

Abstract
The establishment of persistent noncytopathic replication by replicon RNAs of a number of positive-strand RNA viruses usually leads to generation of adaptive mutations in nonstructural genes. Some of these adaptive mutations (e.g., in hepatitis C virus) increase the ability of RNA replication to resist the antiviral action of alpha/beta interferon (IFN-α/β); others (e.g., in Sindbis virus) may also lead to more efficient IFN production. Using puromycin-selectable Kunjin virus (KUN) replicon RNA, we identified two adaptive mutations in the NS2A gene (producing Ala30-to-Pro and Asn101-to-Asp mutations in the gene product; for simplicity, these will be referred to hereafter as Ala30-to-Pro and Asn101-to-Asp mutations) that, when introduced individually or together into the original wild-type (wt) replicon RNA, resulted in ∼15- to 50-fold more efficient establishment of persistent replication in hamster (BHK21) and human (HEK293 and HEp-2) cell lines. Transfection with a reporter plasmid carrying the luciferase gene under the control of the IFN-β promoter resulted in ∼6- to 7-fold-higher luciferase expression in HEp-2 cells stably expressing KUN replicon RNA with an Ala30-to-Pro mutation in the NS2A gene compared to that observed in HEp-2 cells stably expressing KUN replicon RNA with the wt NS2A gene. Moreover, cotransfection of plasmids expressing individual wt or Ala30-to-Pro-mutated NS2A genes with the IFN-β promoter reporter plasmid, followed by infection with Semliki Forest virus to activate IFN-β promoter-driven transcription, showed ∼7-fold inhibition of luciferase expression by the wt but not by the Ala30-to-Pro-mutated NS2A protein. The results show for the first time a role for the flavivirus nonstructural protein NS2A in inhibition of IFN-β promoter-driven transcription and identify a single-amino-acid mutation in NS2A that dramatically reduces this inhibitory activity. The findings determine a new function for NS2A in virus-host interactions, extend the range of KUN replicon vectors for noncytopathic gene expression, and identify NS2A as a new target for attenuation in the development of live flavivirus vaccines.
Kunjin virus (KUN) is an Australian flavivirus closely related to other members of the Japanese encephalitis virus subgroup, including the New York strain of West Nile virus (WNV). The KUN genome consists of single-stranded RNA of positive polarity comprising 11,022 nucleotides (29), with one long open reading frame coding for 3,433 amino acids in three structural proteins (C, prM, and E) and seven nonstructural (NS) proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5) (7). The gene order of KUN genome RNA is 5′-C-(pr)M-E-NS1-NS2A-NS2B-NS3-NS4A-NS4B-NS5-3′. We previously constructed the first flavivirus replicons based on KUN cDNA by deleting the majority of the genomic region including structural genes (30) and used them extensively for the development of a gene expression system (2, 19, 28, 30, 44, 45). KUN replicons were also used extensively in RNA replication and complementation studies (23-27, 32) that contributed substantially to generating a comprehensive model for the formation and operation of the flavivirus RNA replication complex (47, 48).
The establishment of persistent noncytopathic replication by replicon RNAs of a number of positive-strand RNA viruses was shown to lead to the generation of adaptive mutations in nonstructural genes that either decreased (alphavirus replicons) or enhanced (hepatitis C replicons) RNA replication efficiency (1, 4, 13, 14, 34, 39, 41, 50). Some of these adaptive mutations (e.g., in the NS5A protein of the hepatitis C replicon) were shown to increase the ability of RNA replication to resist the antiviral action of alpha/beta interferon (IFN-α/β) (41), while others (e.g., in the nsP2 protein of Sindbis virus) were shown to lead to more efficient IFN production (14). IFN response is the first line of cell defense against viral infections, and a majority of viruses have developed various strategies to overcome it, by either inhibiting IFN production or blocking IFN signaling (15, 22, 35, 43). Recent studies of dengue virus have indicated that flaviviruses may interfere with early steps of IFN signaling and have implied roles for the small nonstructural proteins NS2A, NS4A, and NS4B in this process (37).
In this study, we describe the identification of adaptive mutations in KUN replicon RNA that confirm an advantage in establishing persistent replication in a number of different cell lines. We also demonstrate that induction of IFN-β promoter-driven transcription is severely inhibited in cells expressing replicon RNA with the native sequence, while no such inhibition is observed in cells expressing replicon RNA with an adaptive mutation in the NS2A protein. Similarly, wild-type (wt) but not mutated NS2A protein, when expressed individually, inhibits induction of IFN-β promoter-driven transcription. Our findings demonstrate a novel role for the flavivirus NS2A protein in inhibition of the cellular antiviral response, extend the range of KUN replicon vectors for noncytopathic gene expression, and identify NS2A as a new target for attenuation in the development of a KUN-based vaccine against a closely related emerging human pathogen, the New York strain of West Nile virus.
MATERIALS AND METHODS
Cells and viruses.
BHK21, Vero, HEK293, and HEp-2 cells were grown in Dulbecco's modification of minimal essential medium (DMEM) (Invitrogen, Carlsbad, Calif.) supplemented with 10% fetal bovine serum at 37°C in a CO2 incubator. BHK and HEp-2 cells stably expressing KUN replicon RNAs were maintained in the same medium supplemented with 1 to 5 μg of puromycin (Sigma Aldrich, St. Louis, Mo.) per ml. Vero cells are known to have a deletion of the IFN-β gene (36), and BHK21 cells also appear to have a defect in IFN production (38).
KUN virus strain MRM61C (46) was propagated in suckling mouse brain and titrated in Vero cells. Semliki Forest virus (SFV) was kindly provided by Nigel McMillan (Center for Immunology and Cancer Research, University of Queensland) and propagated and titrated in Vero cells.
Construction of the plasmids.
A construct, repPAC-β-Gal, encoding KUN replicon cDNA with the wild-type KUN sequence, the puromycin resistance gene (PAC), and a reporter gene, β-galactosidase (β-Gal), was described previously (32) and was used to generate replicons with adaptive mutations in NS2A (Ala30 to Pro and Asn101 to Asp) and in NS5 (Pro270 to Ser) by site-directed PCR mutagenesis using high-fidelity Pfu DNA polymerase (Stratagene, La Jolla, Calif.) and appropriate primers. Mutagenesis was initially performed using intermediate plasmids containing smaller fragments from NS2A or NS5 gene regions cloned in the pUC18 vector. After confirmation of the introduced mutations by sequencing analysis, the fragments from the intermediate plasmids containing the individual mutations were used to replace corresponding fragments in repPAC-β-Gal to generate the plasmids NS2A/A30P, NS2A/N101D, and NS5/P270S, respectively. The plasmids NS2A/A30P/N101D, NS2A/A30P_NS5/P270S, and NS2A/A30P/N101D_NS5/P270S containing combined mutations in the NS2A and NS5 genes were generated from the plasmids containing individual mutations by substituting corresponding fragments. All mutations in the resulting constructs were confirmed by sequencing. Plasmid pcDNA3-wtNS2A expressing the wt NS2A cassette, containing the last 22 codons of E, the first 3 and the last 50 codons of NS1, and the full-length NS2A gene fused at the C terminus with 9 codons of the c-Myc epitope, was generated by cloning the corresponding DNA fragment obtained by PCR from FLSDXdNS1.1 DNA (26) into the pcDNA3 vector (Invitrogen) using high-fidelity Pfu DNA polymerase and appropriate primers. Plasmid pcDNA3-NS2A/A30P was constructed from pcDNA3-wtNS2A by site-directed PCR mutagenesis using high-fidelity Pfu DNA polymerase and appropriate primers. Further details of the plasmid constructions can be obtained upon request.
RNA transcription, electroporation, and generation of cell lines stably expressing KUN replicon RNA.
All RNA transcripts were prepared with SP6 RNA polymerase from XhoI-linearized plasmid DNAs and electroporated into BHK21 cells, essentially as described previously (30). Briefly, 10 μg of in vitro-transcribed RNAs were electroporated into 2 × 106 BHK21 (normal BHK) cells in 400 μl in a 0.2-cm electrode gap cuvette (Bio-Rad) in a Bio-Rad Gene Pulser II apparatus. The electroporated cells were split into two aliquots, and one of the aliquots was seeded undiluted into a 60-mm-diameter dish. Two days after electroporation, the cells were harvested to isolate total RNA in order to analyze transient RNA replication and accumulation by Northern blotting. The cells in a second aliquot were serially (10-fold) diluted with 10% fetal calf serum-DMEM and seeded onto 60-mm-diameter dishes. Two days after electroporation, 5 μg of puromycin/ml was added to the medium, and the cells were propagated for an additional 7 days. Puromycin-resistant cell colonies in one of the dishes with appropriate dilution were visualized by staining them with crystal violet, while cell colonies in a parallel dish were isolated by using cloning cylinders (Medos, Brisbane, Australia) and propagated further in the puromycin-containing medium to generate cell lines stably expressing KUN replicon RNA (Fig. 1). In other experiments (see Fig. 3 to 5) stable cell lines were generated by the selection of a total population of RNA-transfected or virus-like particle (VLP)-infected cells in the puromycin-containing medium.
FIG. 1.
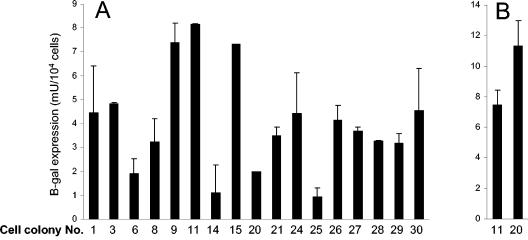
β-Galactosidase expression from KUN replicon RNA in different puromycin-resistant BHK21 cell clones. BHK21 cells were electroporated with the repPAC-β-Gal replicon RNA and subjected to selection during passages in medium containing 5 μg of puromycin/ml as described in Materials and Methods. Individual puromycin-resistant cell colonies were isolated and propagated in puromycin-containing DMEM. The cells were lysed and assayed for β-galactosidase expression at passage 4 (A) and passage 7 (B). The error bars represent standard deviations calculated from the β-Gal expression determined in two wells used for each β-Gal assay.
FIG. 3.
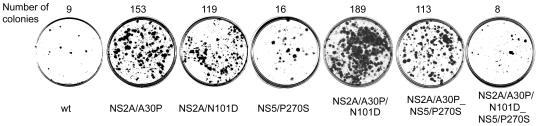
Cell-adaptive mutations confer an advantage in establishing persistent replication of KUN replicon RNA in BHK21 cells. BHK21 cells were electroporated with replicon RNAs containing different cell-adaptive mutations (as in Fig. 2). Forty-eight hours after electroporation, medium with 5 μg of puromycin/ml was added, and the cells were propagated for an additional 7 days. Puromycin-resistant cell colonies were fixed in 4% formaldehyde and stained by crystal violet in phosphate-buffered saline.
FIG. 5.
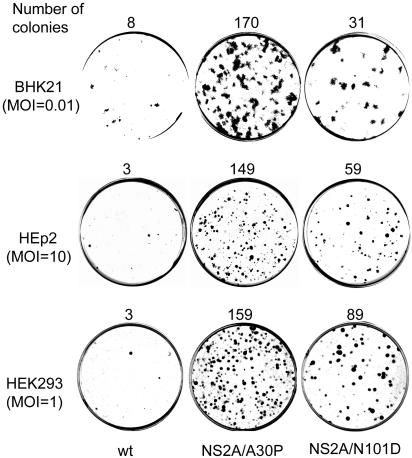
Adaptive mutations confer advantage in establishing persistent replication of KUN replicon RNA in BHK21, HEp-2, and HEK293 cells after infection with replicon VLPs. BHK21, HEK293, and HEp-2 cells were infected with wild-type repPAC-β-Gal replicon VLPs or each of the NS2A mutants at MOIs of 0.01, 1, and 10, respectively. At 48 h postinfection, 1 (HEK293 and HEp-2) and 5 (BHK21) μg of puromycin/ml were added to the medium, and the cells were propagated for an additional 7 days. Puromycin-resistant cell colonies were fixed in 4% formaldehyde and stained either with crystal violet (BHK21) or with X-Gal (HEK293 and HEp-2).
X-Gal staining, β-galactosidase assay, and Northern blotting.
Detection of replication and expression of mutated replicon RNAs in BHK-21 cells was performed by in situ staining with 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal), by β-Gal assay of lysed cells using a β-galactosidase enzyme assay kit (Promega, Madison, Wis.) essentially as described by the manufacturer or by Northern blotting with 32P-labeled probes specific for β-Gal and β-actin nucleotide sequences, as described previously (32).
Packaging of KUN replicon RNAs into virus-like particles.
In vitro-transcribed KUN replicon RNAs containing adaptive mutations were electroporated into the tetKUNCprME packaging cell line essentially as described previously (20). Electroporated cells were cultured in 100-mm-diameter dishes, and the cell culture fluid was harvested 2, 4, 6, and 8 days after electroporation. The titers of secreted VLPs containing encapsidated replicon RNAs were determined by infecting Vero cells and staining them with X-Gal 48 h after infection.
RNA isolation, RT-PCR, and sequencing.
Total RNAs from KUN replicon-expressing cell lines were purified by using an Absolutely RNA reverse transcription (RT)-PCR miniprep kit (Stratagene) as described by the manufacturer. cDNA synthesis and PCR amplification were performed with a SuperScript one-step RT-PCR kit (Invitrogen) using 1 μg of purified total RNA and appropriate primers. Briefly, The RT-PCR mixture (50 μl) containing all necessary components was incubated at 50°C for 30 min for cDNA synthesis by reverse transcriptase, and the following 25 cycles of PCR amplification were performed: 95°C for 30 s, 56°C for 30 s, and 72°C for 2 to 4 min according to the length of the PCR fragments. Sequencing of plasmid DNAs and of PCR fragments was performed by using a DNA-sequencing kit (Perkin-Elmer, Wellesley, Mass.) as described by the manufacturer.
RESULTS
Adaptation of Kunjin replicon RNA to persistent replication in BHK21 cells resulted in the selection of replicons with mutations in NS2A and NS5 genes.
In previous studies, it was noticed that KUN replicons with the native KUN sequence induced some cytopathicity in BHK cells compared to our originally developed noncytopathic replicons (2) that were later found to contain a mutation in the NS1 gene producing a Pro250-to-Leu mutation in NS1 (17) (for simplicity, such mutations will be referred to hereafter as, e.g., Pro250-to-Leu mutations). This mutation was shown to abolish NS1 dimer formation, to delay virus replication in Vero cells, and to attenuate virus replication in weanling mice (17). Incorporation of this mutation in the puromycin-selectable and β-galactosidase-encoding KUN replicon RNA repPAC-β-Gal (32) showed some delay in RNA replication in transient transfections and a moderate (two- to threefold) increase in the number of puromycin-resistant cell colonies after puromycin selection (data not shown). In this study, we aimed at identifying additional mutations in the KUN nonstructural proteins that would allow more efficient establishment of persistent replication of KUN replicon RNA. To achieve this, we established a panel of repPAC-β-Gal-expressing BHK cell clones that were able to grow in the presence of 5 μg of puromycin per ml. The efficiencies of RNA replication and expression in each of the 17 established cell clones at passage 4 were analyzed by β-Gal expression. The β-Gal expression varied from 0.95 to 8.15 mU/104 cells between the individual cell clones (Fig. 1A), and a noticeable difference in the cell growth rates between different cell clones was observed (data not shown). X-Gal staining also showed variation in the intensity of staining, with clone 20 exhibiting the highest intensity in individual cells (not shown). We selected the rapidly growing cell clone 11 (with the highest β-Gal expression) (Fig. 1A) and the slowly growing clone 20 (with the most intensive X-Gal staining) for further passaging and compared levels of β-Gal expression at passage 7. In contrast to the results at passage 4, by passage 7, clone 20 showed ∼1.5-fold-higher β-Gal expression than clone 11 (Fig. 1B). Both clones at passage 7 now propagated with similar growth rates (not shown). Sequencing analysis of the entire KUN replicon cDNA (excluding the β-Gal gene) obtained by reverse transcription and PCR of total cell RNA isolated from these cell clones at passage 7 identified two mutations (Ala30 to Pro in NS2A and Pro270 to Ser in NS5) in clone 11 (Table 1) and one mutation (Asn101 to Asp in NS2A) in clone 20 (Table 1). It is possible that the differences between the levels of β-Gal expression in these two cell lines observed at passages 4 and 7 could be due to a better adaptation of propagated cells to replicating KUN replicon RNA, leading to more rapid cell growth and thus to higher expression and accumulation of β-Gal. It is also possible that the variations in expression levels between the passages could be explained by the different stages of the cell cycle at the time of harvesting.
TABLE 1.
Identification of cell-adaptive mutations in KUN replicon RNA
| Gene | Nucleotide position in genome | Amino acid position in gene | Wild-type nucleotide/amino acid | Clone 11 nucleotide/amino acid | Clone 20 nucleotide/amino acid |
|---|---|---|---|---|---|
| NS2A | 3613 | 30 | G/Ala | C/Pro | |
| NS2A | 3826 | 101 | A/Asn | G/Asp | |
| NS5 | 8488 | 270 | C/Pro | T/Ser |
Effects of adaptive mutations on initiation and efficiency of replicon RNA replication in BHK cells in transient-transfection studies.
Each of the adaptive mutations was introduced individually or in various combinations into the original repPAC-β-Gal replicon, and their effects on initiation and the level (efficiency) of RNA replication were initially tested in transient (2-day) transfection experiments by using X-Gal staining and Northern blot analyses. Ala30-to-Pro mutation in NS2A (NS2A/A30P, as in clone 11) did not significantly affect the number of positively stained cells and the level of accumulated replicon RNA (Fig. 2). In fact, the relative levels of accumulated KUN RNA normalized to the levels of host cell (β-actin) mRNA were the same for transfected wt and NS2A/A30P RNAs (100 and 104%, respectively). Asn101-to-Asp mutation in NS2A (NS2A/N101D, as in clone 20), however, resulted in some decrease in the number of positively stained cells (Fig. 2A) and a corresponding decrease in the level of accumulated replicon RNA (72% of the wt RNA) (Fig. 2B). A combination of adaptive mutations in NS2A resulted in some further decrease in the number of X-Gal-positive cells and in the level of accumulated RNA (55% of the wt RNA) (Fig. 2). Since the number of positively stained cells after transfection of the last two mutant RNAs was also lower than after transfection with the wt or Ala30-to-Pro-mutated RNAs (Fig. 2A), it is likely that Asn101-to-Asp mutation in NS2A affected the initiation of replicon RNA replication, thus leading to a corresponding decrease in the total levels of accumulated RNA detected in Northern blotting. The Pro270-to-Ser mutation in NS5 (NS5/P270S, as in clone 11) had the most profound inhibitory effect on initiation of replicon RNA replication, as judged by the substantially lower number of positive cells and the low total level of accumulated RNA (only 12% of the wt RNA) (Fig. 2).
FIG. 2.
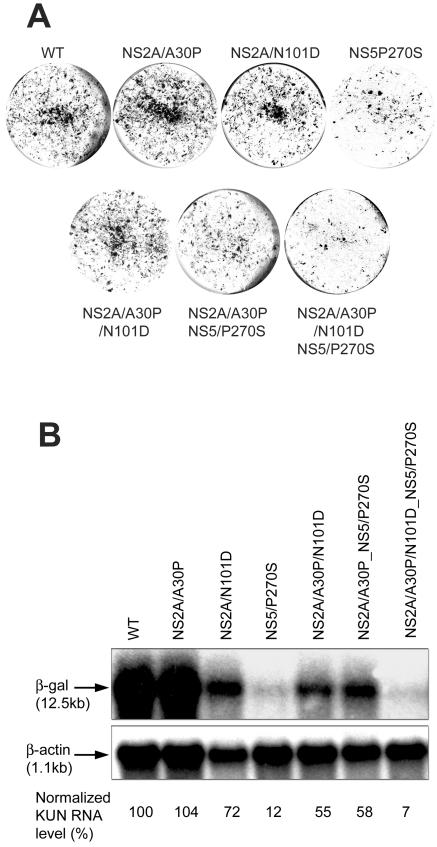
Comparison of replication efficiencies of KUN replicon RNAs with different cell-adaptive mutations in transient transfections. BHK21 cells were electroporated with replicon RNAs with different cell-adaptive mutations. (A) Forty-eight hours after electroporation, the cells were stained with X-Gal to detect cells positive for expression of β-galactosidase from KUN replicon RNA. (B) Total RNA (∼10 μg) isolated from electroporated cells was used for analysis of the accumulation of replicating RNA by Northern blotting with 32P-labeled probes specific for KUN replicon RNA (top) or for β-actin mRNA (bottom). The arrows indicate positions in the gel of RNAs of 12.5 (top row) and 1.1 (bottom row) kb determined relative to migration in the same gel of the ethidium bromide-stained 1-kb Plus ladder (Invitrogen). Normalized KUN RNA levels show percentages of RNA levels from the wt RNA (100%) normalized to the levels of β-actin mRNA.
When NS5 mutation was combined with the other two mutations in NS2A, the resulting replicon RNA was far less efficient in initiation of RNA replication, as judged from the results of X-Gal staining (Fig. 2A) and Northern blot analysis (Fig. 2B). Somewhat surprisingly, combination of Pro270-to-Ser mutation in NS5 with a single Ala30-to-Pro mutation in NS2A did not produce a similar profound inhibitory effect, as would be expected from the presence of a highly inhibitory NS5 mutation (Fig. 2). This suggests that Ala30-to-Pro mutation in NS2A may have some compensatory effect on mutation in NS5. Similarly, different degrees of cooperativity of various adaptive mutations in increasing the efficiency of replication of HCV replicon RNAs in transient-transfection studies were observed (33). The results show that (i) Ala30-to-Pro adaptive mutation in NS2A did not affect the initiation of replication of KUN RNA, (ii) Asp101-to-Asn adaptive mutation in NS2A slightly reduced the efficiency of initiation of RNA replication, (iii) Pro270-to-Ser adaptive mutation in NS5 significantly reduced the efficiency of initiation of RNA replication, and (iv) different combinations of the adaptive mutations resulted in various and not always predictable reductions of efficiency of initiation of RNA replication.
Replicon RNAs with adaptive mutations in NS2A are more efficient in establishing persistent replication in BHK cells.
The dilutions of BHK cells corresponding to 20 ng of each replicon RNA containing either individual or combined adaptive mutations in NS2A and NS5 (from the experiment shown in Fig. 1) were propagated in medium with 5 μg of puromycin/ml for 7 days, followed by staining with crystal violet. Seventeen- and 13-fold increases in the number of puromycin-resistant cell colonies relative to transfections with the wild-type KUN replicon were observed for RNAs with NS2A/A30P and NS2A/N101D mutations, respectively (Fig. 3). Interestingly, combination of two adaptive mutations in NS2A resulted in a slightly higher (21-fold) increase in the number of puromycin-resistant colonies compared to the increase when these mutations were introduced separately (Fig. 3). It is reasonable to assume that the substantial increase in the number of puromycin-resistant cell colonies observed for RNAs with individual or combined mutations in NS2A is due to the decreased cytopathicity of mutated RNAs, since the initial number of X-Gal-positive cells prior to puromycin selection were either similar (NS2A/A30P and NS2A/A30P_N101D) or only slightly lower (NS2A/N101D) than that of the wt RNA (Fig. 1A).
Only a 1.8-fold increase was observed for RNA with the NS5/P270S mutation (Fig. 3). Given the substantially lower number of X-Gal-positive cells detected after transfection of NS5/P270S RNA prior to puromycin selection compared to that with the wt RNA (Fig. 1A), the recovery of a similar or slightly higher number of puromycin-resistant cell clones suggests that despite having a defect in the initiation of RNA replication, NS5/P270S replicon RNA also appears to be less cytopathic. Therefore, Pro270-to-Ser adaptive mutation in NS5 appears to decrease both the efficiency of initiation of RNA replication and its cytopathicity.
Combined adaptive mutations in NS2A and NS5 produced variable effects on the efficiency of establishment of persistent replication. For example, a combination of NS2A/A30P and NS5/P270S mutations (as in clone 11) resulted in a 12.5-fold increase in the number of puromycin-resistant colonies (Fig. 3), while replicon RNA with all three adaptive mutations (two in NS2A and one in NS5) was not more efficient than the wt RNA in establishing persistent replication (Fig. 3). It is not clear why some combinations of adaptive mutations had a cooperative effect (e.g., Ala 30 to Pro and Asn101 to Asp in NS2A) while other combinations did not (e.g., Asn101 to Asp in NS2A and Pro270 to Ser in NS5, or all three mutations). Interestingly, various combinations of adaptive mutations in HCV replicon RNA also did not always cooperate in improving the efficiency of establishing persistent replication, as expected from the effects of individual mutations (34). Nevertheless, our results clearly demonstrated an advantage of adaptive mutations in NS2A, either alone or in combination, in establishing persistent replication of KUN replicon RNA in BHK cells.
Replication efficiencies of RNAs with adaptive mutations are similar in established stable BHK cell lines.
BHK cells (total cell population) transfected with the wt or each of the mutated replicon RNAs were propagated in medium with 5 μg of puromycin/ml for four passages during a total period of 20 days of culture, and the efficiency of RNA replication and expression in established cells was examined by Northern blotting of total cell RNA and by β-Gal assay of cell lysates, respectively. In contrast to significant differences in replication and expression efficiencies between different mutant RNAs observed in transient RNA transfection studies of 2 days duration, the efficiencies of RNA replication and expression in established stable cell lines cultured for 20 days at passage 4 were relatively similar, with a maximum of only a 1.5-fold difference in β-Gal expression between different cell lines (Fig. 4). RT-PCR and sequencing analysis of replicon RNAs confirmed the retention of introduced adaptive mutations (not shown), thus confirming the genetic stability of RNAs with introduced adaptive mutations during the selection process. Similarly, RT-PCR and sequencing analysis of replicon RNA isolated from puromycin-resistant cells established from a total population of cells transfected with the wt KUN replicon RNA showed no mutations in the NS2A and NS5 genes in which adaptive mutations were previously identified. It seems likely that the dominant RNA species initially present in the RNA pool used for transfections are maintained during the selection process when a total cell population rather than the clonal cell selection is used for establishing stably expressing replicon cell lines. However, further sequencing analysis is required to reach a definite conclusion about the absence of any additional adaptive mutations in the remaining replicon sequence which may arise during the selection process. The results so far suggest that in the process of selection, cells down- or up-regulated KUN RNA replication and expression to a certain threshold level that does not harm cellular machinery. Once stable expression is established, this equilibrium between replicon RNA and the surviving cell appears to be maintained by ensuring the replication of different replicon RNAs (with or without adaptive mutations) at a similar level.
FIG. 4.
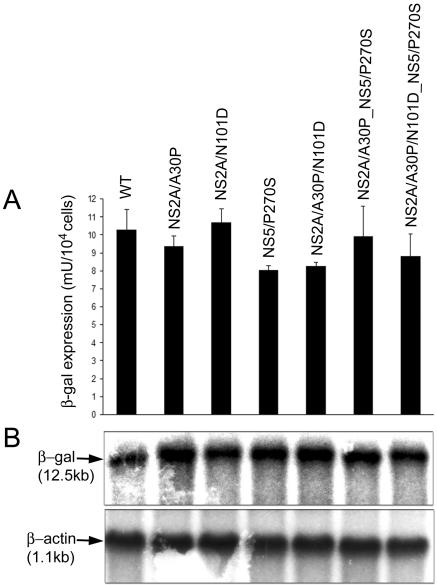
Characterization of replication and expression of KUN replicon RNAs with different adaptive mutations in established puromycin-resistant BHK cell lines. Stably expressing BHK21 cell lines were established after electroporation with KUN replicon RNAs containing different adaptive mutations as described in Materials and Methods. At passage 4, cells from each established cell line were analyzed for β-galactosidase expression (A) and for accumulation of replicating KUN RNA by Northern blotting with 32P-labeled probes specific for KUN replicon RNA (B, top) or for β-actin mRNA (B, bottom). The error bars in panel A represent standard deviations calculated from the β-Gal expression determined in two wells used for each β-Gal assay. In panel B, ∼10 μg of total RNA was used for hybridization; the arrows indicate positions in the gel of RNAs of 12.5 (top) and 1.1 (bottom) kb determined relative to migration in the same gel of the ethidium bromide-stained 1-kb ladder (Invitrogen).
Packaging of KUN replicon RNAs with adaptive mutations into virus-like particles.
A previous report showed that Sindbis virus and SFV replicon RNAs with some of the adaptive (noncytopathic) mutations in nsP2 could not be packaged efficiently into VLPs, while those RNAs with other adaptive mutations in nsP2 could (39). We examined the packaging abilities of KUN replicon RNAs with adaptive mutations by transfecting them into a recently reported tetracycline-inducible packaging BHK cell line, tetKUNCprME (20). The secreted VLPs were harvested every 2 days for 6 to 8 days, and the VLP titers were determined as described in Materials and Methods. Replicon RNA with the NS2A/A30P mutation was packaged with efficiency similar to that of the wt RNA; the other mutant RNAs suffered a 50- to 500-fold decrease in packaging efficiency on day 2, but all except the RNA with combined mutations in NS2A recovered packaging efficiency close to the wild type by day 6 (Table 2). The RNA with combined mutations in NS2A was still packaged 40-fold less efficiently than the wild-type RNA by day 8. RT-PCR and sequencing analysis of KUN replicon RNAs isolated from secreted VLPs harvested on day 6 after transfection showed retention of all the corresponding adaptive mutations, thus excluding reversion to the wt sequence as a possible reason for the improved packaging efficiency later in transfection. It is therefore likely that the improved packaging efficiency later in transfection (days 6 to 8) of the initially inefficiently packaged replicon RNAs was largely due to the spread and accumulation of VLPs in packaging cells. Since the number of cells does not increase once they reach confluency, less efficiently packaged replicons will eventually catch up with more efficiently packaged replicons by spreading to all available packaging cells via VLP infection. At the same time, the number of VLPs will not increase for the more efficiently packaged replicons, since once they spread to all the cells early in transfection, there will be no more cells available for VLPs to spread to. However, the possibility of the occurrence of additional adaptive mutations elsewhere in the replicon genome that may improve the packaging efficiency of initially poorly packaged RNAs cannot be completely ruled out without further confirmation by sequencing analysis of entire replicon genomes. Nevertheless, we can conclude that at least the NS2A/A30P mutation did not affect the packaging efficiency of replicon RNA, while other mutations appeared to inhibit the packaging efficiencies of replicon RNAs early in transfection and replication.
TABLE 2.
Packaging efficiencies of KUN replicon RNAs with different cell-adaptive mutations
| Mutation | Packaging efficiency (PFu/ml) on day:
|
|||
|---|---|---|---|---|
| 2 | 4 | 6 | 8 | |
| Wt | 5 × 106 | 1.3 × 108 | 1.8 × 108 | 3.3 × 108 |
| NS2A/A30P | 4 × 106 | 1 × 108 | 3.4 × 108 | 6 × 108 |
| NS2A/N101D | 4.4 × 104 | 1.3 × 107 | 4 × 107 | NDa |
| NS5/P270S | 1.0 × 105 | 6 × 107 | 1.3 × 108 | ND |
| NS2A/A30P/N101D | 1 × 104 | 1.1 × 105 | 1.2 × 106 | 9 × 106 |
ND, not determined.
Adaptive mutations in NS2A confer an advantage in establishing persistent replication in human cell lines.
We next examined whether the adaptive mutations in NS2A shown to provide an advantage in establishing persistent replication in the hamster cell line, BHK21, would also provide a similar advantage in other cell lines, particularly human cell lines. Monolayers of two human cell lines, HEK293 and HEp-2, were infected with VLPs containing packaged wt and mutated replicon RNAs at multiplicities of infection (MOIs) of 1 and 10, respectively (titrated on Vero cells), and propagated for 7 days in medium with 1 μg of puromycin/ml. X-Gal staining of puromycin-resistant colonies showed a ∼50-fold increase in the number of colonies relative to the wild-type replicon for the NS2A/A30P mutant and ∼20-fold increase for the NS2A/N101D mutant in both HEK293 and HEp-2 cells (Fig. 5). Similar differences between the numbers of puromycin-resistant colonies in the wt and NS2A-mutated replicon RNAs were observed in BHK cells after infection with replicon VLPs at an MOI of 0.01 (Fig. 5). Interestingly, infection of HEK293 and HEp-2 cells required 10- and 100-fold more VLPs, respectively, to produce numbers of puromycin-resistant colonies similar to those produced in BHK21 cells (Fig. 5). Similar differences between these cell lines were observed in the efficiency of replication of wild-type KUN (not shown). In separate experiments, ∼20-fold more efficient replication of wild-type KUN was observed in Vero cells than in BHK cells (results not shown). It is not clear at present whether the differences in efficiencies of replicon and virus replication in different cell lines are due to differences in initial attachment of VLPs and virions to the cell surface or in the initiation and/or efficiency of RNA replication. Nevertheless, the results confirmed the advantage of replicon RNAs with adaptive mutations in NS2A in establishing persistent replication in different cell lines. Further propagation of replicon VLP-infected BHK, Vero, HEK293, and HEp-2 cells (total cell population) in selective medium with puromycin resulted in the establishment of cells stably expressing wt and NS2A-mutated replicon RNAs, with retention of mutations confirmed by sequencing (not shown). In all the experiments with BHK, Vero, HEK293, and HEp-2 cells, the NS2A/A30P mutation allowed more efficient and quicker establishment of stably expressing cell lines (results not shown). In agreement with the results in BHK cells (Fig. 4), the efficiencies of RNA replication and β-Gal expression in established puromycin-resistant cell lines in HEp-2 and HEK293 cells stably expressing different replicon RNAs were also similar (results not shown).
Activation of IFN-β promoter-driven transcription is inhibited in cells stably or transiently transfected with KUN replicon RNAs containing the wt but not the mutated NS2A gene.
Previous studies showed that infection of NIH 3T3 cells with Sindbis virus containing an adaptive mutation in nsP2 that reduced the cytopathicity of Sindbis replicon RNA led to high levels of alpha/beta IFN production in contrast to the low levels of IFN produced by infection with virus containing wt nsP2 (14), thus indicating a role for wild-type nsP2 in inhibiting IFN production. To examine whether KUN NS2A may have a similar function, HEp-2 cells stably expressing KUN replicons with the wt or mutated (Ala30-to-Pro) NS2A were transfected with a reporter plasmid, pIFΔ(−125/+72)Lucter (31), carrying the luciferase gene under the control of the IFN-β promoter, and luciferase expression was determined 55 h after transfection. HEp-2 cells stably transfected with a replicon containing a mutated NS2A gene exhibited a high level of luciferase expression that was ∼6- to 7-fold greater than that observed in cells stably transfected with wt replicon RNA (Fig. 6A), thus clearly demonstrating a role for wt NS2A in the inhibition of IFN-β promoter-driven transcriptional activation in response to KUN replicon RNA replication. To confirm the results obtained with HEp-2 cells stably transfected with replicon RNAs, we performed similar studies with Vero cells transiently expressing replicon RNAs after infection with replicon VLPs. The results were similar (Fig. 6B), although the difference in luciferase expression between the wt and the mutated NS2A-encoding replicons was not as profound in transiently transfected cells as it was in stably transfected cells (Fig. 6). Nevertheless, >50% inhibition of IFN-β promoter-driven transcription was achieved in replicon VLP-infected cells. Factors that could contribute to a less profound difference in IFN-β promoter-driven transcription in cells transiently transfected with replicon RNAs than in stably transfected cells may include a lower percentage of replicon-expressing cells (∼85 versus 100%), a shorter period of replicon RNA replication and expression (16 versus 55 h), and a possible difference between the IFN-β promoter-driven transcription efficiencies in Hep2 cells (stable expression) and Vero cells (transient expression). Note that in both stably and transiently transfected cells the corresponding replication and expression efficiencies in the wt and the NS2A-mutated replicon RNAs were similar within the same experiment (Fig. 6).
FIG. 6.
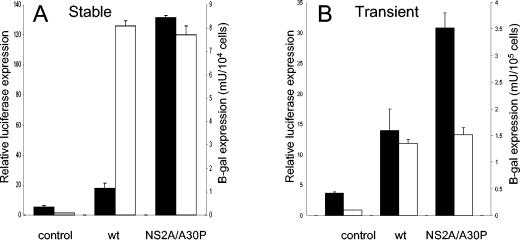
Activation of IFN-β promoter-driven transcription in cells stably or transiently expressing KUN replicon RNAs with the wild-type or the mutated (Ala30 to Pro) NS2A proteins. (A) Normal HEp-2 cells and HEp-2 cells stably expressing wild-type or NS2A-mutated KUN replicon RNAs were transfected with an IFN-β promoter-controlled luciferase reporter gene plasmid, pIFΔ(−125/+72)Lucter (31), and the pGreen Lantern-1 control plasmid (Invitrogen). Fifty-five hours after transfection, the cells were analyzed for luciferase expression to detect activation of IFN-β promoter-controlled transcription, for green fluorescent protein (GFP) fluorescence by fluorescence-activated cell sorting to normalize luciferase expression between transfected samples, and for β-galactosidase expression to compare replication and expression efficiencies of replicon RNAs. The error bars represent standard deviations calculated from the luciferase or β-galactosidase expression determined in two wells used for each assay. (B) Vero cells were transfected with the above-mentioned plasmids and, 32 h after transfection, were infected with KUN VLPs containing packaged wt or NS2A-mutated replicon RNAs. Twenty hours after infection, the cells were assayed for luciferase activity, GFP fluorescence, and β-galactosidase expression as in panel A. The solid bars represent luciferase expression, and the open bars represent β-galactosidase expression.
Individually expressed wt but not mutated NS2A protein inhibits activation of IFN-β promoter-driven transcription.
To determine the abilities of individually expressed wt NS2A protein and NS2A protein with an Ala30-to-Pro mutation to inhibit activation of IFN-β promoter-driven transcription, we cloned corresponding gene cassettes into the pcDNA3 vector. In order to allow proper translocation of NS2A across the membrane of the endoplasmic reticulum and its proper release from the preceding NS1 protein (7), wt and mutated NS2A genes were expressed as cassettes with the signal sequence for the NS1 protein followed by a large deletion of 295 amino acids, leaving intact the first 3 and the last 50 codons of NS1 (as in dNS1.1 [26]) and the full-length NS2A gene with the addition of a C-terminal c-Myc tag. In previous studies, KUN RNA with this coding deletion of 295 amino acids in the NS1 protein was efficiently complemented in BHK cells stably expressing KUN replicon RNA as a helper (26), suggesting that proper processing of the NS1-NS2A cassette occurred in these experiments. Plasmid DNAs expressing wt and mutated NS2A gene cassettes were cotransfected with the IFN-β promoter reporter plasmid pIFΔ(−125/+72)Lucter into Vero cells, which are normally defective in IFN-β synthesis (36). IFN-β promoter-driven transcription was activated by SFV infection with an MOI of 10 32 h after transfection, and the cells were analyzed for luciferase expression 16 h later. The results clearly showed that in cells expressing wt NS2A, IFN-β promoter-driven transcription was strongly inhibited (by ∼7- to 8-fold) compared to that in the control cells transfected with pcDNA3 vector DNA (Fig. 7A). In contrast, expression of mutated NS2A did not inhibit activation of IFN-β promoter-driven transcription in response to SFV infection (Fig. 7A). Both wt and mutated NS2A proteins were expressed at similar levels and processed correctly, as judged by immunofluorescence (Fig. 7B) and Western blot (Fig. 7C) analyses using monoclonal antibodies to the c-Myc tag. The results clearly demonstrate for the first time a role for the flavivirus NS2A protein in inhibition of the activation of IFN-β promoter-driven transcription.
FIG. 7.
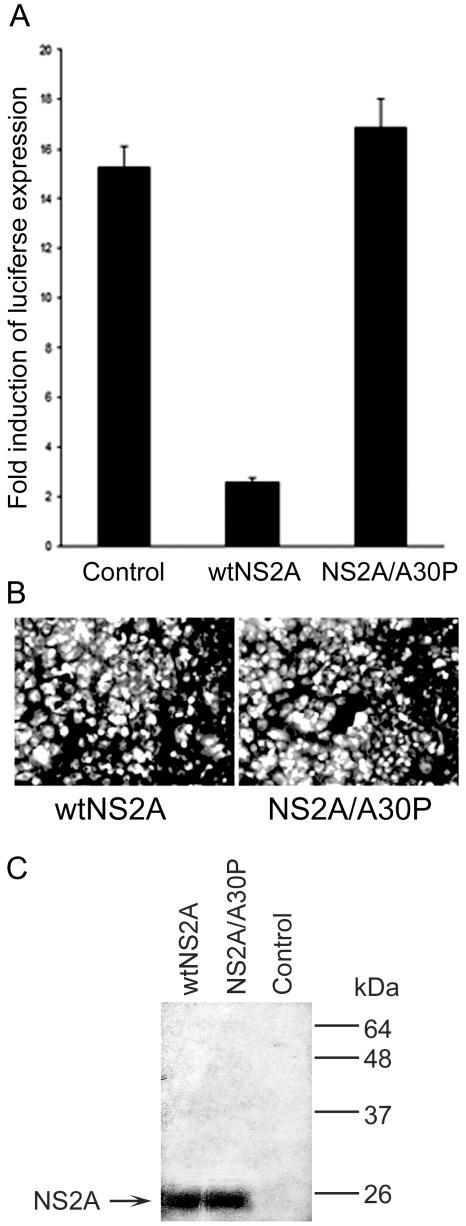
KUN wild-type but not Ala30-to-Pro-mutated NS2A protein inhibits the activation of IFN-β promoter-driven transcription. (A) Vero cells in 24-well plates (two wells per transfection mixture) were cotransfected with an IFN-β promoter-controlled luciferase reporter gene plasmid, pIFΔ(−125/+72)Lucter (31); the control β-galactosidase-expressing plasmid pCMV-β (Invitrogen); and the plasmid pcDNA3-wtNS2A or pcDNA3-NS2A/A30P expressing either the wild-type NS2A gene or the Ala30-to-Pro-mutated NS2A gene, respectively. Forty hours after transfection, the cells were infected with SFV at an MOI of 10 for 16 h and lysed to determine luciferase activity. The activation of IFN-β promoter-driven transcription is shown as induction of luciferase expression in SFV-infected cells relative to luciferase expression in uninfected cells. The error bars represent standard deviations calculated from the luciferase expression determined in two wells used for the luciferase assay. Luciferase expression was normalized to the expression of β-galactosidase from the cotransfected pCMV-β plasmid. (B and C) Vero cells were transfected with pcDNA3 plasmids expressing the wt NS2A and NS2A/A30P mutant proteins fused to the c-Myc epitope. Forty-eight hours after transfection, the cells were either fixed with cold acetone and used in indirect immunofluorescence analysis with anti-c-Myc monoclonal antibodies (21) or lysed and assayed for expression of NS2A-c-Myc fusion proteins by Western blot analysis with the ECL Plus Western-blotting kit (Amersham Biosciences, Castle Hill, New South Wales, Australia) essentially as described by the manufacturer. Control in panel C represents a sample from untransfected Vero cells.
DISCUSSION
We have described identification of the adaptive mutations in the KUN NS2A gene that allow more efficient establishment of noncytopathic, persistent replication of KUN replicon RNA in different cell lines. A replicon with the Ala30-to-Pro adaptive mutation in NS2A was 2- to 5-fold more efficient than a replicon with the Asn101-to-Asp adaptive mutation and 15- to 50-fold more efficient than the original wild-type replicon RNA in establishing persistent replication in BHK, HEK293, and HEp-2 cells. Comparison of the abilities of cells stably expressing KUN replicon RNAs to induce IFN-β promoter-driven transcription with those of cells expressing the wild-type NS2A or the NS2A/A30P mutant genes demonstrated for the first time a role for the flavivirus (wild-type) NS2A in inhibition of IFN-β promoter-driven transcription. Finally, the role of NS2A in inhibition of IFN-β promoter-driven transcription was confirmed by showing that the activation of IFN-β promoter-directed transcription of a reporter gene in response to SFV infection was strongly inhibited by the individually expressed wt NS2A, whereas expression of the NS2A/A30P mutant protein had no such inhibitory effect.
Adaptive mutations, RNA replication, and cytopathicity.
It was shown for the highly cytopathic alphavirus replicons that adaptation of their replication to particular cell lines using antibiotic-selectable replicons led to generation of mutations that when introduced into the original replicons conferred significant advantage in establishing noncytopathic, persistent replication (1, 13, 39). All the adaptive mutations in Sindbis virus and SFV replicons were located in the nsP2 protein, with some of them down-regulating RNA replication and others not. Selection of noncytopathic pestivirus using a similar approach (puromycin resistance) resulted in the identification of a single-amino-acid substitution in NS4B protein; the mutation did not down-regulate RNA replication (42). Persistent replication of hepatitis C virus (HCV) replicon RNA in Huh7 cells also resulted in the generation of adaptive mutations located in different nonstructural proteins; however in contrast to alphavirus replicons, these adaptive mutations generally enhanced HCV replicon RNA replication (4, 34, 41, 50). It was therefore reasonable to assume that establishment of persistent replication of KUN replicons in cells using a similar approach would allow the selection of KUN replicons with adaptive mutations in the nonstructural proteins. Indeed, sequencing of the entire replicon RNA isolated from two puromycin-resistant cell clones identified three adaptive mutations, two in NS2A and one in NS5 (Table 1). Adaptive mutation in NS5 when introduced into the original wt KUN replicon RNA only marginally improved the ability of RNA to establish persistent replication while resulting in dramatic down-regulation of initiation and/or efficiency of RNA replication in transient studies. In contrast, both adaptive mutations in NS2A provided an advantage in establishing persistent replication while not significantly inhibiting the initiation and efficiency of RNA replication. Thus, it seems likely from our results that adaptation of KUN RNA to persistent replication may proceed without down-regulation of KUN RNA replication.
KUN NS2A protein inhibits IFN-β promoter-driven transcription.
Virus infections trigger a massive antiviral response that leads to activation of a wide range of antiviral genes, including those for IFNs. IFN-β is one of the first antiviral genes whose transcription is induced by viral infections. Transcription factors involved in activation of IFN-β gene transcription may be induced by viruses through a number of different pathways, with the interferon regulatory factor 3 (IRF-3) pathway being the most prominent (11, 15, 22, 43). Members of the family Flaviviridae, HCV and bovine viral diarrhea virus, have been shown to block the IRF-3 pathway by either preventing IRF-3 phosphorylation (HCV) (11) or directly preventing transcriptional activity of phosphorylated IRF-3 (bovine viral diarrhea virus) (3). Most recent studies with the more closely related flavivirus WNV (New York 2000 strain) showed direct involvement of the IRF-3 pathway in controlling cell-to-cell spread of the virus (12). However, the actions of the virus-induced IRF-3 pathway were insufficient to prevent WNV replication (12). Our experiments showed that antiviral response was severely compromised in Hep2 cells stably transfected with KUN replicon RNA encoding the wt NS2A gene. The cells showed a dramatic decrease in activation of the transcription of a reporter gene from the IFN-β promoter. Furthermore, expression of wt NS2A protein alone was able to inhibit activation of transcription from the IFN-β promoter in response to exogenous virus stimulus. It is possible that wt NS2A protein may be down-regulating IFN-β promoter-driven transcription through inhibition of one of the components of the IRF-3 pathway, thus allowing virus to escape the host antiviral response. Alternatively, activation of other antiviral factors, such as PKR or 2′-5′-oligoadenylate synthetase, may be inhibited by the wt NS2A protein.
In contrast to our findings, recent studies with dengue virus did not show inhibition of IFN-β promoter-driven transcription by any of the dengue virus NS proteins, including NS2A, when they were expressed individually in 293T cells (37). It is possible that dengue virus NS2A expressed individually from cDNA encoding the NS2A gene alone, without the preceding sequence from NS1 required for its proper processing (9, 10, 40), could not function properly in these assays. In our experiments, KUN NS2A was expressed as a cassette with the preceding NS1 sequences and was shown to be processed properly to release the native NS2A protein. It is also possible that substantial differences between the amino acid sequences of KUN and dengue virus NS2A proteins and/or differences in cell lines used for their expression (293T cells for dengue virus and Vero cells for KUN) could affect the NS2A function in inhibition of IFN-β promoter-driven transcription. For example, only a single-amino-acid change in KUN NS2A (Ala30 to Pro) was sufficient to completely abolish its function in inhibition of the activation of transcription from the IFN-β promoter (see below). Similarly, a single-amino-acid mutation in the simian virus 5 V protein was shown to be responsible for the difference in its ability to block interferon signaling in human and murine cell lines (49).
Adaptive mutations, IFN response, and persistent replication.
As mentioned above, Ala30-to-Pro mutation in NS2A completely abolished its function in inhibiting the activation of IFN-β promoter-driven transcription either when expressed alone or in the context of KUN replicon RNA. It is not clear how this mutation, which actually up-regulated the cellular antiviral response by not blocking the activation of IFN-β promoter-driven transcription in response to viral-RNA replication, could provide an advantage to KUN replicon RNA in establishing persistent replication. Studies with Sindbis virus showed that the adaptive mutation in Sindbis virus nsP2 protein (Pro726 to Leu) that provided an advantage in establishing persistent replication for the replicon RNA also resulted in efficient production of IFN-α/β when introduced into a Sindbis virus infectious clone, while the wt virus was deficient in IFN-α/β production (14). Apparently, the resulting accumulation of IFN-α/β in the medium of nsP2-mutated Sindbis virus-infected cells led to protection of neighboring uninfected cells, as well as to down-regulation of virus replication in infected cells. Interestingly, Pro726-to-Leu mutation in nsP2 also significantly reduced RNA replication efficiency (13), presumably by inhibiting nsP2-associated activities as part of a replication complex. Other studies with Sindbis virus and SFV replicons reported different adaptive mutations in nsP2 that provided an advantage in establishing persistent replication without affecting RNA replication efficiency (39). Neither of the studies, however, showed the effects of these adaptive mutations on IFN-α/β production either when introduced into the replicon RNA or when present in individually expressed nsP2 protein. It is therefore not clear from the alphavirus replicon studies whether the establishment of persistent (noncytopathic) replication is associated with the abilities of adaptive mutations to alter IFN response or to simply down-regulate RNA replication or both.
Ala30-to-Pro mutation in KUN NS2A provided an advantage in establishing persistent replication without inhibiting KUN replicon RNA replication. At the same time, the mutation was shown to remove the block in activation of IFN-β promoter-driven transcription characteristic of the wt NS2A protein either in response to KUN RNA replication when present in replicon RNA (replicon-expressing cells) or in response to exogenous stimulus (SFV challenge) when present in individually expressed NS2A protein. Although wt and NS2A-mutated replicons clearly differed in their abilities to down-regulate the activation of IFN-β promoter-driven transcription, the antiviral activities of IFN-α/β were inhibited with similar efficiencies in cells stably expressing wt and NS2A-mutated replicons (W. J. Liu and A. A. Khromykh, unpublished results). It was shown recently that once established, KUN replicon RNA replication was resistant to the antiviral activity of IFN-α (16). Most recently, it has also been shown that KUN and WNV blocked IFN-α/β signaling (W. J. Liu, X. J. Wang, V. V. Mokhonov, P.-Y. Shi, R. Randall, and A. A. Khromykh, submitted for publication). Therefore, any increase in the amount of IFN-α/β produced as a consequence of up-regulated IFN-β promoter-driven transcription by NS2A-mutated replicon RNA should not affect ongoing replication of replicon RNA. Hence, it is possible that the effect of an adaptive mutation(s) in NS2A on IFN response may not be related to the more efficient establishment of persistent replication, and thus the decreased cytopathicity of NS2A-mutated KUN replicon RNA may be caused by other mechanisms. These may include inhibition of double-stranded-RNA-induced apoptosis and reduction in NS2A cytotoxicity (6). Clearly, further studies are required to elucidate the mechanisms allowing flavivirus replicon RNAs to establish persistent (noncytopathic) replication.
It is becoming apparent that removing or mutating the genes in RNA viruses responsible for inhibition of IFN production or signaling leads to significant virus attenuation, and this approach has been employed in developing a number of safe vaccine candidates (5, 8). It is therefore likely that incorporation of the Ala30-to-Pro mutation in KUN NS2A that abolishes NS2A activity in inhibition of the antiviral response will lead to further attenuation of KUN and thus provide the basis for an even safer infectious KUN DNA-based vaccine against the New York strain of WNV (18). Studies to determine the in vivo effects of Ala30-to-Pro mutation in NS2A on the attenuation and efficacy of infectious KUN DNA-based WNV vaccine are under way.
Acknowledgments
We are grateful to Richard Randall and Stephen Goodbourn for the generous gift of the IFN-β promoter reporter plasmid and for helpful discussions on designing and interpreting IFN-related experiments.
This work was supported by grants to A.A.K. from the National Health and Medical Research Council of Australia.
Footnotes
This is publication number 206 from the Clinical Medical Virology Centre and the Sir Albert Sakzewski Virus Research Centre.
REFERENCES
- 1.Agapov, E. V., I. Frolov, B. D. Lindenbach, B. M. Pragai, S. Schlesinger, and C. M. Rice. 1998. Noncytopathic Sindbis virus RNA vectors for heterologous gene expression. Proc. Natl. Acad. Sci. USA 95:12989-12994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anraku, I., T. J. Harvey, R. Linedale, J. Gardner, D. Harrich, A. Suhrbier, and A. A. Khromykh. 2002. Kunjin virus replicon vaccine vectors induce protective CD8+-T-cell immunity. J. Virol. 76:3791-3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baigent, S. J., G. Zhang, M. D. Fray, H. Flick-Smith, S. Goodbourn, and J. W. McCauley. 2002. Inhibition of beta interferon transcription by noncytopathogenic bovine viral diarrhea virus is through an interferon regulatory factor 3-dependent mechanism. J. Virol. 76:8979-8988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blight, K. J., A. A. Kolykhalov, and C. M. Rice. 2000. Efficient initiation of HCV RNA replication in cell culture. Science 290:1972-1974. [DOI] [PubMed] [Google Scholar]
- 5.Bossert, B., and K. K. Conzelmann. 2002. Respiratory syncytial virus (RSV) nonstructural (NS) proteins as host range determinants: a chimeric bovine RSV with NS genes from human RSV is attenuated in interferon-competent bovine cells. J. Virol. 76:4287-4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang, Y. S., C. L. Liao, C. H. Tsao, M. C. Chen, C. I. Liu, L. K. Chen, and Y. L. Lin. 1999. Membrane permeabilization by small hydrophobic nonstructural proteins of Japanese encephalitis virus. J. Virol. 73:6257-6264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coia, G., M. D. Parker, G. Speight, M. E. Byrne, and E. G. Westaway. 1988. Nucleotide and complete amino acid sequences of Kunjin virus: definitive gene order and characteristics of the virus-specified proteins. J. Gen. Virol. 69:1-21. [DOI] [PubMed] [Google Scholar]
- 8.Donelan, N. R., C. F. Basler, and A. Garcia-Sastre. 2003. A recombinant influenza A virus expressing an RNA-binding-defective NS1 protein induces high levels of beta interferon and is attenuated in mice. J. Virol. 77:13257-13266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Falgout, B., R. Chanock, and C. J. Lai. 1989. Proper processing of dengue virus nonstructural glycoprotein NS1 requires the N-terminal hydrophobic signal sequence and the downstream nonstructural protein NS2a. J. Virol. 63:1852-1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Falgout, B., and L. Markoff. 1995. Evidence that flavivirus NS1-NS2A cleavage is mediated by a membrane-bound host protease in the endoplasmic reticulum. J. Virol. 69:7232-7243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Foy, E., K. Li, C. Wang, R. Sumpter, Jr., M. Ikeda, S. M. Lemon, and M. Gale, Jr. 2003. Regulation of interferon regulatory factor-3 by the hepatitis C virus serine protease. Science 300:1145-1148. [DOI] [PubMed] [Google Scholar]
- 12.Fredericksen, B. L., M. Smith, M. G. Katze, P. Y. Shi, and M. Gale, Jr. 2004. The host response to West Nile virus infection limits viral spread through the activation of the interferon regulatory factor 3 pathway. J. Virol. 78:7737-7747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frolov, I., E. Agapov, T. A. Hoffman, Jr., B. M. Pragai, M. Lippa, S. Schlesinger, and C. M. Rice. 1999. Selection of RNA replicons capable of persistent noncytopathic replication in mammalian cells. J. Virol. 73:3854-3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frolova, E. I., R. Z. Fayzulin, S. H. Cook, D. E. Griffin, C. M. Rice, and I. Frolov. 2002. Roles of nonstructural protein nsP2 and alpha/beta interferons in determining the outcome of Sindbis virus infection. J. Virol. 76:11254-11264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goodbourn, S., L. Didcock, and R. E. Randall. 2000. Interferons: cell signalling, immune modulation, antiviral response and virus countermeasures. J. Gen. Virol. 81:2341-2364. [DOI] [PubMed] [Google Scholar]
- 16.Guo, J. T., Q. Zhu, and C. Seeger. 2003. Cytopathic and noncytopathic interferon responses in cells expressing hepatitis C virus subgenomic replicons. J. Virol. 77:10769-10779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hall, R. A., A. A. Khromykh, J. M. Mackenzie, J. H. Scherret, T. I. Khromykh, and J. S. Mackenzie. 1999. Loss of dimerisation of the nonstructural protein NS1 of Kunjin virus delays viral replication and reduces virulence in mice, but still allows secretion of NS1. Virology 264:66-75. [DOI] [PubMed] [Google Scholar]
- 18.Hall, R. A., D. J. Nisbet, K. B. Pham, A. T. Pyke, G. A. Smith, and A. A. Khromykh. 2003. DNA vaccine coding for the full-length infectious Kunjin virus RNA protects mice against the New York strain of West Nile virus. Proc. Natl. Acad. Sci. USA 100:10460-10464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harvey, T. J., I. Anraku, R. Linedale, D. Harrich, J. Mackenzie, A. Suhrbier, and A. A. Khromykh. 2003. Kunjin replicon vectors for human immunodeficiency virus vaccine development. J. Virol. 77:7796-7803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harvey, T. J., W. J. Liu, X. J. Wang, R. Linedale, M. Jacobs, A. Davidson, T. T. Le, I. Anraku, A. Suhrbier, P. Y. Shi, and A. A. Khromykh. 2004. Tetracycline-inducible packaging cell line for production of flavivirus replicon particles. J. Virol. 78:531-538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hilpert, K., G. Hansen, H. Wessner, G. Kuttner, K. Welfle, M. Seifert, and W. Hohne. 2001. Anti-c-myc antibody 9E10: epitope key positions and variability characterized using peptide spot synthesis on cellulose. Protein Eng. 14:803-806. [DOI] [PubMed] [Google Scholar]
- 22.Katze, M. G., Y. He, and M. Gale, Jr. 2002. Viruses and interferon: a fight for supremacy. Nat. Rev. Immunol. 2:675-687. [DOI] [PubMed] [Google Scholar]
- 23.Khromykh, A. A., M. T. Kenney, and E. G. Westaway. 1998. trans-Complementation of flavivirus RNA polymerase gene NS5 by using Kunjin virus replicon-expressing BHK cells. J. Virol. 72:7270-7279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khromykh, A. A., H. Meka, K. J. Guyatt, and E. G. Westaway. 2001. Essential role of cyclization sequences in flavivirus RNA replication. J. Virol. 75:6719-6728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khromykh, A. A., P. L. Sedlak, K. J. Guyatt, R. A. Hall, and E. G. Westaway. 1999. Efficient trans-complementation of the flavivirus kunjin NS5 protein but not of the NS1 protein requires its coexpression with other components of the viral replicase. J. Virol. 73:10272-10280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khromykh, A. A., P. L. Sedlak, and E. G. Westaway. 2000. cis- and trans-acting elements in flavivirus RNA replication. J. Virol. 74:3253-3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khromykh, A. A., P. L. Sedlak, and E. G. Westaway. 1999. trans-Complementation analysis of the flavivirus Kunjin ns5 gene reveals an essential role for translation of its N-terminal half in RNA replication. J. Virol. 73:9247-9255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khromykh, A. A., A. N. Varnavski, and E. G. Westaway. 1998. Encapsidation of the flavivirus Kunjin replicon RNA by using a complementation system providing Kunjin virus structural proteins in trans. J. Virol. 72:5967-5977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Khromykh, A. A., and E. G. Westaway. 1994. Completion of Kunjin virus RNA sequence and recovery of an infectious RNA transcribed from stably cloned full-length cDNA. J. Virol. 68:4580-4588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Khromykh, A. A., and E. G. Westaway. 1997. Subgenomic replicons of the flavivirus Kunjin: construction and applications. J. Virol. 71:1497-1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.King, P., and S. Goodbourn. 1994. The beta-interferon promoter responds to priming through multiple independent regulatory elements. J. Biol. Chem. 269:30609-30615. [PubMed] [Google Scholar]
- 32.Liu, W. J., P. L. Sedlak, N. Kondratieva, and A. A. Khromykh. 2002. Complementation analysis of the flavivirus Kunjin NS3 and NS5 proteins defines the minimal regions essential for formation of a replication complex and shows a requirement of NS3 in cis for virus assembly. J. Virol. 76:10766-10775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lohmann, V., S. Hoffmann, U. Herian, F. Penin, and R. Bartenschlager. 2003. Viral and cellular determinants of hepatitis C virus RNA replication in cell culture. J. Virol. 77:3007-3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lohmann, V., F. Korner, A. Dobierzewska, and R. Bartenschlager. 2001. Mutations in hepatitis C virus RNAs conferring cell culture adaptation. J. Virol. 75:1437-1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mahalingam, S., J. Meanger, P. S. Foster, and B. A. Lidbury. 2002. The viral manipulation of the host cellular and immune environments to enhance propagation and survival: a focus on RNA viruses. J. Leukoc. Biol. 72:429-439. [PubMed] [Google Scholar]
- 36.Mosca, J. D., and P. M. Pitha. 1986. Transcriptional and posttranscriptional regulation of exogenous human beta interferon gene in simian cells defective in interferon synthesis. Mol. Cell. Biol. 6:2279-2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Munoz-Jordan, J. L., G. G. Sanchez-Burgos, M. Laurent-Rolle, and A. Garcia-Sastre. 2003. Inhibition of interferon signaling by dengue virus. Proc. Natl. Acad. Sci. USA 100:14333-14338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Otsuki, K., J. Maeda, H. Yamamoto, and M. Tsubokura. 1979. Studies on avian infectious bronchitis virus (IBV). III. Interferon induction by and sensitivity to interferon of IBV. Arch. Virol. 60:249-255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Perri, S., D. A. Driver, J. P. Gardner, S. Sherrill, B. A. Belli, T. W. Dubensky, Jr., and J. M. Polo. 2000. Replicon vectors derived from Sindbis virus and Semliki Forest virus that establish persistent replication in host cells. J. Virol. 74:9802-9807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pethel, M., B. Falgout, and C. J. Lai. 1992. Mutational analysis of the octapeptide sequence motif at the NS1-NS2A cleavage junction of dengue type 4 virus. J. Virol. 66:7225-7231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pflugheber, J., B. Fredericksen, R. Sumpter, Jr., C. Wang, F. Ware, D. L. Sodora, and M. Gale, Jr. 2002. Regulation of PKR and IRF-1 during hepatitis C virus RNA replication. Proc. Natl. Acad. Sci. USA 99:4650-4655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Qu, L., L. K. McMullan, and C. M. Rice. 2001. Isolation and characterization of noncytopathic pestivirus mutants reveals a role for nonstructural protein NS4B in viral cytopathogenicity. J. Virol. 75:10651-10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sen, G. C. 2001. Viruses and interferons. Annu. Rev. Microbiol. 55:255-281. [DOI] [PubMed] [Google Scholar]
- 44.Varnavski, A. N., and A. A. Khromykh. 1999. Noncytopathic flavivirus replicon RNA-based system for expression and delivery of heterologous genes. Virology 255:366-375. [DOI] [PubMed] [Google Scholar]
- 45.Varnavski, A. N., P. R. Young, and A. A. Khromykh. 2000. Stable high-level expression of heterologous genes in vitro and in vivo by noncytopathic DNA-based Kunjin virus replicon vectors. J. Virol. 74:4394-4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Westaway, E. G. 1973. Proteins specified by group B togaviruses in mammalian cells during productive infections. Virology 51:454-465. [DOI] [PubMed] [Google Scholar]
- 47.Westaway, E. G., J. M. Mackenzie, and A. A. Khromykh. 2003. Kunjin RNA replication and applications of Kunjin replicons. Adv. Virus Res. 59:99-140. [DOI] [PubMed] [Google Scholar]
- 48.Westaway, E. G., J. M. Mackenzie, and A. A. Khromykh. 2002. Replication and gene function in Kunjin virus. Curr. Top. Microbiol. Immunol. 267:323-351. [DOI] [PubMed] [Google Scholar]
- 49.Young, D. F., N. Chatziandreou, B. He, S. Goodbourn, R. A. Lamb, and R. E. Randall. 2001. Single amino acid substitution in the V protein of simian virus 5 differentiates its ability to block interferon signaling in human and murine cells. J. Virol. 75:3363-3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhu, Q., J. T. Guo, and C. Seeger. 2003. Replication of hepatitis C virus subgenomes in nonhepatic epithelial and mouse hepatoma cells. J. Virol. 77:9204-9210. [DOI] [PMC free article] [PubMed] [Google Scholar]