

Abstract
Rhabdoviruses such as rabies virus (RV) encode only five multifunctional proteins accomplishing viral gene expression and virus formation. The viral phosphoprotein, P, is a structural component of the viral ribonucleoprotein (RNP) complex and an essential cofactor for the viral RNA-dependent RNA polymerase. We show here that RV P fused to enhanced green fluorescent protein (eGFP) can substitute for P throughout the viral life cycle, allowing fluorescence labeling and tracking of RV RNPs under live cell conditions. To first assess the functions of P fusion constructs, a recombinant RV lacking the P gene, SAD ΔP, was complemented in cell lines constitutively expressing eGFP-P or P-eGFP fusion proteins. P-eGFP supported the rapid accumulation of viral mRNAs but led to low infectious-virus titers, suggesting impairment of virus formation. In contrast, complementation with eGFP-P resulted in slower accumulation of mRNAs but similar infectious titers, suggesting interference with polymerase activity rather than with virus formation. Fluorescence microscopy allowed the detection of eGFP-P-labeled extracellular virus particles and tracking of cell binding and temperature-dependent internalization into intracellular vesicles. Recombinant RVs expressing eGFP-P or an eGFP-P mutant lacking the binding site for dynein light chain 1 (DLC1) instead of P were used to track interaction with cellular proteins. In cells expressing a DsRed-labeled DLC1, colocalization of DLC1 with eGFP-P but not with the mutant P was observed. Fluorescent labeling of RV RNPs will allow further dissection of virus entry, replication, and egress under live-cell conditions as well as cell interactions.
Rabies virus (RV) of the Lyssavirus genus and related members of the Rhabdoviridae, such as vesicular stomatitis virus (VSV, Vesiculovirus genus), are composed of only five multifunctional viral proteins, namely, nucleoprotein (N), phosphoprotein (P), matrix protein (M), glycoprotein (G), and a large (L) RNA-dependent RNA polymerase. The viral RNA is enwrapped with N and associated with P and L to form a typical helical ribonucleoprotein (RNP) complex, which is active in RNA synthesis. During RV assembly, highly condensed RNPs of the typical rhabdovirus or bullet shape are enwrapped into an envelope containing the M and G proteins (31, 32). Entry of rhabdoviruses into cells involves receptor-mediated endocytosis, pH-dependent fusion of the viral and endosomal membranes (21), release into the cytoplasm, and uncoating of RNPs from M (35) such that gene expression can resume. Although these basic principles of the rhabdovirus infection pathway have been known for some time, details of many particular steps during entry, uncoating, gene expression, and virus egress await illumination. The possibility of real-time visualization of the entire infection pathway of rhabdoviruses and of tracking virus components such as RNPs in living cells is one highly desirable tool for the study of the rhabdovirus life cycle. A widely used approach in imaging of proteins or viruses in live cells is to fuse autofluorescent proteins such as green fluorescent protein (GFP) to either the amino or the carboxy terminus of the protein of interest and to examine fluorescence within the cells over time. However, in view of the multiple functions of all rhabdovirus RNPs, their multiple interactions with viral and cellular proteins, and possible structural constraints, there may be uncertainty as to the success of this approach.
We here assessed the feasibility of labeling RV RNPs and rabies virus virions by N- and C-terminal fusion of enhanced GFP (eGFP) to the phosphoprotein P. P is not just a structural component of the RNP but is crucially involved in numerous events during the virus life cycle, including proper formation of viral RNPs and virus particles and viral RNA synthesis. In rhabdovirus-infected cells, P is present in the form of oligomers (20). Binding of P to N is thought to chaperone N such that it specifically encapsidates viral RNA (9, 23, 26), making it a suitable template for RNA synthesis by the viral polymerase complex. In addition, P is an essential cofactor of the polymerase complex itself and directly binds the catalytic L protein (6). During RNA synthesis, RV RNPs or the polymerase complex interact with the M protein, which is involved in regulating the balance of mRNA transcription and RNP replication (17, 18). Moreover, RV P protein may also be involved in interactions with cellular factors important for in vivo infection. P was shown to bind to dynein light chain 1 (DLC1), suggesting a function in the transport of RNPs by motor proteins (27, 33, 34). In addition, an N-terminally truncated form of P has been found to enter the nucleus and to colocalize with promyelotic leukemia bodies (1).
Since it was not clear whether the fusion of eGFP to the N or C terminus of RV P affected its essential functions, recombinant P fusion proteins were first used for complementation of a recombinant RV lacking the P gene. Successful complementation was possible with N- and C-terminally fused P proteins, but differential activity in RNA synthesis and virus formation were noticed. The eGFP-P fusion protein allowed effective production of fluorescent extracellular virions that could be detected by conventional epifluorescence microscopy or laser-scanning microscopy and could be tracked during attachment to cells and internalization. The recovery of a viable recombinant RV expressing eGFP-P instead of P and its interaction with the cellular DLC1 protein further suggested that such recombinant viruses represent promising and reliable tools to study diverse aspects of rhabdovirus biology in living cells.
MATERIALS AND METHODS
Cells and viruses.
Viruses were grown on BHK-21 clone BSR cell monolayers maintained in Glasgow minimal essential medium supplemented with 10% newborn calf serum. BSR T7/5 cells constitutively expressing bacteriophage T7 RNA polymerase (2) was used for recovery of RV from transfected cDNA. BSR cells that constitutively express the authentic RV P protein were generated by cotransfection of 2.0 μg of p4P plasmid containing the P gene after the simian virus 40 promoter and of 0.2 μg of pMamNeo containing the neomycin resistance gene and subsequent cultivation in the presence of G418 (1 mg/ml). Cells expressing P fused to the N or C terminus of eGFP (BSR T7-eGFP-P and BSR T7-P-eGFP) were selected after cotransfection of BSR T7/5 cells with 5.0 μg of pEGFP-P or pP-EGFP and 0.5 μg of pTRE-Hyg (Clontech) and subsequent cultivation in the presence of hygromycin (1 mg/ml). The expression plasmids pEGFP-P and pP-EGFP are derivates of pEGFP-C3 and pEGFP-N3 (Clontech), with different linkers of 14 and 16 amino acids, respectively. BSR T7/5 cells that constitutively express DLC1 fused to the N terminus of a modified red fluorescent protein from Discosoma sp. (DsRed) were selected after cotransfection of the expression vector pDLC1-DsRed and pTRE-Hyg and selection as described above for eGFP-P cells. pDLC1-DsRed was constructed by an insertion of the coding sequence of DLC1 from human 293 cells into the pDsRed-Express-N1 vector (Clontech).
Recombinant RV lacking P expression, SAD ΔP, was generated from pSAD ΔP, which is a cDNA derivative of pSAD L16 (36). A deletion of 934 bp in the P-gene sequence (SAD B19 [7] nucleotides 1500 to 2434) resulted in the complete deletion of the P-protein-encoding sequence. In SAD eGFP-P, the authentic P coding sequence was replaced with the eGFP-P fusion encoding sequence from pEGFP-P. For generation of SAD eGFP-PI, the P protein coding sequence in pTIT-P (16) was mutated with the mutagenesis primer (5′-CCAGGAAAGTCTTCAGCAGCGGCAGCGGCAGCGGCAGCGGGCCGAGAGCTCAAGAAG-3′) using the Chameleon mutagenesis kit (Stratagene) as specified by the supplier. In pTIT-PI, 8 amino acids (142-EDKSTQTT-149) were thus replaced by alanine residues, deleting the DLC1 binding motif. The corresponding recombinant SAD eGFP-PI was generated by insertion of the mutated P gene sequence into the pSAD eGFP-P cDNA clone. A detailed description of all cloning steps and the final sequences are available from the authors on request by e-mail.
cDNA rescue experiments.
cDNA plasmids were transfected into cells after calcium phosphate precipitation (mammalian transfection kit; Stratagene) as specified by the suppliers. Vaccinia virus-free rescue of recombinant RV was performed as described previously (16) by transfection of 10 μg of full-length cDNA and plasmids pTIT-N (5 μg), pTIT-P (2.5 μg), and pTIT-L (2.5 μg) in 106 BSR T7/5 cells grown in 8-cm2 culture dishes. For recovery and subsequent amplification of the P-deficient RV SAD ΔP, culture supernatants from transfected cells were transferred onto BSR P cells at 3 days postinfection and incubated until almost 100% of the cells were infected.
Density gradient centrifugation.
Supernatants from 3 × 106 virus-infected cells were harvested on day 2 after infection at a multiplicity of infection (MOI) of 1, and the virions were pelleted through 20% sucrose in TEN buffer (50 mM Tris-HCl, 1 mM EDTA, 150 mM NaCl [pH 7.4]) on a 60% sucrose cushion. The interphase fraction was diluted in TEN buffer and was loaded on a 12-ml 20 to 40% iodixanol (Optiprep; Axis-Shield) density gradient. After 18 h of centrifugation (Beckman SW28 instrument; 27,000 rpm at 4°C), 12 fractions of 1 ml each were collected and the quality of the density gradients was controlled by determination of the refraction index of each fraction. After polyacrylamide PAA gel electrophoresis and Western blotting, fractions were analyzed with RV N, P, and M protein-specific sera or with a polyclonal serum recognizing eGFP.
Cell binding and virus uptake.
To observe binding of virus to the surface of cells, BSR T7/5 cells were trypsinized and the suspension was incubated with virus for 2 h at 4°C. The cells were then pelleted, resuspended in ice-cold phosphate-buffered saline and transferred onto the slides. After fixation and counterstaining with propidium iodide, the cells were mounted (Vectashield) and analyzed by confocal laser-scanning microscopy. To monitor virus uptake, the cells were incubated at 37°C for 30 min after incubation of cells for 2 h at 4°C.
Fluorescence microscopy.
Epifluorescence microscopy was performed on an inverse Zeiss Axiovert 200M microscope with 20× and 63× (NA 1.4) objectives, using Zeiss Filtersets (FS) 10 for GFP (excitation, BP450 to 490 nm; emission, BP515 to 565 nm) and FS 00 (excitation, BP530 to 585 nm; emission, LP615 nm) for DsRed. Images were taken with a Zeiss Axiocam HRm microscope using the Axiovision 3.1 software. Confocal laser-scanning microscopy was performed with the Leica TCS NT laser system, using a Leica DM IRB microscope or with a Zeiss LSM510 Meta laser system using a Zeiss Axivert200 microscope.
RNA analysis.
RNA from cells was isolated with the RNeasy mini kit (Qiagen). Northern blot analyses and hybridizations with [α-32P]dCTP-labeled cDNAs recognizing the RV N or P gene sequences were performed as described previously (8). Hybridization signals were quantified by PhosphorImager analysis (Molecular Dynamics Storm).
RESULTS
Recovery of a P-deficient RV and complementation with eGFP fusion proteins.
To establish a versatile system for assaying activities of different RV P constructs, we constructed an RV cDNA in which the complete P open reading frame was deleted (Fig. 1A). Rescue of the cDNA into recombinant RV SAD ΔP was achieved after transfection of BSR T7/5 cells with plasmids encoding RV N, P, L, and SAD ΔP antigenome RNA as described previously (16, 36). For further propagation of the newly generated viruses, supernatants from transfected cells were then transferred to a cell line constitutively expressing RV P (BSR-P) that was generated as described in Materials and Methods. The low levels of P expressed in BSR-P cells (not shown) supported the amplification of SAD ΔP; however, infectious-virus titers did not exceed 105 infectious units (IU) per ml of cell culture supernatant (not shown).
FIG. 1.
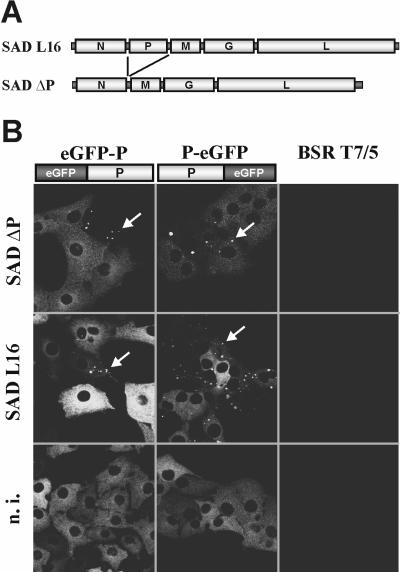
Complementation of a P-deficient RV in cells expressing eGFP-tagged P proteins. (A) Genome organization of SAD ΔP, lacking the entire P open reading frame, and the parental SAD L16. (B) Recruitment of cellular eGFP-tagged P proteins into RV inclusion bodies. Cells expressing the indicated P fusion proteins were infected with SAD ΔP or SAD L16. The appearance of fluorescent inclusion bodies at 1 day postinfection demonstrated nucleoprotein gene expression from SAD ΔP. Both eGFP-P and P-eGFP proteins also colocalized to inclusion bodies of wt SAD L16. In noninfected cells (n.i.), diffuse fluorescence was exclusively observed.
To test whether P proteins to which fluorescent eGFP was fused at the N or C terminus (eGFP-P or P-eGFP, respectively) would support the growth of SAD ΔP and provide a possibility to label virus RNPs and virus particles, two cell lines constitutively expressing the fluorescent proteins were generated. Expression of both fusion proteins led to a diffuse green cytoplasmic fluorescence with no significant signal in the nucleus of cells (Fig. 1B, bottom). However, when BSR eGFP-P and BSR P-eGFP cells were infected with the P-deficient SAD ΔP or the standard RV SAD L16 at an MOI of 0.1, fluorescence was concentrated in granular structures similar to the typical inclusion bodies that are usually observed in RV-infected cells (Fig. 1B, top and middle). Since coexpression of the RV N and P proteins is sufficient for formation of the inclusion bodies (5), the appearance of these fluorescent dots in SAD ΔP-infected cells demonstrated successful viral N gene expression by the activity of the two fluorescent P fusion proteins. In addition, it indicated typical interaction of eGFP-P and P-eGFP with N. Indeed, immunostaining with antibodies recognizing RV N protein revealed colocalization with the fusion proteins. In wild-type (wt) RV infection, this interaction took place in the presence of excess authentic P expressed from the virus. Moreover, similar to SAD L16, further spread of SAD ΔP in the cell cultures could be directly observed by the appearance the autofluorescent inclusion bodies in previously homogenously fluorescing cells (data not shown). This suggested that infectious-virus particle formation may also be supported by the eGFP-P fusions.
The effects of the P fusion proteins on virus gene expression and protein synthesis were further investigated by Western and Northern blot analyses (Fig. 2). In both cell lines the cell-derived GFP fusions with an expected size of approximately 62 kDa were detectable with a serum recognizing RV P protein (Fig. 2A). Infection with SAD ΔP led to the synthesis of considerable amounts of N protein only in the cell lines expressing the tagged P proteins. A faint signal for N was also detectable in noncomplementing BSR T7/5 cells, which could represent viral input N and newly expressed N protein through primary transcription of input virus. Notably, although eGFP-P was apparently present at higher levels than P-eGFP, similar levels of N protein accumulated in the two cell lines, suggesting a somewhat higher specific activity of P-eGFP in viral gene expression (Fig. 2A). This was supported by the finding of lower levels of cellular P-eGFP mRNA than of eGFP-P mRNA (Fig. 2B, bottom), combined with slightly more abundant N mRNAs in SAD ΔP-infected P-eGFP cells (Fig. 2B, top). Thus, it appears that P-eGFP is less impaired than eGFP-P in its function in viral mRNA synthesis. Moreover, expression of eGFP-P had a considerable negative effect on RNA synthesis and protein expression of wt RV, illustrated by reduced N and P mRNA and protein levels (Fig. 2, right panels), whereas viral RNA synthesis in P-eGFP cells was more similar to that in the control BSR T7/5 cells.
FIG. 2.
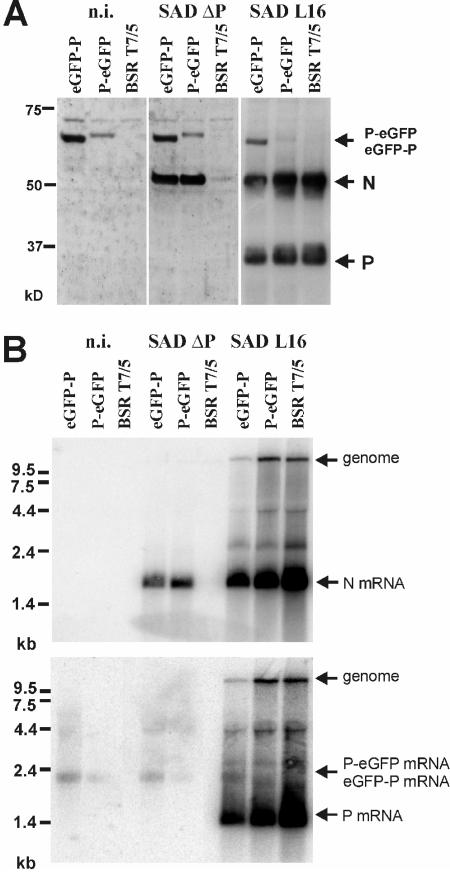
Gene expression of SAD ΔP and SAD L16 in eGFP-P and P-eGFP protein-expressing cells. Cells were infected with the indicated viruses at an MOI of 0.5 for 2 days and processed for analysis of viral proteins and RNA. (A) Western blot with a serum recognizing RV N and P proteins. For SAD L16-infected cells, three times less extract was loaded. (B) Northern blot hybridization with N- and P-specific cDNA probes. n.i., not infected.
Incorporation of eGFP-P into virions.
Although SAD ΔP virus gene expression was readily detected in the complementing cell lines, the yield of infectious virus was low, reaching maximum titers of 7 × 105 IU per ml of cell culture supernatant, i.e., 1,000-fold lower than standard virus SAD L16 titers in BSR cells. Most interestingly, infectious titers on P-eGFP cells were the same as or lower than those on eGFP-P cells, in spite of the obviously more rapid accumulation of viral RNAs in P-eGFP cells. To study virus formation and incorporation of the P-fusion proteins, supernatant virions from SAD ΔP-infected cells were enriched 4 days after infection at an MOI of 0.1 by centrifugation through 20% sucrose on a 60% sucrose cushion. Western blot analysis with sera recognizing GFP or RV N and P revealed the presence of both P fusion proteins along with N and suggested the formation of virus particles containing the P fusion proteins (Fig. 3A). Consistently, supernatants from eGFP-P cells were found to contain more viral protein, suggesting that its poor transcription activity is compensated by better permitting particle formation. Further purification by iodixanol density gradient centrifugation confirmed the formation of typical virus particles containing eGFP-P. A common protein peak of RV N, M, and the cell-derived 62-kDa eGFP-P protein, which also represented the peak of infectious virus (not shown), was demonstrated by Western blot analyses using sera against viral proteins and GFP (Fig. 3B, fractions 3 to 5). The complemented SAD ΔP(eGFP-P) virions completely lacked the authentic 37-kDa RV P protein abundantly present in particles from SAD L16-infected cells (Fig. 3C). As indicated by the appearance of a faint band representing eGFP-P in virions from e-GFP-expressing cells infected with SAD L16, incorporation of eGFP-P into virions was possible, although at low efficiency, in the presence of excess amounts of authentic viral P protein (Fig. 3C).
FIG. 3.
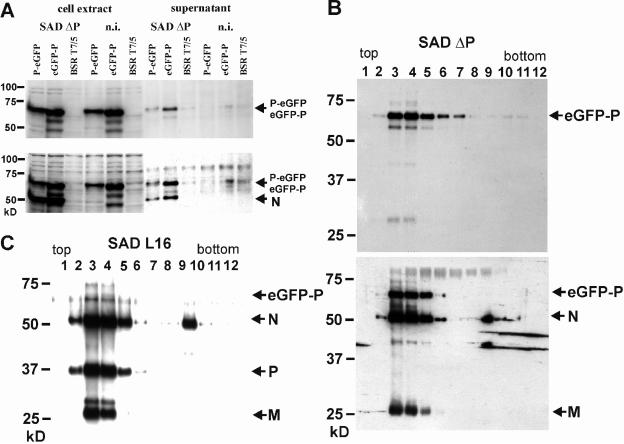
Incorporation of eGFP-P into RV virions. (A) Supernatants from cells expressing the indicated P fusion proteins and infected for 4 days with SAD ΔP virus were centrifuged through 20% sucrose and analyzed by Western blotting using sera recognizing eGFP (upper blot) and RV N and P proteins (lower blot). n.i., not infected. (B) Purification of supernatant virions from SAD ΔP-infected eGFP-P cells in a 20 to 40% iodixanol density gradient. Fractions were analyzed by Western blotting with sera recognizing GFP (upper blot) or RV N, P, and M proteins (lower blot). (C) Incorporation of eGFP-P into gradient-purified SAD L16 virions from eGFP-P expressing cells. Fractions were analyzed with RV N, P, and M protein-specific sera.
Detection of labeled virus particles by fluorescence microscopy.
To check whether the amount of virus-incorporated eGFP-P is sufficient for detection of virions by fluorescence microscopy, density gradient-purified SAD ΔP(eGFP-P) virions were transferred to glass coverslips and, without further treatment, were analyzed by confocal laser-scanning microscopy. Bright punctiform green fluorescence was observed, with individual dots of rather uniform size (Fig. 4A). Such typical patterns were not observed with purified supernatants from noninfected eGFP-P-expressing cells (Fig. 4E) or with wt virions (Fig. 4F), indicating that the fluorescent dots represent single extracellular, labeled virions. To further characterize the fluorescent structures observed, immunostaining of the unfixed and unpermeabilized virions was performed with antibodies specific for different virus proteins and Cy3-labeled secondary antibodies. Incubation with an antibody recognizing the viral surface glycoprotein G resulted in costaining (yellow) of most of the dots (Fig. 4B). Antibodies to the internal matrix protein M, which is located underneath the viral lipid bilayer, costained only a few dots (Fig. 4C), most probably as a result of disruption of the viral membrane during the course of virus purification. A similar staining was observed for the nucleoprotein N, a component of the inner RNP (Fig. 4D). These data strongly suggest that the punctuate structures observed by confocal microscopy represented intact fluorescent RV particles.
FIG. 4.
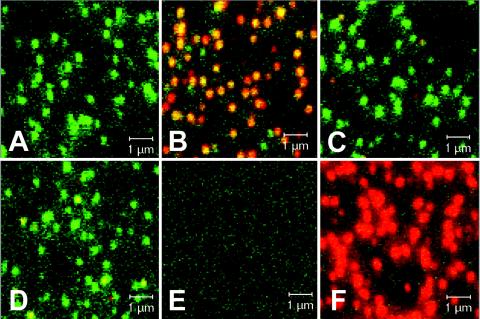
Autofluorescence of eGFP-P-containing SAD ΔP virions and coimmunostaining for viral proteins. (A to D) Density gradient-purified SAD ΔP virions complemented with eGFP-P were adhered to glass coverslips and directly analyzed by confocal laser-scanning microscopy (A) or were immunostained (red fluorescence) with sera recognizing RV G (B), M (C), or N (D). (E) Negative control: no virus, anti-RV G immunostain. (F) SAD L16 virions stained with anti-G antibodies. Overlays of the eGFP-P fluorescence (green) and of the immunostainings (red) are shown.
Cell binding and uptake of fluorescent virions.
To further investigate the biological behavior of green fluorescent virions, cell infection experiments were performed. To monitor binding to target cells, BSR cells grown in suspension were incubated with gradient-purified SAD ΔP(eGFP-P) for 2 h at 4°C, thereby preventing receptor-mediated endocytosis. Typical green dots associated with the surface of propidium iodide-counterstained cells could be visualized by confocal microscopy (Fig. 5A, top). The dots were of homogenous size, less than 1 μm, and were present exclusively at the surface of cells. These dots were not present in preparations of mock-incubated cells (Fig. 5A, bottom), suggesting that they represent virus particles. Notably, when virus-neutralizing antibodies were added prior to incubation with the cells, fluorescent signals were also associated with the surface of cells. However, the fluorescent structures were much larger (>1 μm), suggesting that they represent cross-linked virus aggregates (Fig. 5A, middle).
FIG. 5.
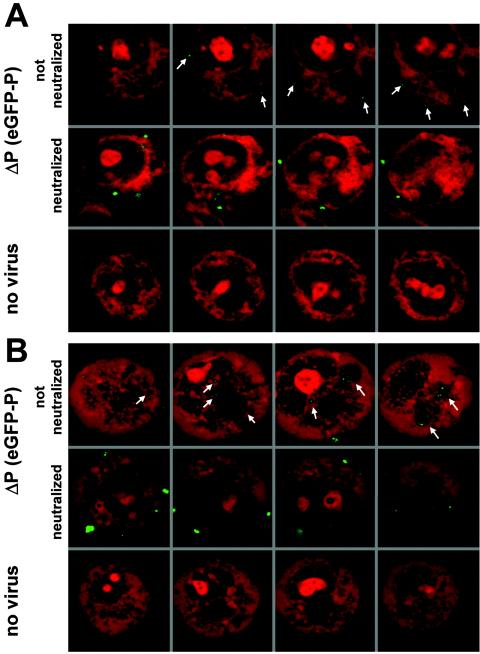
Cell binding and internalization of fluorescent virus. (A) Binding at 4°C of density gradient-purified SAD ΔP(eGFP-P) to the cell surface. BSR cells were incubated for 2 h at 4°C with virus, fixed, and counterstained with propidium iodide. Small green dots (<1 μm) were detectable at the cell surface (top row). Neutralization with anti-G antibodies prior to incubation with cells resulted in larger aggregates (>1 μm [middle row]) that were still able to attach. Analysis of multiple sections of cells revealed that fluorescent signals were detectable exclusively at the cell surface. No comparable signals were obtained in the absence of virus (bottom row). (B) Internalization of bound SAD ΔP virus particles into large vesicular structures was observed after subsequent temperature shift to 37°C (top row). Most neutralized virus (anti-G antibodies) remained at the cell surface; however, some virus aggregates were also internalized (middle row). No green fluorescence was observed in cells not infected with SAD ΔP(eGFP-P) virus (bottom row). All laser scans shown in the figure were made at identical parameter settings.
To further monitor the fate of fluorescent virions during cell entry, binding assays were performed as above, followed by a temperature shift to 37°C, which should allow receptor-mediated endocytosis. After 30 min at 37°C, the nonneutralized green dots were found internalized into intracellular vesicles (Fig. 5B, top). In contrast, the vast majority of the larger cross-linked complexes remained bound at the cell surface, although some of the complexes were also internalized (Fig. 5B, middle). Such temperature-dependent internalization and accumulation of dots in large vesicular structures indicate that eGFP-labeled virus particles were internalized by receptor-mediated endocytosis as described for natural RV.
Recombinant RV expressing eGFP-P fusion protein: live interaction of eGFP-P with DLC1.
Successful complementation of SAD ΔP in the eGFP-P expressing cell line to yield fluorescent intact virions led us to construct a full-length RV cDNA encoding the eGFP-P fusion protein instead of P (Fig. 6A). Successful rescue of viable virus from cDNA in standard transfection experiments was indicated by the appearance of the typical fluorescent inclusion bodies in foci of transfected BSR T7/5 cells at 5 days posttransfection and their further spread throughout the culture. In standard virus stock preparations, SAD eGFP-P virus yielded infectious titers of 2 × 106 focus-forming units (FFU)/ml, which is about 100-fold lower than those of the parental SAD L16 virus. Infection of cells and spread in cell culture could be monitored by fluorescence microscopy (Fig. 6B). Control immunostaining of the RV N protein revealed that all infected cells expressed eGFP-P protein (not shown), confirming the absence of virus in which the GFP sequence had been deleted during virus amplification. In live cells, green fluorescence was confined to the cytoplasm and was not observed in the nucleus, as is the case with a virus expressing GFP from an extra gene (not shown).
FIG. 6.
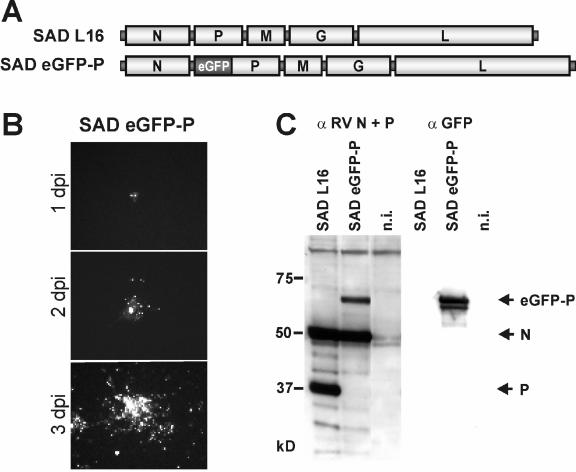
Recovery of autonomously replicating RV expressing the eGFP-P fusion. (A) Genome organization of SAD L16 and SAD eGFP-P in which eGFP is fused to the N terminus of P. (B) Spread of SAD eGFP-P in noncomplementing BSR T7/5 infected at a MOI of 0.001 observed in live cells by fluorescence microscopy at the indicated time points. (C) Expression of eGFP-P in cells infected with SAD ΔP at a MOI of 1 after 2 days by Western blots with the indicated sera. n.i., not infected.
Interactions of the virus-derived fluorescent eGFP-P fusion with other virus proteins were analyzed in more detail by immune staining of the RV N, P, or L protein and examination by confocal laser-scanning microscopy. Both N and L most exclusively colocalized with eGFP-P in the typical inclusion bodies (Fig. 7A). As expected, G protein was located predominantly in Golgi-endoplasmic reticulum compartments and at the cytoplasm membrane and did not co-localize with eGFP-P.
FIG. 7.
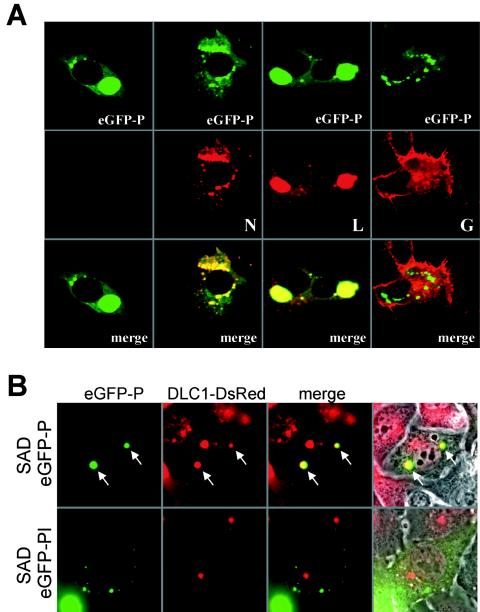
Interaction of eGFP-P with viral proteins (A) and cellular DLC1 (B). (A) SAD eGFP-P-infected BSR T7/5 cells were fixed at 2 days postinfection, immunostained with sera recognizing the RV N, G, or L proteins (red fluorescence), and analyzed by confocal laser-scanning microscopy. As a negative control, the infected cells were only incubated with fluorophor-conjugated secondary antibody (first column). (B) A cell line constitutively expressing DsRed-tagged DLC1 was infected with SAD eGFP-P and a mutant virus lacking the DLC1 binding site, SAD eGFP-PI. Autofluorescence of eGFP-P (green) and DsRed-DLC1 (red) was observed in live cells 1 day postinfection by conventional epifluorescence microscopy. Whereas DsRed perfectly colocalized with the SAD eGFP-P inclusion bodies (upper lane, arrows), no colocalization was observed in cells infected with the inclusion bodies of SAD eGFP-PI. Right-hand column, fluorescence and phase contrast.
RV P protein interacts with cellular DLC1. To check whether eGFP-P has retained this feature, a cell line expressing DLC1 fused to red fluorescent protein DsRed was generated as described in Materials and Methods and was infected with SAD eGFP-P virus. At 1 day postinfection, the distribution of DLC1-DsRed in living cells was analyzed (Fig. 7B, top). Most eGFP-P inclusion bodies showed red fluorescence, demonstrating interaction of the DLC1 and P subunits of the fusion proteins. To confirm the specificity of the interaction, an SAD eGFP-P mutant virus was generated in which the binding motif for DLC1 was replaced by a stretch of 8 alanine residues, SAD eGFP-PI. On infection of DLC1-dsRED cells with the mutant, costaining of inclusion bodies was no longer observed (Fig. 7B, bottom), demonstrating that eGFP-PI has lost the specific ability of eGFP-P to bind to DLC1-DsRed.
DISCUSSION
The availability of proteins tagged with GFP or other autofluorescent proteins, combined with progress in optical imaging, has recently allowed us to address the dynamics of many biological processes in living cells (15). This technology is particularly attractive to monitor virus infection in vitro and in vivo. Compared to chemical labeling, which allows input virus to be tracked (11, 22, 28, 37, 38), genetic labeling of virus proteins allows us to study all aspects of the virus life cycle in real time. For instance, successful labeling by GFP has been reported for human immunodeficiency virus, herpes simplex virus, and rotavirus particles (3, 12, 29). Structural and functional constraints of highly organized viruses, such as rhabdoviruses, however, may preclude the inclusion of entire fluorescent proteins or protein domains. We report here the successful construction of a viable, fluorescent RV which behaves like natural virus in many aspects and which allows live tracking of virus and virus components throughout the viral live cycle. Of note, even single virus particles were detectable by conventional live fluorescence microscopy, which is highly preferable over integrated imaging for kinetic studies.
Several reasons argued for the strategy to select the P protein for fusion with eGFP. Since P is a component of the viral RNP, fluorescent P therefore allows tracing not only of extracellular virions but also of RNPs following entry into cells and uncoating. Of the RNPs, P is of intermediate abundance. With approximately 450 copies of P present in VSV RNPs (40), it greatly exceeds the 120 molecules of eGFP required to visualize single rotavirus particles (3). Previously, an elegant study described successful labeling of the L protein of measles virus (Paramyxoviridae) by inserting GFP into an internal flexible loop of the protein and without disrupting its basic functions in RNA synthesis (14). However, single virus particles have not been observed, probably due to the lower content of L in RNPs. Approximately 50 copies of L were reported to be present in VSV RNPs (40). Approaches to labeling rhabdovirus proteins have so far been confined to the G protein of VSV. However, GFP fused to the cytoplasmic tail of VSV G allowed the generation of recombinant virus only when the authentic G protein was coexpressed (10).
Since RV P is instrumental in several essential virus functions, we first wanted to assess the functions of P proteins with N- and C-terminal eGFP extensions in virus complementation assays. We therefore constructed a recombinant P-deficient RV (SAD ΔP) and a P-cell line in which this virus could be amplified. The subsequent establishment of cell lines that express either P-eGFP or eGFP-P fusion proteins allowed versatile testing of protein functions related to virus gene expression and formation of virions.
RV gene expression could be visualized simply by infection and subsequent fluorescence microscopy. The redistribution of the diffuse cytoplasmic green fluorescence into the typical inclusion bodies demonstrated successful expression of N from the SAD ΔP genome (Fig. 1). Moreover, the appearance of inclusion bodies, first in single cells and later in neighboring cells, illustrated virus formation and spread (not shown) and provided strong evidence that both eGFP-P and P-eGFP were able to support the entire virus life cycle. This was confirmed by the detection of infectious virions in the cell culture and their repeated passage in the complementing cells.
Although both P fusion proteins were able to rescue the P deficiency of SAD ΔP, both were obviously disrupted in P functions. Moreover, fusion of eGFP to either the N terminus or C terminus resulted in different phenotypes, as may have been expected from the complex organization of the RV P protein and, in particular, the importance of the terminal regions of the P protein. The first 19 residues of P represent a binding site for L (6), whereas the C terminus of P is involved in binding to N (5, 6).
It was therefore interesting that SAD ΔP mRNA transcription was more effective in P-eGFP-expressing cells than in eGFP-P expressing cells, in spite of higher levels of the latter protein (Fig. 2). The decreased efficiency of eGFP-P in gene expression may therefore be due to steric hindrance, by the N-terminal GFP, of proper formation of the viral RNA polymerase complex, which, in VSV, is thought to contain three P molecules (19). The interesting observation of impaired gene expression of SAD L16 in eGFP-P-expressing cells may also be explained by a dominant negative effect of the eGFP-P fusion on the polymerase complex. Either incorporation of an eGFP-P molecule in the multimeric polymerase complex, leading to reduced activity, or a reduced binding of the viral polymerase to N/eGFP-P complexes might be responsible. In addition, the N-terminal extension of eGFP-P should abolish the expression of a series of N-terminally truncated P proteins (P2, P3, and P4) which are expressed in wt RV-infected cells from the P gene by translation initiation at downstream in-frame initiation codons (4). However, the recovery of viable SAD eGFP-P virus illustrated that these truncated forms of P do not represent essential virus proteins. Moreover, the dominant negative effect of eGFP-P on RNA synthesis of SAD L16, which does express the truncated P forms (not shown), strongly suggests that the intrinsic functions of the eGFP-P full-length protein rather than the lack of truncated P proteins is responsible for the poor RNA synthesis of SAD eGFP-P.
Compared to eGFP-P, the C-terminally extended P-eGFP showed less impairment in RNA synthesis; however, it was less effective in supporting infectious-virus formation. Comparison of supernatant virions from complementing cell lines indicated a less efficient release of virus particles from SAD ΔP-infected P-eGFP-expressing cells (Fig. 3A). The C-terminal moiety of RV P is involved in N binding. In particular, the C-terminal residues aa 268 to 297 were reported to strongly bind N (5). Thus, the defect of P-eGFP in virus particle formation might be due to an impairment of N binding. If so, however, it is notable that this defect in N binding did not greatly affect RNA replication, which is thought to depend on proper P/N interaction and interaction of the P/L polymerase with the RNP template. The poor formation of virus particles in P-eGFP-expressing cells is most probably due to its poor activity in assembly (see above; Fig. 2). In addition, the expression levels may be limiting, thus contributing to the low-efficiency virus production.
In the approach of generating fluorescent RV virions similar to wt RV, the phenotype of eGFP-P with reduced RNA synthesis and reasonable virus formation was preferable to that of P-eGFP, where viral RNA and protein accumulation was expected due to more severe defects in virus assembly. Therefore, eGFP-complemented SAD ΔP virus and recombinant SAD eGFP-P virus were used for further analysis of fluorescent RV. Comigration of eGFP-P with the virion marker proteins N and M in density gradients demonstrated the formation of typical RV virions and efficient incorporation of the eGFP-tagged P protein. As already suggested by the occurrence of strong Western blot signals (Fig. 3B), the level of incorporated eGFP-P was high enough to enable detection of extracellular virus particles by confocal laser-scanning microscopy (Fig. 4) and conventional epifluorescence microscopy (not shown). Moreover, a series of different experiments demonstrated that even single virus particles could be identified. Untreated purified virions appeared as green individual dots of homogenous size. Staining with antibodies against external (G) and internal (M and N) virus proteins supported the identity of the dots as individual virus particles rather than protein aggregates. Virtually all dots stained for the surface G protein; some, in which the membrane was probably disrupted, stained for M; and virtually none stained for the RNP protein P. This is in agreement with the proposed structure of RV, with a tight M protein layer located underneath the lipid envelope, completely protecting the RNP from access by antibodies (32).
Results of binding and internalization assays also suggest that we have observed single and intact virus particles which behave like natural virus. RV enters the cells by receptor-mediated endocytosis (39). Binding of fluorescent dots of uniform size to the surface of cells was demonstrated at low temperature. In the presence of neutralizing antibodies, fluorescent aggregates of larger and different size were present, some of which were also bound to the cell surface. Increasing the temperature to 37°C then allowed internalization of the individual dots into vesicles, within 30 min, reflecting virus uptake by receptor-mediated endocytosis (Fig. 5B). Also, some of the large aggregates formed by neutralizing antibodies were internalized. These observations are in full agreement with previous studies showing that binding of antibody-cross-linked RV to cells is still possible and internalization is not completely prevented (13). This indicates typical attachment and entry of SAD eGFP-P into target cells, suggesting that this virus is a promising tool to further dissect molecular and kinetic details of RV binding, uptake, membrane fusion, and uncoating.
Further experiments with SAD eGFP-P were aimed at validating whether some prominent function of authentic P in the interaction with host cell proteins is maintained by eGFP-P. RV P has been described to efficiently bind DLC1 (27, 34). Although deletion of the DLC1 binding site of P does not affect virus transcription and growth in standard cell culture (30, 33; unpublished data), a role of wt P in the intracellular retrograde transport of RNPs along microtubules was suggested, with a potentially important impact on the spread and pathogenesis of RV. To examine the interaction of P with DLC1 in live cells, a cell line was generated that constitutively expressed DsRed-labeled DLC1 as well as a SAD eGFP-P mutant virus in which the DCL1 binding motif was replaced with a stretch of alanine residues (SAD eGFP-PI; see Materials and Methods). This modification in P did not change the growth characteristics of the virus in BSR cultures (not shown). On infection with SAD eGFP-P, DsRed DCL1 colocalized with viral inclusion bodies. eGFP-P was exclusively responsible for recruitment of DsRed-DLC1 into the inclusion bodies, since in SAD eGFP-PI-infected cells, colocalization was never observed (Fig. 7). The availability of recombinant RV expressing fluorescent P now also provides the possibility of directly monitoring the movement of virus or viral RNPs in more specialized cell cultures such as neurons, or in vivo, and addressing the disputed question whether dynein motor transport of intracellular RNPs or transport of vesicles containing entire virus makes the greater contribution to the famous retrograde axonal transport of RV to the central nervous system.
In summary, tagging of the RV P with eGFP-P appears to allow many aspects of the entire RV life cycle. For instance, monitoring of RV binding to cells and neurons in vitro and in vivo may facilitate investigations of the distribution and nature of RV receptors, virus endocytosis, and membrane fusion. As viral RNPs are labeled, release of the RNP into the cytoplasm, uncoating of the RNP from the M layer (35), and onset of RNA synthesis may be tracked as well. As for studies on the mechanisms involved in entry, approaches to inhibit virus entry are more easily done with fluorescent virions and may be automated in the future. Further work will reveal whether the assembly and egress of rhabdovirus, which seems to usurp cellular vesicle transport mechanisms and budding (24, 25), can also be visualized in live cells.
Acknowledgments
We thank N. Hagendorf for perfect technical assistance.
This work was supported by the Deutsche Forschungsgemeinschaft, SFB 455 A3.
REFERENCES
- 1.Blondel, D., T. Regad, N. Poisson, B. Pavie, F. Harper, P. P. Pandolfi, H. de The, and M. K. Chelbi-Alix. 2002. Rabies virus P and small P products interact directly with PML and reorganize PML nuclear bodies. Oncogene 21:7957-7970. [DOI] [PubMed] [Google Scholar]
- 2.Buchholz, U. J., S. Finke, and K. K. Conzelmann. 1999. Generation of bovine respiratory syncytial virus (BRSV) from cDNA: BRSV NS2 is not essential for virus replication in tissue culture, and the human RSV leader region acts as a functional BRSV genome promoter. J. Virol. 73:251-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Charpilienne, A., M. Nejmeddine, M. Berois, N. Parez, E. Neumann, E. Hewat, G. Trugnan, and J. Cohen. 2001. Individual rotavirus-like particles containing 120 molecules of fluorescent protein are visible in living cells. J. Biol. Chem. 276:29361-29367. [DOI] [PubMed] [Google Scholar]
- 4.Chenik, M., K. Chebli, and D. Blondel. 1995. Translation initiation at alternate in- frame AUG codons in the rabies virus phosphoprotein mRNA is mediated by a ribosomal leaky scanning mechanism. J. Virol. 69:707-712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chenik, M., K. Chebli, Y. Gaudin, and D. Blondel. 1994. In vivo interaction of rabies virus phosphoprotein (P) and nucleoprotein (N): existence of two N-binding sites on P protein. J. Gen. Virol. 75:2889-2896. [DOI] [PubMed] [Google Scholar]
- 6.Chenik, M., M. Schnell, K. K. Conzelmann, and D. Blondel. 1998. Mapping the interacting domains between the rabies virus polymerase and phosphoprotein. J. Virol. 72:1925-1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Conzelmann, K. K., J. H. Cox, L. G. Schneider, and H. J. Thiel. 1990. Molecular cloning and complete nucleotide sequence of the attenuated rabies virus SAD B19. Virology 175:485-499. [DOI] [PubMed] [Google Scholar]
- 8.Conzelmann, K. K., J. H. Cox, and H. J. Thiel. 1991. An L (polymerase)-deficient rabies virus defective interfering particle RNA is replicated and transcribed by heterologous helper virus L proteins. Virology 184:655-663. [DOI] [PubMed] [Google Scholar]
- 9.Curran, J., J. B. Marq, and D. Kolakofsky. 1995. An N-terminal domain of the Sendai paramyxovirus P protein acts as a chaperone for the NP protein during the nascent chain assembly step of genome replication. J. Virol. 69:849-855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dalton, K. P., and J. K. Rose. 2001. Vesicular stomatitis virus glycoprotein containing the entire green fluorescent protein on its cytoplasmic domain is incorporated efficiently into virus particles. Virology 279:414-421. [DOI] [PubMed] [Google Scholar]
- 11.Da Poian, A. T., A. M. Gomes, R. J. Oliveira, and J. L. Silva. 1996. Migration of vesicular stomatitis virus glycoprotein to the nucleus of infected cells. Proc. Natl. Acad. Sci. USA 93:8268-8273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Desai, P., and S. Person. 1998. Incorporation of the green fluorescent protein into the herpes simplex virus type 1 capsid. J. Virol. 72:7563-7568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dietzschold, B., M. Tollis, M. Lafon, W. H. Wunner, and H. Koprowski. 1987. Mechanisms of rabies virus neutralization by glycoprotein-specific monoclonal antibodies. Virology 161:29-36. [DOI] [PubMed] [Google Scholar]
- 14.Duprex, W. P., F. M. Collins, and B. K. Rima. 2002. Modulating the function of the measles virus RNA-dependent RNA polymerase by insertion of green fluorescent protein into the open reading frame. J. Virol. 76:7322-7328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ehrhardt, D. 2003. GFP technology for live cell imaging. Curr. Opin. Plant Biol. 6:622-628. [DOI] [PubMed] [Google Scholar]
- 16.Finke, S., and K. K. Conzelmann. 1999. Virus promoters determine interference by defective RNAs: selective amplification of mini-RNA vectors and rescue from cDNA by a 3′ copy-back ambisense rabies virus. J. Virol. 73:3818-3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Finke, S., and K. K. Conzelmann. 2003. Dissociation of rabies virus matrix protein functions in regulation of viral RNA synthesis and virus assembly. J. Virol. 77:12074-12082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Finke, S., R. Mueller-Waldeck, and K. K. Conzelmann. 2003. Rabies virus matrix protein regulates the balance of virus transcription and replication. J. Gen. Virol. 84:1613-1621. [DOI] [PubMed] [Google Scholar]
- 19.Gao, Y., N. J. Greenfield, D. Z. Cleverley, and J. Lenard. 1996. The transcriptional form of the phosphoprotein of vesicular stomatitis virus is a trimer: structure and stability. Biochemistry 35:14569-14573. [DOI] [PubMed] [Google Scholar]
- 20.Gao, Y., and J. Lenard. 1995. Cooperative binding of multimeric phosphoprotein (P) of vesicular stomatitis virus to polymerase (L) and template: pathways of assembly. J. Virol. 69:7718-7723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gaudin, Y. 2000. Rabies virus-induced membrane fusion pathway. J. Cell Biol. 150:601-612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Georgi, A., C. Mottola-Hartshorn, A. Warner, B. Fields, and L. B. Chen. 1990. Detection of individual fluorescently labeled reovirions in living cells. Proc. Natl. Acad. Sci. USA 87:6579-6583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Green, T. J., S. Macpherson, S. Qiu, J. Lebowitz, G. W. Wertz, and M. Luo. 2000. Study of the assembly of vesicular stomatitis virus N protein: role of the P. protein. J. Virol. 74:9515-9524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harty, R. N., M. E. Brown, J. P. McGettigan, G. Wang, H. R. Jayakar, J. M. Huibregtse, M. A. Whitt, and M. J. Schnell. 2001. Rhabdoviruses and the cellular ubiquitin-proteasome system: a budding interaction. J. Virol. 75:10623-10629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harty, R. N., J. Paragas, M. Sudol, and P. Palese. 1999. A proline-rich motif within the matrix protein of vesicular stomatitis virus and rabies virus interacts with WW domains of cellular proteins: implications for viral budding. J. Virol. 73:2921-2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Horikami, S. M., J. Curran, D. Kolakofsky, and S. A. Moyer. 1992. Complexes of Sendai virus NP-P and P-L proteins are required for defective interfering particle genome replication in vitro. J. Virol. 66:4901-4908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jacob, Y., H. Badrane, P. E. Ceccaldi, and N. Tordo. 2000. Cytoplasmic dynein LC8 interacts with lyssavirus phosphoprotein. J. Virol. 74:10217-10222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lakadamyali, M., M. J. Rust, H. P. Babcock, and X. Zhuang. 2003. Visualizing infection of individual influenza viruses. Proc. Natl. Acad. Sci. USA 100:9280-9285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McDonald, D., M. A. Vodicka, G. Lucero, T. M. Svitkina, G. G. Borisy, M. Emerman, and T. J. Hope. 2002. Visualization of the intracellular behavior of HIV in living cells. J. Cell Biol. 159:441-452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mebatsion, T. 2001. Extensive attenuation of rabies virus by simultaneously modifying the dynein light chain binding site in the P protein and replacing Arg333 in the G protein. J. Virol. 75:11496-11502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mebatsion, T., M. Konig, and K. K. Conzelmann. 1996. Budding of rabies virus particles in the absence of the spike glycoprotein. Cell 84:941-951. [DOI] [PubMed] [Google Scholar]
- 32.Mebatsion, T., F. Weiland, and K. K. Conzelmann. 1999. Matrix protein of rabies virus is responsible for the assembly and budding of bullet-shaped particles and interacts with the transmembrane spike glycoprotein G. J. Virol. 73:242-250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Poisson, N., E. Real, Y. Gaudin, M. C. Vaney, S. King, Y. Jacob, N. Tordo, and D. Blondel. 2001. Molecular basis for the interaction between rabies virus phosphoprotein P and the dynein light chain LC8: dissociation of dynein-binding properties and transcriptional functionality of P. J. Gen. Virol. 82:2691-2696. [DOI] [PubMed] [Google Scholar]
- 34.Raux, H., A. Flamand, and D. Blondel. 2000. Interaction of the rabies virus P protein with the LC8 dynein light chain. J. Virol. 74:10212-10216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rigaut, K. D., D. E. Birk, and J. Lenard. 1991. Intracellular distribution of input vesicular stomatitis virus proteins after uncoating. J. Virol. 65:2622-2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schnell, M. J., T. Mebatsion, and K. K. Conzelmann. 1994. Infectious rabies viruses from cloned cDNA. EMBO J. 13:4195-4203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Seisenberger, G., M. U. Ried, T. Endress, H. Buning, M. Hallek, and C. Brauchle. 2001. Real-time single-molecule imaging of the infection pathway of an adeno- associated virus. Science 294:1929-1932. [DOI] [PubMed] [Google Scholar]
- 38.Suomalainen, M., M. Y. Nakano, S. Keller, K. Boucke, R. P. Stidwill, and U. F. Greber. 1999. Microtubule-dependent plus- and minus end-directed motilities are competing processes for nuclear targeting of adenovirus. J. Cell Biol. 144:657-672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Superti, F., M. Derer, and H. Tsiang. 1984. Mechanism of rabies virus entry into CER cells. J. Gen. Virol. 65:781-789. [DOI] [PubMed] [Google Scholar]
- 40.Thomas, D., W. W. Newcomb, J. C. Brown, J. S. Wall, J. F. Hainfeld, B. L. Trus, and A. C. Steven. 1985. Mass and molecular composition of vesicular stomatitis virus: a scanning transmission electron microscopy analysis. J. Virol. 54:598-607. [DOI] [PMC free article] [PubMed] [Google Scholar]