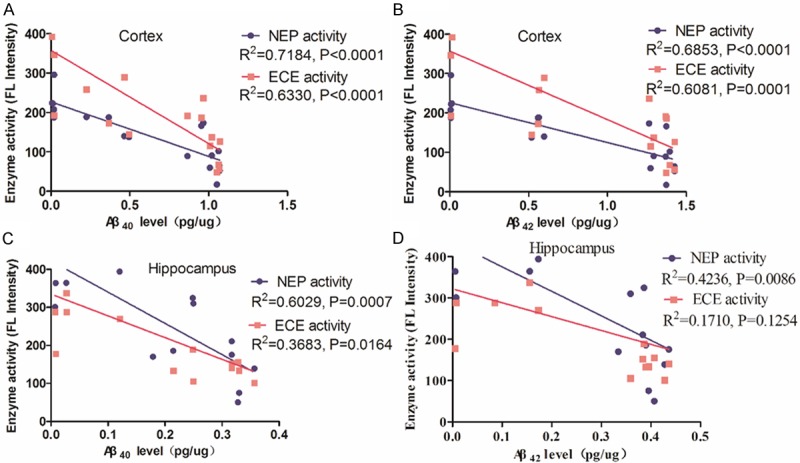
Relationship between NEP or ECE activity and Aβ levels in the cerebral cortex and hippocampus of APP/PS1 mice. A and B: Correlation analysis between NEP or ECE activity and Aβ40 or Aβ42 levels was observed in the cerebral cortex of APP/PS1 mice, respectively. (R2=0.7184, P<0.0001 and R2=0.6330, P<0.0001; or R2=0.6853, P<0.0001 and R2=0.6081, P=0.0001). C and D: Correlation analysis between NEP or ECE activity and Aβ40 or Aβ42 levels was observed in the hippocampus of APP/PS1 mice, respectively. (R2=0.7184, P<0.0001 and R2=0.6330, P<0.0001; or R2=0.6853, P<0.0001 and R2=0.6081, P=0.0001).
Discussion
Alzheimer’s disease is a progressive disease and characterized by cognitive deficits and impair motor abilities clinically. Although the pathogenesis of AD remains unclear and are extensively debated [4,36], most attention has still considered that Aβ metabolism plays a major role in AD development [4,13].
In our study, Morris water maze was performed to evaluate the cognitive function of APP/PS1 and WT mice. As previous studies, APP/PS1 mice revealed obvious cognition deficits at 9 month, showing significant decrease in the time spent and swimming distance in the target quadrant when compared to WT mice during probe trial (Figure 1). The cerebral cortex and hippocampus was the regions susceptible to amyloid accumulation [4,21]. Thus, we examined the levels of Aβ40 and Aβ42 in the cerebral cortex and hippocampus of APP/PS1 and WT mice. Both of Aβ40 and Aβ42 levels exhibited a significant increase with age in APP/PS1 mice. Aβ40 and Aβ42 levels in the cerebral cortex and hippocampus increased gradually in APP/PS1 mice older than 6 month of age, where Aβ40 levels increased approximately 40% and 90%, and Aβ42 levels were about 50% and 130% enhanced in the cerebral cortex (Figure 2A). Moreover, Aβ40 and Aβ42 levels in the hippocampus of APP/PS1 mice displayed a similar to the results of the cerebral cortex (Figure 2B). In addition, Aβ42 has been reported constituted the vast majority of the deposition of Aβ in brain [32,37,38]. Agree with the other’s reports, levels of Aβ42 were significantly higher than Aβ40 in the same age of APP/PS1 mice cortex and hippocampus after 6 month in our study (Figure 2), implied the importance of Aβ42 in amyloid deposition [20,32]. These data showing the typical characteristics of Alzheimer’s disease demonstrated the mouse model mimics human AD pretty well.
Evidences have demonstrated that the role of Aβ degradation is an important process in Aβ clearance [39-42]. Until now, as Aβ degrading enzyme, NEP and its homologs ECE have been caused extensively concern. NEP and ECE levels and activity correlated well with Aβ accumulation and the sporadic AD [3,6,12,43], and data in vitro and in vivo revealed Aβ is a physiologically relevant substrate of NEP and ECE [6,24]. Previously we found that mRNA, protein and activity of NEP were decreased, whereas ECE-1 mRNA, protein and activity tended an increase in AD patients [27]. Here, we indicated age- and region-related alternations in the levels of NEP and ECE in the cerebral cortex and hippocampus. NEP protein and activity in the cerebral cortex and hippocampus was significantly decrease in the APP/PS1 mice older than 6 month of age (Figures 3A and 3B, 5A and 5C), which highly matched the increase of Aβ indicated by Western blotting, immunofluorescence staining and ELISA assay. Meanwhile, ECE protein expression was up-regulated with age in APP/PS1 mice elder than 6 month (Figure 3A and 3B), while ECE activity displayed an increase at 6 month and then reduced both in cortex and hippocampus. For APP/PS1 transgenic mice, overproduction of Aβ play the key role in the Aβ deposits. The Aβ degrading enzymes, however, play crucial roles to resist the Aβ deposit. As we speculated, there was no Aβ increase in the APP/PS1 brains younger than 3 month. Meanwhile, a significant increase of ECE protein and activity was detected which can compensated resist the accumulation of overproduced Aβ. These data is consistent with our previous study in AD patients [27]. In addition, double immunofluorescence staining also demonstrated NEP and ECE highly colocalized in cytoplasmic and membrane (Figure 4I and 4J), and consistent with ECE protein expression, the statistically analysis showed ECE immunoreactivity were dramatically increased with age in APP/PS1 mice (Figure 4), but no significant changes in NEP immunoreactivity.
Furthermore in vitro study revealed that NEP and ECE mRNA, protein and activity were significantly up-regulated by treatment with HNE or Aβ in cultured SH-SY5Y cells. Oxidative modification of Aβ degrading enzymes could somewhat inactivate their activity [25,28], which might explain the evidence that ECE protein increased significantly but activity decreased in present study. Moreover, correlation analysis between NEP or ECE activity and Aβ40 or Aβ42 revealed significant negative correlations both in the cerebral cortex and hippocampus of APP/PS1 mice, implying the significance of NEP and ECE activity in the Aβ accumulation in AD mice, and NEP may play more important role in the AD progression.
In conclusion, NEP rather than ECE plays more important role to resist Aβ accumulation which compensatory secured of Aβ metabolism and normal neuronal functions in infant and juvenile mice, and might provide a clue for Alzheimer’s disease treatment.
Acknowledgements
This project was supported by the grants (to Rui Wang) from National Natural Science Foundation of China 81072627; the 111 Project (Grant No. B07023) from Ministry of Education; Pujiang talent project (11PJ1402300); Key project from Shanghai Science and Technology Committee (12431900901).
Disclosure of conflict of interest
None.
References
- 1.El-Amouri SS, Zhu H, Yu J, Marr R, Verma IM, Kindy MS. Neprilysin: an enzyme candidate to slow the progression of Alzheimer’s disease. Am J Pathol. 2008;172:1342–1354. doi: 10.2353/ajpath.2008.070620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zheng L, Calvo-Garrido J, Hallbeck M, Hultenby K, Marcusson J, Cedazo-Minguez A, Terman A. Intracellular localization of amyloid-beta peptide in SH-SY5Y neuroblastoma cells. J Alzheimers Dis. 2013;37:713–733. doi: 10.3233/JAD-122455. [DOI] [PubMed] [Google Scholar]
- 3.Carty N, Nash K, Lee D, Mercer M, Gottschall PE, Meyers C, Muzyczka N, Gordon MN, Morgan D. Adeno-Associated Viral Vector Mediated Gene Delivery of Endothelin Converting Enzyme Reduces Aβ Deposits in APP+PS-1 Transgenic Mice. Mol Ther. 2008;16:1580–1586. doi: 10.1038/mt.2008.148. [DOI] [PubMed] [Google Scholar]
- 4.Wang DS, Dickson DW, Malter JS. β-Amyloid Degradation and Alzheimer’s Disease. J Biomed Biotech. 2006;2006:58406. doi: 10.1155/JBB/2006/58406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scheltens P, Blennow K, Breteler MM, de Strooper B, Frisoni GB, Salloway S, Van der Flier WM. Alzheimer’s disease. Lancet. 2016;388:505–17. doi: 10.1016/S0140-6736(15)01124-1. [DOI] [PubMed] [Google Scholar]
- 6.Eckman EA, Watson M, Marlow L, Sambamurti K, Eckman CB. Alzheimer’s disease beta-amyloid peptide is increased in mice deficient in endothelin-converting enzyme. J Biol Chem. 2003;278:2081–2084. doi: 10.1074/jbc.C200642200. [DOI] [PubMed] [Google Scholar]
- 7.Jones MB, Spencer B, Michael S, Castello NA, Agazaryan AA, Davis JL, Josef Muller F, Loring FJ, Maslish E, LaFerla FM. Neural stem cells genetically-modified to express neprilysin reduce pathology in Alzheimer transgenic models. Stem Cell Res Ther. 2014;5:46. doi: 10.1186/scrt440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun X, Chen WD, Wang YD. beta-Amyloid: the key peptide in the pathogenesis of Alzheimer’s disease. Front Pharmacol. 2015;6:221. doi: 10.3389/fphar.2015.00221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eckman EA, Adams SK, Troendle FJ, Stodola BA, Kahn MA, Fauq AH, Xiao HD, Bernstein KE, Eckman CB. Regulation of steady-state β -amyloid level s in the brain by neprilysin and endothelin-converting enzyme but not angiotensin-converting enzyme. J Biol Chem. 2006;281:30471–30478. doi: 10.1074/jbc.M605827200. [DOI] [PubMed] [Google Scholar]
- 10.Pacheco-Quinto J, Herdt A, Eckman CB, Eckman EA. Endothelin-converting enzymes and related metalloproteases in Alzheimer’s disease. J Alzheimers Dis. 2013;33(Suppl 1):S101–10. doi: 10.3233/JAD-2012-129043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bohm C, Chena F, Sevalle J, Qamar S, Doddb R, Li Y, Schmitt-Ulms G, Fraser PE, St George-Hyslop PH. Current and future implications of basic and translational research on amyloid-β peptide production and removal pathways. Mol Cellular Neurosci. 2015;66:3–11. doi: 10.1016/j.mcn.2015.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choi DS, Wang D, Yu GQ, Zhu GF, Kharazia VN, Paredes JP, Chang WS, Deitchman JK, Mucke L, Messing RO. PKCε increases endothelin converting enzyme activity and reduces amyloid plaque pathology in transgenic mice. Proc Natl Acad Sci U S A. 2006;103:8215–8220. doi: 10.1073/pnas.0509725103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gough M, Parr-Sturgess C, Parkin E. Zinc metalloproteinases and amyloid Beta-Peptide metabolism: the positive side of proteolysis in Alzheimer’s disease. Biochem Res Int. 2011;2011:721463. doi: 10.1155/2011/721463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hellstrom-Lindahl E, Ravid R, Nordberg A. Age-dependent decline of neprilysin in Alzheimer’s disease and normal brain: inverse correlation with A beta levels. Neurobiol Aging. 2008;29:210–221. doi: 10.1016/j.neurobiolaging.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 15.Wang R, Malter JS, Wang DS. N-acetylcysteine prevents 4-hydroxynonenal- and amyloid-beta-induced modification and inactivation of neprilysin in SH-SY5Y cells. J Alzheimers Dis. 2010;19:179–189. doi: 10.3233/JAD-2010-1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grimm MO, Mett J, Stahlmann CP, Haupenthal VJ, Zimmer VC, Hartmann T. Neprilysin and Aβ Clearance: Impact of the APP Intracellular Domain in NEP Regulation and Implications in Alzheimer’s Disease. Front Aging Neurosci. 2013;5:98. doi: 10.3389/fnagi.2013.00098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Relton JM, Gee NS, Matsas R, Turner AJ, Kenny AJ. Purification of endopeptidase-24.11 (‘enkephalinase’) from pig brain by immunoadsorbent chromatography. Biochem J. 1983;215:519–523. doi: 10.1042/bj2150519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Malfroy B, Kuang WJ, Seeburg PH, Mason AJ, Schofield PR. Molecular cloning and amino acid sequence of human enkephalinase (neutral endopeptidase) FEBS Lett. 1988;229:206–210. doi: 10.1016/0014-5793(88)80828-7. [DOI] [PubMed] [Google Scholar]
- 19.Nalivaeva NN, Belyaev ND, Zhuravin IA, Turner AJ. The Alzheimer’s amyloid-degrading peptidase, neprilysin: can we control it? Int J Alzheimers Dis. 2012;2012:383796. doi: 10.1155/2012/383796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou L, Wei CS, Huang W, Bennett DA, Dickson DW, Wang R, Wang DS. Distinct subcellular patterns of neprilysin protein and activity in the brains of Alzheimer’s disease patients, transgenic mice and cultured human neuronal cells. Am J Transl Res. 2013;5:608–621. [PMC free article] [PubMed] [Google Scholar]
- 21.Caccamo A, Oddo S, Sugarman MC, Akbari Y, LaFerla FM. Age- and region-dependent alterations in Aβ -degrading enzymes: implications for Aβ-induced disorders. Neurobiol Aging. 2005;26:645–654. doi: 10.1016/j.neurobiolaging.2004.06.013. [DOI] [PubMed] [Google Scholar]
- 22.Yasojima K, Akiyama H, McGeer EG, McGeer PL. Reduced neprilysin in high plaque areas of Alzheimer brain_ a possible relationship to deficient degradation of β-amyloid peptide. Neurosci Lett. 2001;297:97–100. doi: 10.1016/s0304-3940(00)01675-x. [DOI] [PubMed] [Google Scholar]
- 23.Iwata N, Tsubuki S, Takaki Y, Morishima MK, Aizawa YS, Iwata K, Lee HJ, Saido TC. Identification of the major Abeta1-42-degrading catabolic pathway in brain parenchyma_ suppression leads to biochemical and pathological deposition. Nat Med. 2000;6:143–150. doi: 10.1038/72237. [DOI] [PubMed] [Google Scholar]
- 24.Marr RA, Guan H, Rockenstein E, Kindy M, Gage FH, Verma I, Masliah E, Hersh LB. Neprilysin regulates amyloid β peptide levels. J Mol Neurosci. 2004;22:5–11. doi: 10.1385/JMN:22:1-2:5. [DOI] [PubMed] [Google Scholar]
- 25.Wang R, Wang S, Malter JS, Wang DS. Effects of 4-Hydroxy-Nonenal and Amyloid-β on Expression and Activity of Endothelin Converting Enzyme and Insulin Degrading Enzyme in SH-SY5Y Cells. J Alzheimers Dis. 2009;17:489–501. doi: 10.3233/JAD-2009-1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eckman EA, Reed DK, Eckman CB. Degradation of the Alzheimer’s amyloid beta peptide by endothelin-converting enzyme. J Biol Chem. 2001;276:24540–24548. doi: 10.1074/jbc.M007579200. [DOI] [PubMed] [Google Scholar]
- 27.Wang S, Wang R, Chen L, Bennett DA, Dickson DW, Wang DS. Expression and functional profiling of neprilysin, insulin-degrading enzyme, and endothelin-converting enzyme in prospectively studied elderly and Alzheimer’s brain. J Neurochem. 2010;115:47–57. doi: 10.1111/j.1471-4159.2010.06899.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang R, Wang S, Malter JS, Wang DS. Effects of HNE-modification induced by Abeta on neprilysin expression and activity in SH-SY5Y cells. J Neurochem. 2009;108:1072–1082. doi: 10.1111/j.1471-4159.2008.05855.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hüttenrauch M, Baches S, Gerth J, Bayer TA, Weggen S, Wirths O. Neprilysin deficiency alters the neuropathological and behavioral phenotype in the 5XFAD mouse model of Alzheimer’s disease. J Alzheimers Dis. 2015;44:1291–1302. doi: 10.3233/JAD-142463. [DOI] [PubMed] [Google Scholar]
- 30.Zhang W, Hao J, Liu R, Zhang Z, Lei GS, Su CJ, Miao JT, Li ZY. Soluble Abeta levels correlate with cognitive deficits in the 12-month-old APPswe/PS1dE9 mouse model of Alzheimer’s disease. Behav Brain Res. 2011;222:342–350. doi: 10.1016/j.bbr.2011.03.072. [DOI] [PubMed] [Google Scholar]
- 31.Morris R. Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Meth. 1984;11:47–60. doi: 10.1016/0165-0270(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 32.Johnson-Wood K, Lee M, Motter R, Hu K, Gordon G, Barbour R, Khan K, Gordon M, Tan H, Games D, Lieberburg I, Schenk D, Seubert P, McConlogue L. Amyloid precursor protein processing and Aβ42 deposition in a transgenic mouse model of Alzheimer disease. Proc Natl Acad Sci U S A. 1997;94:1550–1555. doi: 10.1073/pnas.94.4.1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Houacine J, Bolmont T, Aeschbach L, Oulad-Abdelghani M, Fraering PC. Selective neutralization of APP-C99 with monoclonal antibodies reduces the production of Alzheimer’s Aß peptides. Neurobiol Aging. 2012;33:2704–2714. doi: 10.1016/j.neurobiolaging.2011.12.033. [DOI] [PubMed] [Google Scholar]
- 34.Westmark CJ, Hervey CM, Berry-Kravis EM, Malter JS. Effect of Anticoagulants on Amyloid ß-Protein Precursor and Amyloid Beta Levels in Plasma. J Alzheimers Dis Parkinsonism. 2011;1:101. doi: 10.4172/2161-0460.1000101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miners JS, Baig S, Palmer J, Palmer LE, Kehoe PG, Love S. Abeta-degrading enzymes in Alzheimer’s disease. Brain Pathol. 2008;18:240–252. doi: 10.1111/j.1750-3639.2008.00132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O’Brien RJ, Wong PC. Amyloid precursor protein processing and Alzheimer’s disease. Annu Rev Neurosci. 2011;34:185–204. doi: 10.1146/annurev-neuro-061010-113613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, Guido T, Hagopian S, Johnson-Wood K, Khan K, Lee M, Leibowitz P, Lieberburg I, Little S, Masliah E, Mcconlogue L, Montoya-Zavala M, Mucke L, Paganini L, Penniman E, Power M, Schenk D, Seubert P, Snyder B, Soriano F, Tan H, Vitale J, Wadsworth S, Wolozin B, Zhao J. Alzheimer-type neuropathology in transgenic mice overexpressing V717F β-amyloid precursor protein. Nat. 1995;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- 38.Barucker C, Harmeier A, Weiske J, Fauler B, Albring KF, Prokop S, Hildebrand P, Lurz R, Heppner FL, Huber O, Multhaup G. Nuclear translocation uncovers the amyloid Peptide Aβ42 as a regulator of gene transcription. J Biol Chem. 2014;289:20182–20191. doi: 10.1074/jbc.M114.564690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saido T, Leissring MA. Proteolytic degradation of amyloid beta-protein. Cold Spring Harb Perspect Med. 2012;2:a006379. doi: 10.1101/cshperspect.a006379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saido TC, Iwata N. Metabolism of amyloid β peptide and pathogenesis of Alzheimer’s disease. Neurosci Res. 2006;54:235–253. doi: 10.1016/j.neures.2005.12.015. [DOI] [PubMed] [Google Scholar]
- 41.Iwata N, Higuchi M, Saido TC. Metabolism of amyloid-β peptide and Alzheimer’s disease. Pharmacol Ther. 2005;108:129–148. doi: 10.1016/j.pharmthera.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 42.Nalivaeva NN, Beckett C, Belyaev ND, Turner AJ. Are amyloid-degrading enzymes viable therapeutic targets in Alzheimer’s disease? J Neurochem. 2012;120:167–185. doi: 10.1111/j.1471-4159.2011.07510.x. [DOI] [PubMed] [Google Scholar]
- 43.Maetzler W, Stoycheva V, Schmid B, Schulte C, Hauser AK, Brockmann K, Melms A, Gasser T, Berg D. Neprilysin activity in cerebrospinal fluid is associated with dementia and amyloid-β42 levels in Lewy body disease. J Alzheimers Dis. 2010;22:933–938. doi: 10.3233/JAD-2010-101197. [DOI] [PubMed] [Google Scholar]