

Abstract
Conditionally replicating adenoviruses (CRAds) represent a promising class of novel anticancer agents that are used for virotherapy. The E1AΔ24 mutation-based viruses, Ad5-Δ24 [CRAd(E3−); E3 region deleted] and infectivity-enhanced Ad5-Δ24RGD [CRAd(E3+)] have been shown to potently eradicate tumor cells. The presence of the E3 region in the latter virus is known to improve cell killing that can be attributed to the presence of the oncolysis-enhancing Ad death protein. The more precise mechanism by which CRAds kill tumor cells is unclear, and the role of the host cell apoptotic machinery in this process has been addressed only in a limited way. Here, we examine the role of several major apoptotic pathways in the CRAd-induced killing of non-small-cell lung cancer H460 cells. As expected, CRAd(E3+) was more potent than CRAd(E3−). No evidence for the involvement of the p53-Bax apoptotic pathway was found. Western blot analyses demonstrated strong suppression of p53 expression and unchanged Bax levels during viral replication, and stable overexpression of human papillomavirus type 16-E6 in H460 cells did not affect killing by both CRAds. CRAd activity was also not hampered by stable overexpression of anti-apoptotic Bcl2 or BclXL, and endogenous Bcl2/BclXL protein levels remained constant during the oncolytic cycle. Some evidence for caspase processing was obtained at late time points after infection; however, the inhibition of caspases by the X-linked inhibitor of apoptosis protein overexpression or cotreatment with zVAD-fmk did not inhibit CRAd-dependent cell death. Analyses of several apoptotic features revealed no evidence for nuclear fragmentation or DNA laddering, although phosphatidylserine externalization was detected. We conclude that despite the known apoptosis-modulating abilities of individual Ad proteins, Ad5-Δ24-based CRAds trigger necrosis-like cell death. In addition, we propose that deregulated apoptosis in cancer cells, a possible drug resistance mechanism, provides no barrier for CRAd efficacy.
Conditionally replicating adenoviruses (CRAds) represent a promising class of biologic agents designed to selectively replicate in and lyse cancer cells, also known as virotherapy (for reviews, see references 2, 23, and 37). Tumor specificity of CRAds has been achieved by modifying a viral gene(s) important for efficient viral replication in normal cells but not in tumor cells that possess complementing genetic defects.
Two types of CRAds can be distinguished, in which either the adenoviral genome is modified by a specific mutation or deletion or tumor-specific promoters are inserted to drive the expression of essential viral genes. For example, Ad5-Δ24 contains a partial deletion in the CR2 domain of the pRb-binding protein E1A (dl922-947) that is complemented in pRb-deficient tumor cells (13). An integrin-directed infectivity-enhanced variant, CRAdΔ24RGD, which is currently being evaluated in clinical trials, demonstrated effective killing of different cancer cells in vitro and in vivo (3, 29, 46, 47).
In animal studies and in the clinic, CRAds appear to be safe and well tolerated; however, their antitumor activity as a single agent is modest, and improvement of the therapy is required (14, 24, 40). Strategies currently explored to enhance the efficacy of these agents include the combined use of CRAds with conventional therapies and the generation of armed CRAds containing cytotoxic payloads. In this respect, the possibility that interactions between the viral genome and host cell factors will determine adenoviral oncolytic efficacy has been left relatively unexplored (28) for wild-type Ad (wtAd) and has not been addressed for CRAdΔ24 variants. Ads normally infect cells of the respiratory tract; however, they are redirected in virotherapy to infect and kill tumor cells. Tumor cells are likely to possess intrinsic genetic differences compared to normal host cells that may also exist among different tumor types, which could either facilitate or hamper CRAd replication and cell killing. Such mechanisms may provide an explanation for observed discrepancies in CRAd efficacy in addition to the known distinct infection efficiencies.
Apoptosis is a specific form of programmed cell death (PCD) that is characterized by several morphological changes that are most prominently visible in the nucleus, including chromosome condensation and nuclear shrinkage and fragmentation. The caspases, members of the family of aspartic acid-specific cysteine-proteases, are the executioners of apoptosis and essential for the disassembly of the cell (for reviews, see references 9, 20, 45, and 48). Two main apoptotic pathways have been identified that induce caspase-dependent apoptosis upon exposure to various stimuli. The intrinsic pathway can be triggered by cytotoxic agents, for example, and involves the destabilization of mitochondria leading to activation of caspase-9. The extrinsic pathway is initiated via death receptors, such as Fas/CD95, that activate caspase-8. Both caspase-8 and caspase-9 can activate the downstream or executor caspase-3, -6, and -7. The activity of caspases can be regulated by members of the inhibitor of apoptosis (IAP) family, such as X-linked IAP (XIAP) (54). The Bcl-2 family of proteins comprise antiapoptotic and proapoptotic members that are key regulators of the mitochondrial pathway (1, 27, 41). Proapoptotic members, including Bax, can induce mitochondrial instability leading to the release of cofactors that allow the activation of caspase-9 assembled in the apoptosome. The activation of the basic apoptotic machinery as briefly outlined above is considered to largely determine the success of cancer therapies, and obstruction of this apoptotic machinery in cancer cells may cause resistance to treatment.
Ads have a rather complex infrastructure in which the viral genes produce numerous proteins that prevent the death of infected cells early after infection and other proteins that favor cell death at later stages (6, 28, 33, 38, 42, 52). Several viral encoded proapoptotic and antiapoptotic proteins are known to maintain temporal control of the Ad on the host cell.
Most notably, E1B-19KD (an Ad Bcl2 homologue), E1B-55KD in complex with E4orf6, and the E3-10.4KD and E3-14.5KD proteins, which are encoded by early transcribed genes, act as antiapoptotic proteins and are responsible for the prevention of premature oncolysis or cause immune suppression, while E1A-12S, E1A-13S, E4orf4, and E3-11.6KD (Ad death protein [ADP]) act as proapoptotic proteins when tested individually. In addition, the mechanism responsible for the oncolysis-enhancing effect of ADP is largely unknown (32, 50). Thus, during Ad infection, the viral proteins act in concert with several host cell-encoded key apoptotic regulators in a sort of cell death-balancing act, leading to the timed delay of cell death and allowing the efficient replication and generation of viral offspring.
Despite the known apoptosis-regulatory function of individual Ad genes, it is currently unknown whether the disruption of cancer cells at the last stage of CRAd infection, named oncolysis, employs the basic apoptotic machinery of the host cell. To explore this, we set out to investigate the role of the basic apoptotic machinery during the CRAdΔ24-induced killing of non-small-cell lung cancer (NSCLC) H460 cells. The involvement of several main apoptotic pathways was studied, including p53, Bcl2, and caspase-mediated cell death, and the morphological and biochemical features associated with CRAdΔ24-induced cell death were examined. Interestingly, CRAd-induced cell death can be classified as a necrosis-like PCD that bypasses the apoptotic machinery of the host cell irrespective of the presence or absence of the E3 region (ADP), suggesting that deregulated apoptotic pathways in cancer are unlikely to have a negative impact on CRAd-induced cell killing.
MATERIALS AND METHODS
Cell culture and treatment.
The human NSCLC NCI H460 parental cell line (p53+ Rb wt+ p16INK4−) (22); the previously generated derivatives H460Bcl2, H460BclXL, and H460XIAP; these derivatives' empty vector controls H460\PEFPGK3 and H460\pcDNA3 (11); and H460-human papillomavirus type 16 (HPV16)-E6 (designated H460HPV16-E6) (53) were cultured in RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum (Invitrogen, Breda, The Netherlands), 50 IU of penicillin/ml, 50 μg of streptomycin/ml, and 1 μg of puromycin/ml or 200 μg of Geneticin/ml, depending on the cell line, and grown at 37°C in a humidified air with 5% CO2. The cells were tested routinely for the absence of mycoplasma before use, and overexpression of Bcl2, BclXL, and XIAP proteins was confirmed by immunohistochemistry prior to use in the experiments. For apoptosis activation, cells were treated with 25 ng of anti-Fas activating antibody (clone CH11; Upstate Biotechnology, Inc., Lake Placid, N.Y.)/ml together with 0.5 μg of cycloheximide (CHX; Sigma, St. Louis, Mo.)/ml for 16 h or with 14 μM cisplatin (CDDP; Bristol-Myers Squibb, Woerden, The Netherlands) for 3 days. Caspase activation was inhibited by exposure of cells to the pan-caspase inhibitor zVAD-fmk at 100 μM (Enzyme Systems Products, Livermore, Calif.).
CRAd infection and viability measurement.
AdΔ24 [CRAd(E3−)] and AdΔ24RGD [CRAd(E3+)] harbor a 24-bp deletion in the pRb-binding CR2 domain in the E1A region; the latter contains the E3 region (E3+) and harbors an RGD motif cloned in the fiber knob that enhances infectivity in a wide range of cancer cells (47). Cells cultured to near-confluence in 96-well plates were incubated with different multiplicities of infection (MOIs) (0.001 to 100 PFU/cell) of CRAd(E3−) or CRAd(E3+) in growth medium (50 μl/well) at 37°C. Two hours postinfection, another volume of virus-free medium was added. Cell viability was measured within 7 days postinfection with WST-1 reagent (Roche Diagnostics, Mannheim, Germany). Briefly, the culture medium was removed and replaced by 100 μl of 10% WST-1 in culture medium.
Depending on cell type and density, the formation of the formazan dye was allowed to proceed for 30 to 60 min at 37°C, and the A450 was measured with a model 550 microplate reader (Bio-Rad Laboratories, Hercules, Calif.). The percentage of growth (WST-1 conversion) of treated cells was expressed as a percentage of the conversion by uninfected control cells after subtraction of background values of WST-1 incubated in the absence of cells. Alternatively, the oncolytic potential of the CRAds in relation to cell viability was determined by crystal violet staining. Cells were washed with phosphate-buffered saline (PBS) (10.9 mM Na2HPO4, 1.8 mM NaH2PO4, 8.2 g of NaCl/liter), fixed for 10 min at room temperature (RT) in 4% (vol/vol) formaldehyde in PBS, and stained using 10 g of crystal violet dye/liter in 70% (vol/vol) ethanol for 20 min at RT. After being washed several times with water, the culture plates were air dried prior to imaging.
Western blotting.
Cells were lysed at 24, 48m and 72 h postinfection as indicated in RIPA buffer (50 mM Tris-HCl, 150 mM NaCl, 0.05% sodium dodecyl sulfate, 0.5% sodium deoxycholate [Fluka Biochemika, Buchs, Switzerland], 1% Igepal [NP-40; Sigma], and 1 mM PEFA block protease inhibitors mixture [Roche Diagnostics]) on ice and stored at −80°C for further use. After determining sample protein concentrations by the bicinchoninic acid protein assay reagent kit (Pierce Biotechnology, Rockfort, Ill.), 20 μg of each sample was separated on a sodium dodecyl sulfate-12.5%polyacrylamide gel electrophoresis gel. Thereafter, the proteins were blotted on a polyvinylidene difluoride membrane (Bio-Rad). The membrane was rinsed in blocking solution containing 5% nonfat milk in TBST (0.1% Tween 20, 150 mM NaCl, and 10 mM Tris-HCl [pH 8]). The following first antibodies, dissolved in 5% bovine serum albumin-TBST, were used: anti-human p53 monoclonal antibody (MAb) (dilution 1:1,000; DAKO, Glostrup, Denmark), anti-Bax MAb (dilution 1:250; BD Transduction Laboratories, Sparks, Md.), rabbit polyclonal anti-BclXL (dilution 1:250; DAKO Diagnostics, Mississauga, Ontario, Canada), anti-Bcl2 MAb (dilution 1:2,000; DAKO), rabbit polyclonal anti-poly(ADP-ribose) polymerase (PARP) (dilution 1:2000; Roche, Almere, The Netherlands), anti-caspase-8 MAb (dilution 1:2,000; Immunotech, Prague, Czech Republic), rabbit anti-caspase-9 (dilution 1:2,000; PharMingen, San Diego, Calif.), anti-caspase-3 MAb (dilution 1:1,000; BD Transduction Laboratories), and anti-β-actin MAb (dilution 1:7,500; Sigma).
After incubation for 1 to 2 h with the primary antibody and washing in TBST, the blots were incubated with peroxidase-conjugated goat anti-rabbit (dilution 1:5,000) or rabbit anti-mouse (dilution 1:2,000) IgG secondary antibody (DAKO). For chemoluminescence detection, blots were immersed in enhanced chemiluminescence solution and exposed to hyperfilm (Amersham Pharmacia UK, Ltd., Buckinghamshire, United Kingdom).
Apoptosis detection assays.
For DNA fragmentation analyses, 5 × 105 cells were harvested 72 h postinfection, and DNA was extracted as described previously (12). In brief, cells were pelleted and lysed with 30 μl of lysis buffer containing 20 mM EDTA, 100 mM Tris (pH 8.0), 0.8% sodium lauryl sarcosinate, and 5 mg of proteinase K/ml. After 2 h of incubation at 50°C, 0.2 mg of RNase A/ml was added, and the lysate was incubated for another 30 min at 37°C, prior to electrophoresis on a 1.5% agarose gel containing ethidium bromide (0.2 μg/ml). Nuclear morphology was examined in treated or untreated cells cultured on coverslips that were washed once with PBS before incubation in the fixative (3.7% formaldehyde in PBS) for 30 min at RT, stained with Hoechst 33342 (Sigma) for 1 h at RT, and mounted with Vectashield (Vector Laboratories Inc., Burlingame, Calif.). Stained nuclei were visualized at a 40× magnification under UV light. Phosphatidylserine (PS) externalization was determined after exposure to the phospholipid-binding protein Annexin V, according to the manufacturer's protocol (Roche). In brief, cells cultured on coverslips were incubated with fluorescein-labeled Annexin V and propidium iodide (PI)-containing buffer (Roche) for 15 min at RT. Annexin V and PI staining were visualized at a 63× magnification under blue and green light, respectively, with an inverted DMIRB/E fluorescence microscope (Leica, Heidelberg, Germany) using Leica Q500MC Quantimet software, version 1.01 (Leica Cambridge, Ltd., Cambridge, United Kingdom).
RESULTS
CRAd-induced killing of H460 cells.
To determine the mode of cell death induced by CRAds, we used two variants: E3 region-deleted virus, CRAd(E3−), and CRAd(E3+), containing the E3 region and possessing integrin-mediated enhanced infectivity (29). In this study, we compared the mechanism of cell killing by these two CRAds, assuming that RGD targeting only leads to enhanced infection efficiency without affecting the underlying mechanism of cell killing. For a model system, we employed the NSCLC H460 cell line. First, we assessed the oncolytic effect of the CRAds in H460 cells by determining cell viability by WST-1 (Fig. 1A) and crystal violet staining assays (Fig. 1B) at 5 and 11 days, respectively, after infection with different MOIs. As expected, both viruses showed MOI-dependent toxicity with CRAd(E3+) that was more potent than with CRAd(E3−) in killing H460 cells; 50% inhibitory concentration values were 3.5 and 30 PFU/cell, respectively.
FIG. 1.

Infectivity-enhanced AdΔ24RGD[CRAd(E3+)] is more potent in killing H460 cells than AdΔ24[CRAD(E3−)]. The viability of H460 cells was determined by the WST-1 assay, with three experiments. Data show means ± standard deviation (A) and crystal violet staining (B) at 5 and 11 days postinfection, at different MOIs.
p53-Independent cell killing by CRAds in H460 cells.
To investigate the role of p53 in mediating cell death triggered by CRAds in H460 cells, the expression of p53 was examined by Western blotting in extracts derived from CRAd-infected cells that were harvested at different time points after infection (Fig. 2A). The levels of p53 progressively decreased within 2 days after CRAd(E3−) infection, which can be attributed to E1B-55KD-mediated degradation (18, 36). The presence of the E3 region accelerated the reduction in p53 levels that may also be caused by accelerated internalization via the integrin-targeting motif RGD. The expression of the p53-inducible proapoptotic Bcl2 family member Bax was also studied, revealing a time-dependent decrease after CRAd(E3−) infection. The presence of the E3 region augmented CRAd-induced Bax reduction at 24 h postinfection, and levels remained low at later time points. As a control, H460 cells were treated with CDDP, which resulted in the induction of Bax and a less appreciable accumulation of p53 (Fig. 2A), which is in line with the expected activation of a p53-dependent route to apoptotic cell death. Activation of a p53-independent death receptor pathway with an agonistic Ab directed against the Fas receptor caused a decrease in the levels of p53 and Bax.
FIG. 2.
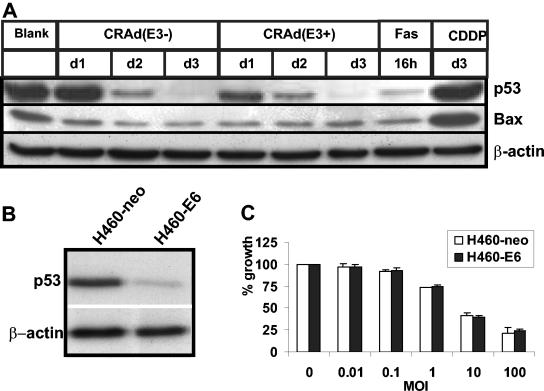
The p53-Bax apoptotic pathway is not involved in CRAd(E3+)- or CRAd(E3−)-induced cell killing of H460 cells. Western blot analyses of p53 and Bax expression in H460 cells infected with CRAd(E3+) or CRAd(E3−) at an MOI of 25 at different time points after infection are shown. As a control, H460 cells were treated with CDDP or with FasL+CHX (Fas). β-Actin serves as a control for loading (A). The reduced expression level of p53 in stable HPV16-E6-overexpressing H460 cells (H460-E6) was confirmed (B). CRAd-induced cell killing at different MOIs was examined in H460-E6 cells and in empty vector-transfected control H460 cells (H460-neo) at 7 days after infection by WST-1 assays. The results obtained for CRAd(E3+) are shown and were similar to those for CRAd(E3−) (C). Values are means ± standard deviation of three experiments.
To further assess the role of p53 in CRAd-induced cell death, H460 cells stably transfected with HPV16-E6, which possess reduced levels of p53 caused by HPV16-E6-mediated degradation of p53 (Fig. 2B), were infected with different MOIs of CRAd(E3+). Cell viability was measured at 7 days postinfection and was compared to CRAd toxicity in empty vector-transfected p53-expressing control cells (H460-neo) (Fig. 2C). The E6-dependent inhibition of p53 did not influence the oncolytic effect of this CRAd. Together, these data indicate that the cell killing effect of both tested CRAds is not dependent on the activation of p53-dependent apoptosis.
Cell death induced by CRAds is triggered independent of caspases and Bcl2.
Next, the contribution of the basic apoptotic machinery to CRAd-induced cell death was evaluated more directly. H460 cells were infected with the two CRAds at an MOI of 25, and the processing of procaspases-9, -8, and -3 and the caspase substrate PARP was determined by Western blotting at various days postinfection. Figure 3A shows that both CRAds produced some PARP cleavage at 24 and 48 h after infection, followed by a decrease in both unprocessed PARP (116 kDa) and cleaved PARP (89 kDa) at 3 days postinfection. The activation of caspases was studied by monitoring the decrease in the band representing the procaspase form that is indicative for caspase activation. Apart from a small decrease in the procaspase bands at 3 days postinfection, there is no clear indication of caspase processing in contrast to the strong activation observed during Fas-induced apoptosis (Fig. 3A) that was used as a positive control for death receptor and caspase-dependent apoptosis. Subsequently, the involvement of the anti-apoptotic Bcl2 family members Bcl2 and BclXL, key regulators of the mitochondrial apoptotic pathway, was studied during CRAd-induced cell death. In Western blotting experiments, the expression levels of Bcl2 and BclXL did not decrease (Fig. 3B); together with the above-demonstrated lack of increase in Bax levels (Fig. 1A), these findings are indicative for the lack of participation of the mitochondrial pathway in mediating cell killing by these CRAds.
FIG. 3.
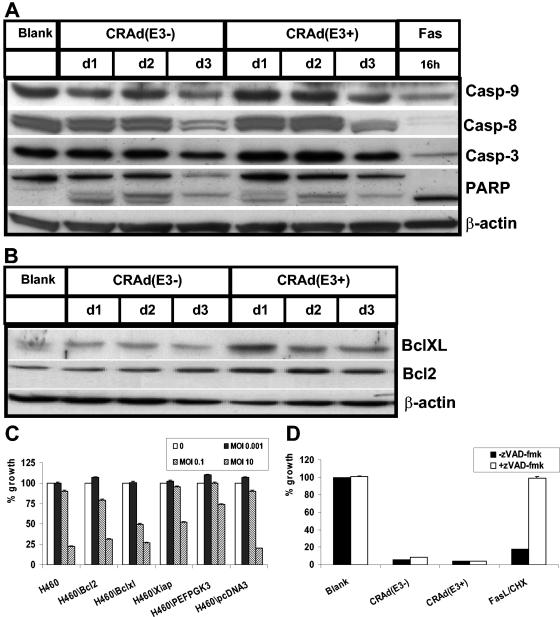
CRAd(E3±)-induced cell death is not mediated by caspases and is not dependent on the activation of the mitochondrial apoptotic pathway regulated by Bcl2. H460 cells were infected at an MOI of 25, and cell extracts were made at different times postinfection and subjected to Western blotting. As a control, H460 cells were treated with agonistic Fas Abs in combination with CHX (Fas). (A) The expression levels of unprocessed procaspase-9, -8, and -3 were assessed together with the cleavage of caspase substrate PARP. (B) Bcl2 and BclXL expression was also determined. (C) CRAd(E3+)-induced cell killing in H460-derived stable transfected cell lines overexpressing antiapoptotic Bcl2 and BclXL or the caspase-inhibitor XIAP when compared to the empty vector controls H460\PEFPGK3 and H460\pcDNA3 was studied. Different MOIs were used, and WST-1 activity was measured at 4 days postinfection. Similar results were obtained for CRAD(E3−). (D) Cotreatment with the broad caspase inhibitor zVAD-fmk failed to protect H460 cells from CRAd(E3±)-induced cell death, whereas it was effective in protecting against apoptosis induced by FasL+CHX cells that served as a positive control. Values are means ± standard deviation of three experiments.
To further test the role of caspases and Bcl2 in CRAd-triggered cell death, we used a panel of H460-derived stable transfectants overexpressing Bcl2, BclXL, and XIAP, the latter an inhibitor of caspase-9- and caspase-3-dependent apoptosis (54). These cell lines have been generated previously and shown to be potent in inhibiting mitochondria- and caspase-dependent apoptosis in H460 cells (11). The stable transfectants were tested for their sensitivity to the oncolytic effect of CRAd(E3+) when compared to parental H460 cells and vector-transfected control H460 cells (Fig. 3C, H460\PEFPGK3 and H460\pcDNA3). No protection was observed by the overexpression of Bcl2, BcXl, or XIAP, and similar results were found for CRAd(E3−) (data not shown).
As a final test to examine the possible involvement of caspases in CRAd-induced cell death, H460 cells were cotreated with the broad caspase inhibitor zVAD-fmk at a concentration of 100 μM, which completely protected against the apoptotic effect of FasL plus CHX (FasL+CHX) (Fig. 3D). In line with the above, CRAd activity was not suppressed in a detectable way by zVAD-fmk administration, further confirming that caspases are not involved in the execution of CRAd-induced cell death.
Analyses of biochemical and morphological features of apoptosis in H460 cells undergoing CRAd-induced cell death.
Finally, CRAd-dependent changes in several parameters of apoptosis were examined. Nuclear morphology was studied by staining H460 cells with Hoechst 33342 at 3 days after infection with both CRAds at an MOI of 25 in the presence or absence of 100 μM zVAD-fmk to assess caspase-dependent phenomena (Fig. 4A). Regardless of the presence of the E3 region, both CRAds seemed to cause some swelling of the nuclei, and regions with more intense staining (i.e., a speckled appearance) were observed that were not affected by zVAD-fmk treatment. In contrast, FasL+CHX exposure clearly induced chromosome condensation, nuclear shrinkage, and/or fragmentation that could be reverted to normal by cotreatment with 100 μM zVAD-fmk, indicative of caspase-dependent apoptosis.
FIG. 4.
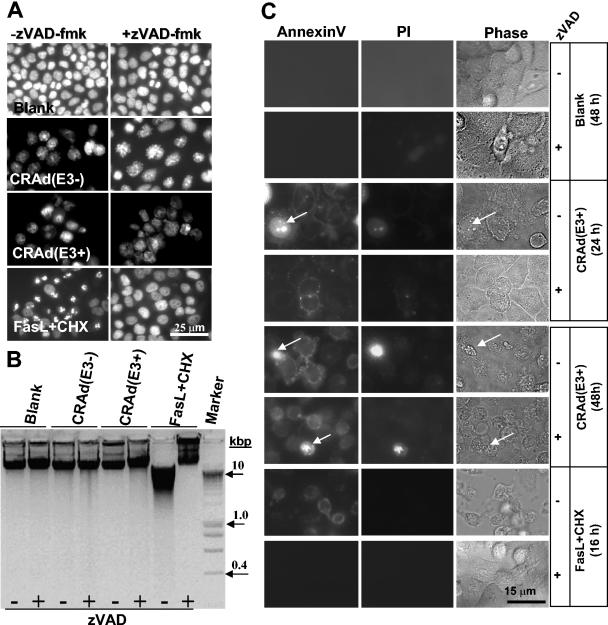
Lack of apoptotic features in H460 cells undergoing cell death triggered by CRAd(E3+) and CRAd(E3−). (A) H460 cells were infected with both CRAds at an MOI of 25 alone or in combination with 100 μM zVAD-fmk for 2 days. As a control, cells were exposed for 16 h to FasL+CHX with or without zVAD-fmk. Nuclei were stained with Hoechst 33342 to visualize chromatin condensation, nuclear shrinkage, or fragmentation that are markers for apoptosis by immunofluorescence microscopy. CRAd-infected cells did not display nuclear apoptotic features, in contrast to Fas+CHX-exposed cells that show condensed and fragmented nuclei reversible by zVAD-fmk. (B) DNA fragmentation was also analyzed, revealing no apoptotic DNA laddering in CRAd-infected cells, whereas FasL+CHX treatment results in clear DNA smearing. (C) Annexin V staining caused by PS externalization was examined 1 and 2 days postinfection and in FasL+CHX-treated H460 cells with and without zVAD-fmk. Cells were cotreated with PI to allow the distinction between Annexin V staining in the presence or absence of an intact membrane detected by immunofluorescence microscopy; the phase-contrast image is also shown (Phase). A minor portion of CRAd-infected cells were PI positive (indicated by arrows). Annexin V staining is caspase independent in CRAd-infected cells and caspase dependent when induced by FasL+CHX. Results are shown for CRAd(E3+)-infected cells; similar findings were obtained for CRAd(E3−)-infected cells.
DNA fragmentation assays were also performed at 3 days after infection with both CRAds. Contrary to the pronounced DNA fragmentation found in FasL+CHX-positive control cells, no DNA fragmentation was observed in CRAd(E3+)- or CRAd(E3−)-infected cells (Fig. 4B). Caspase inhibition by 100 μM zVAD-fmk completely protected against FasL+CHX-induced DNA laddering.
The last marker of apoptosis that we examined was the loss of phospholipid symmetry and externalization of PS residues in cells with an intact membrane that is indicative for apoptosis (10, 21) and which was determined by AnnexinV staining within 2 days after CRAd infection. The cells were costained with PI in order to discriminate apoptotic cells (Annexin V positive and PI negative) from necrotic ones (Annexin V and PI positive due to membrane permeabilization). The results obtained were similar for both CRAds and are only shown for CRAd(E3+), which induced a somewhat more rapid PS externalization than CRAd(E3−), in line with its faster rate of oncolysis. Untreated cells remained negative for Annexin V staining for the duration of the experiment (Fig. 4C). The majority of CRAd-infected cells showed Annexin V-positive membrane staining in the absence of PI staining. In a small portion of CRAd-infected cells, PS externalization was accompanied by PI staining; this may represent a minority of cells that became leaky during virus propagation. Coexposure with zVAD-fmk did not alter the Annexin V staining pattern in the infected cells. FasL+CHX-treated cells displayed Annexin V-positive and PI-negative cells, and PS exposure was completely prevented by zVAD-fmk (Fig. 4C). This indicates that CRAds trigger PS externalization via a caspase-independent mechanism, in contrast to FasL+CHX treatment, which induces caspase-dependent PS exposure.
DISCUSSION
The eradication of cancer cells by gene therapy approaches that are based on the molecular characteristics of cancer cells provides a promising new therapeutic platform. In this respect, CRAds represent rationally designed agents for the selective killing of cancer cells, while leaving normal cells intact. Despite exciting laboratory results with CRAds, several practical obstacles need to be overcome before virotherapy can fulfill its goals in the clinic.
In this study, we focused on the possibility that the host cell apoptotic machinery may either facilitate or suppress CRAd-induced cell killing in cancer cells, as this may cause resistance to CRAds and lead to intratumor variation in CRAd efficacy. In various cellular and biochemical assays, we showed that CRAd(E3−) and CRAd(E3+) kill H460 NSCLC cells independent from the basic apoptotic machinery. At late time points after infection, some evidence for procaspase processing was found (Fig. 3); however, the pan-caspase-inhibitor zVAD-fmk and overexpressed XIAP did not influence CRAd-mediated H460 cell killing, indicating that the observed caspase cleavage is a cophenomenon and not instrumental for CRAd toxicity. In line with this, the small proportion of cleaved PARP may reflect to some extent the apoptosis-regulatory activity of the adenoviral proteins in preventing the cell from dying early before completion of the reproductive viral cycle. Rather than inducing PARP cleavage, PARP expression fades away at 3 days postinfection, which is suggestive of caspase-independent cell death. The lack of effect of caspase inhibition by zVAD-fmk on cell death triggered by these CRAds was also observed in NSCLC A549 and SW1574 cells (unpublished data).
The apparent lack of involvement of the basic apoptotic machinery in mediating CRAd-induced cell death was confirmed in H460 cells overexpressing Bcl2 or BclXL that failed to protect against the oncolytic effect of CRAds. In this context, the presence or absence of the E3 region did not affect the mode of cell death, in agreement with the observed nonapoptotic cell death induced by ADP (6, 50).
Previously, it has been speculated that ADP that is expressed in large amounts at late stages after infection facilitates oncolyses in a stoichiometric manner by targeting the nuclear membrane where it either binds and/or deactivates antiapoptotic proteins like cellular Bcl2 or viral E1B-19KD or acts by disrupting nuclear membrane (5, 15). Our data showing that ADP did not engage the apoptotic machinery is more in favor with the latter hypothesis that ADP mainly targets the nuclear membrane rather than modulating antiapoptotic factors to enhance oncolysis.
The role of p53 in determining the oncolytic activity of replicating Ads is somewhat controversial. After Ad infection, p53 accumulates, which is counteracted by E1B-55KD together with E4orf6, acting as E3 ubiquitin ligase, which complexes with p53, resulting in its degradation and preventing p53-dependent apoptosis. On one hand, it has been demonstrated that p53 in a complex with E1B-55KD-E4orf6 is required for efficient (rapid) oncolysis (8, 16) and that p53 overexpression (19, 44, 51) enhances the oncolytic effect of CRAds; in other studies, the presence or absence of functional p53 did not alter replication and release of Ad (26). A possible drawback in these latter studies is that tumor cell lines derived from distinct sources were compared, which does not rule out the possibility that other genetic differences apart from p53 status may influence Ad propagation. By using the isogenic cell lines H460 and H460HPV16-E6, we sought to overcome this. Our experiments with AdΔ24-based CRAd(E3+) and CRAd(E3−) did not show an effect of overexpression of the p53-sequestering HPV16-E6 protein on CRAd-induced cell killing in H460 cells, which supports the notion that p53 status does not influence CRAd-induced cell death. Our results are in agreement with reports of Harada and Berk (17) and others (18, 45), who showed comparable adenoviral production (replication and release) in cancer cells independent from p53 status, including in p53-null NSCLC cell lines (e.g., H1299) and p53-wild-type counterparts (17, 43). However, in our experiments we cannot rule out the possibility that even in H460HPV16-E6 cells only a very small portion of p53/E1B-55KD/E4orf6 is formed that is sufficient for normal oncolysis. In this scenario, p53 triggers an as-yet-unknown cell death mechanism that is distinct from classic p53-dependent apoptosis via the intrinsic (mitochondrion-mediated) route. Yet we can rule out that the E1A mutation specific to the Ad5-Δ24-based viruses plays a role in rendering CRAd(E3+)- and CRAd(E3−)-induced cell killing p53 independent, since wtAd5 induces a similar p53-independent cell killing in HPV16-E6-overexpressing H460 cells (data not shown).
Apart from addressing the role of the apoptotic machinery in CRAd-induced cell death, we also monitored a panel of morphological parameters that allows the more precise classification of the mode of cell death that can range from apoptosis to necrosis. Apoptosis, as a distinct PCD process, is mainly characterized by a combination of hallmark morphological and biochemical changes, including DNA fragmentation, chromatin condensation, cytoplasmic shrinkage, and PS externalization, which can be associated with molecular markers such as the activation of caspases (20). More recently, it has been reviewed that other types of PCD exist in which caspases are not activated despite the presence of apoptotic features, such as DNA fragmentation and chromatin condensation; this type of PCD is named apoptosis-like PCD (31). In the absence of chromatin condensation, this type of PCD is termed necrosis-like PCD, which can still be associated with apoptotic markers such as PS externalization. CRAd-induced oncolysis, regardless of the presence of the E3 region, was not associated with apoptotic DNA fragmentation caused by internucleosomal DNA cleavage (Fig. 4B). Nuclear staining revealed small aggregates (speckled staining) spread in the nucleus of CRAd-infected cells (Fig. 4A), which is difficult to assign either to aggregates of Ad DNA or to irregular chromatin condensation. By visual observation, CRAd infection seemed to increase the size of the nucleus to some extent, in contrast to the nuclear shrinkage and fragmentation observed with FasL+CHX-treated cells. Finally, Annexin V staining assays showed that CRAd-induced cell death was associated with PS externalization that could not be blocked by zVAD-fmk, in contrast to PS exposure-induced by FasL+CHX that was caspase dependent. Regardless of the observed loss of membrane integrity in a portion of CRAd-infected cells, PS exposure was the only apoptotic marker found in this study. In Table 1, the results are summarized whereby CRAd-induced phenomena are compared to apoptosis induced by FasL+CHX treatment and four forms of cell death. It can be concluded that the detected molecular and cellular features that characterize CRAd-induced cell death rule out apoptotic or necrotic cell death and that necrosis-like PCD fits CRAd(E3+)- and CRAd(E3−)-induced cell death best, although this is inevitably somewhat arbitrary.
TABLE 1.
Summary of apoptotic features found in H460 cells infected with AdΔ24-based CRAds and mode of cell death
| Cellular and molecular changes | Cell death
|
Induced cell death
|
||||
|---|---|---|---|---|---|---|
| Apoptosis | Apoptosis-like PCD | Necrosis-like PCD | Necrosis | CRAds-induced oncolysis | FasL+CHX | |
| Chromatin condensation | + | + | − | − | +a | + |
| DNA laddering | + | −/+ | − | − | − | + |
| Cytoplasmic shrinkage | + | −/+ | −/+ | − | − | + |
| PS exposure | + | + | + | − | + | + |
| Caspase activation | + | −/+ | −/+ | −/+ | − | + |
Chromatin condensation occurs but is distinct from “apoptotic condensation” (see text).
The found lack of involvement of the basic apoptotic machinery in mediating CRAd(E3+)- and CRAd(E3−)-induced cell death corresponds with previous data showing that wtAd type 5 (wtAd5)-induced cell kill was not associated with DNA fragmentation or the formation of apoptotic nuclei (35, 49) and also indicates that the Δ24 mutation in E1A does not alter these features. This is remarkable when considering the clear apoptotic or antiapoptotic activities of the various characterized individual adenoviral proteins (reviewed in references 6, 38, and 52). For example, E1A13S-induced apoptosis is associated with caspase-3 activation and PARP cleavage (4); Lavoie and coworkers (30) showed that E4orf4-induced p53-independent cell death was associated with loss of mitochondrial membrane potential and chromatin condensation but without caspase-3 activation or PARP cleavage. However, as we demonstrate here, when Ad proteins act in concert, the net outcome is the activation of a caspase-independent mode of cell death. This may be explained by assuming that Ad proteins that trigger caspase-independent and nonclassical apoptotic cell death dominate this process, such as the previously mentioned E4orf4 protein (7, 25). In the literature, cell death induced by replicating Ads is often referred to as apoptosis, which in light of this study is incorrect. It should be noted that different assays are used in these studies to determine apoptosis that are not suitable for making the distinction between apoptotic and nonapoptotic forms of cell death. More importantly, it should be realized that depending on the particular combination of replicating Ad vector and host cell line used, the outcome may vary. For example, the use of an Ad2-based mutant that lacks E1B-19KD (a Bcl2 homologue) expression was found to have an enhanced cytopathic effect in colon cancer HCT116 cells with a functional Bax gene when compared to non-Bax-expressing counterparts, which was interpreted as evidence for Bax contributing to Ad-induced apoptosis (34). This finding may be true for a virus with a deleted E1B-19KD gene in a Bax-positive background, but it is not relevant for a CRAd expressing the Bax-inhibiting E1B-19KD protein. Similarly, the concept that induction of cell death in Ad-infected cancer cells may enhance progeny virus release and spread has been shown for a particular combination of host cells and Ad. Mi and coworkers (39) found that in HeLa cells infected with Ad E1/E3-deleted viruses that overexpress IκB, which sensitizes cells for tumor necrosis factor α (TNF-α)-induced apoptosis, the administration of TNF-α after the virion assembly stage enhanced viral release.
For the generation of more effective CRAds, interfering with the early apoptosis-regulating properties of the Ad should be avoided when possible, to prevent adverse effects on optimal virus production. Correctly timed expression of proapoptotic genes is therefore essential and can be achieved (for example) by production from the E3 region, as reported for TNF-α (42). The expression of diffusible apoptosis-inducing proteins could also have therapeutic benefit by their ability to act at a distance from the infected cells, thereby circumventing possible barriers in the tumor mass that may hinder virus spread.
Our study indicates that deregulated apoptosis in cancer cells will likely not oppose effective CRAd-induced cell death. Their mechanism of tumor cell killing is therefore different from conventional therapies in which apoptosis activation contributes to cell death and may provide an explanation for the potency of CRAds in killing chemo-resistant tumor cells. It seems that during evolution, Ads evolved a mechanism for disrupting the host cell at the final stage of the viral cycle that does not require the activation of the basic apoptotic machinery. Currently, we are attempting to identify pathways that are involved in the activation of CRAd-induced cell death by employing microarray-based approaches. The elucidation of host cell mechanisms that affect CRAd-dependent oncolysis will help to identify CRAd-resistance mechanisms and allow the development of rationalized strategies to circumvent such hurdles.
Acknowledgments
We are grateful to D. T. Curiel (Birmingham, Ala.) for kindly providing Ad-Δ24RGD and W. El Deiry (Philadelphia, Pa.) for H460HPV16-E6 cells. We are also grateful to H. M. Pinedo for his continuous interest and support.
REFERENCES
- 1.Adams, J. M., and S. Cory. 2001. Life-or-death decisions by the Bcl-2 protein family. Trends Biochem. Sci. 26:61-66. [DOI] [PubMed] [Google Scholar]
- 2.Alemany, R., C. Balague, and D. T. Curiel. 2000. Replicative adenoviruses for cancer therapy. Nat. Biotechnol. 18:723-727. [DOI] [PubMed] [Google Scholar]
- 3.Bauerschmitz, G. J., J. T. Lam, A. Kanerva, K. Suzuki, D. M. Nettelbeck, I. Dmitriev, V. Krasnykh, G. V. Mikheeva, M. N. Barnes, R. D. Alvarez, P. Dall, R. Alemany, D. T. Curiel, and A. Hemminki. 2002. Treatment ovarian cancer with a tropism modified oncolytic adenovirus. Cancer Res. 62:1266-1270. [PubMed] [Google Scholar]
- 4.Boulakia, C. A., G. Chen, F. W. Ng, J. G. Teodoro, P. E. Branton, D. W. Nicholson, G. G. Poirier, and G. C. Shore. 1996. Bcl-2 and adenovirus E1B 19 kDA protein prevent E1A-induced processing of CPP32 and cleavage of poly(ADP-ribose) polymerase. Oncogene 12:529-535. [PubMed] [Google Scholar]
- 5.Boyd, J. M., S. Malstrom, T. Subramanian, L. K. Venkatesh, U. Schaeper, B. Elangovan, C. D'Sa-Eipper, and G. Chinnadurai. 1994. Adenovirus E1B 19 kDa and Bcl-2 proteins interact with a common set of cellular proteins. Cell 79:341-351. [DOI] [PubMed] [Google Scholar]
- 6.Braithwaite, A. W., and I. A. Russell. 2001. Induction of cell death by adenoviruses. Apoptosis 6:359-370. [DOI] [PubMed] [Google Scholar]
- 7.Branton, P. E., and D. E. Roopchand. 2001. The role of adenovirus E4orf4 protein in viral replication and cell killing. Oncogene 20:7855-7865. [DOI] [PubMed] [Google Scholar]
- 8.Dix, B. R., S. J. O'Carroll, C. J. Myers, S. J. Edwards, and A. W. Braithwaite. 2000. Efficient induction of cell death by adenoviruses requires binding of E1B55k and p53. Cancer Res. 60:2666-2672. [PubMed] [Google Scholar]
- 9.Earnshaw, W. C., L. M. Martins, and S. H. Kaufmann. 1999. Mammalian Caspases: structure, activation, substrates, and functions during apoptosis. Annu. Rev. Biochem. 68:383-424. [DOI] [PubMed] [Google Scholar]
- 10.Fadok, V. A., A. de Cathelineau, D. L. Daleke, P. M. Henson, and D. L. Bratton. 2001. Loss of phospholipid asymmetry and surface exposure of phosphatidylserine is required for phagocytosis of apoptotic cells by macrophages and fibroblasts. J. Biol. Chem. 276:1071-1077. [DOI] [PubMed] [Google Scholar]
- 11.Ferreira, C. G., S. W. Span, G. J. Peters, F. A. E. Kruyt, and G. Giaccone. 2000. Chemotherapy triggers apoptosis in a caspase-8-dependent and mitochondria-controlled manner in the NSCLC cell line NCI-H460. Cancer. Res. 60:7133-7141. [PubMed] [Google Scholar]
- 12.Ferrer, M., T. Izeboud, C. G. Ferreira, S. W. Span, G. Giaccone, and F. A. E. Kruyt. 2003. Cisplatin triggers apoptotic or nonapoptotic cell death in Fanconi anemia lymphoblasts in a concentration-dependent manner. Exp. Cell Res. 286:381-395. [DOI] [PubMed] [Google Scholar]
- 13.Fueyo, J., C. Gomez-Manzano, R. Alemany, P. S. Lee, T. J. McDonnell, P. Mitlianga, Y. X. Shi, V. A. Levin, W. K. Yung, and A. P. Kyritsis. 2000. A mutant oncolytic adenovirus targeting the Rb pathway produces anti-glioma effect in vivo. Oncogene 19:2-12. [DOI] [PubMed] [Google Scholar]
- 14.Ganly, I., D. Kirn, S. G. Eckhardt, G. I. Rodriguez, D. S. Soutar, R. Otto, A. G. Robertson, O. Park, M. L. Gulley, C. Heise, D. D. Von Hoff, and S. B. Kaye. 2000. A phase I study of Onyx-015, an E1B attenuated adenovirus, administered intratumorally to patients with recurrent head and neck cancer. Clin Cancer Res. 6:798-806. [PubMed] [Google Scholar]
- 15.Givol, I., I. Tsarfaty, J. Resau, S. Rulong, P. P. da Silva, G. Nasioulas, J. DuHadaway, S. H. Hughes, and D. L. Ewert. 1994. Bcl-2 expressed using a retroviral vector is localized primarily in the nuclear membrane and the endoplasmic reticulum of chicken embryo fibroblasts. Cell Growth Differ. 5:419-429. [PubMed] [Google Scholar]
- 16.Hall, A. R., B. R. Dix, S. J. O'Carroll, and A. W. Braithwaite. 1998. p53-dependent cell death/apoptosis is required for a productive adenovirus infection. Nat. Med. 4:1068-1072. [DOI] [PubMed] [Google Scholar]
- 17.Harada, J. N., and A. J. Berk. 1999. p53-Independent and -dependent requirements for E1B-55K in adenovirus type 5 replication. J. Virol. 73:5333-5344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harada, J. N., A. Shevchenko, A. Shevchenko, D. C. Pallas, and A. J. Berk. 2002. Analysis of the adenovirus E1B-55K-anchored proteome reveals its link to ubiquitination machinery. J. Virol. 76:9194-9206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haviv, Y. S., K. Takayama, J. N. Glasgow, J. L. Blackwell, M. Wang, X. Lei, and D. T. Curiel. 2002. A model system for the design of armed replicating adenoviruses using p53 as a candidate transgene. Mol. Cancer Ther. 1:321-328. [PubMed] [Google Scholar]
- 20.Hengartner, M. O. 2000. The biochemistry of apoptosis. Nature 407:770-776. [DOI] [PubMed] [Google Scholar]
- 21.Hoffmann, P. R., A. M. deCathelineau, C. A. Ogden, Y. Leverrier, D. L. Bratton, D. L. Daleke, A. J. Ridley, V. A. Fadok, and P. M. Henson. 2001. Phosphatidylserine (PS) induces PS receptor-mediated macropinocytosis and promotes clearance of apoptotic cells. J. Cell Biol. 155:649-660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kataoka, M., S. Wiehle, F. Spitz, G. Schumacher, J. A. Roth, and R. J. Cristiano. 2000. Down-regulation of bcl-2 is associated with p16INK4-mediated apoptosis in non-small cell lung cancer cells. Oncogene 19:1589-1595. [DOI] [PubMed] [Google Scholar]
- 23.Kirn, D. 2000. Replication-selective oncolytic adenoviruses: virotherapy aimed at genetic targets in cancer. Oncogene 19:6660-6669. [DOI] [PubMed] [Google Scholar]
- 24.Kirn, D. 2001. Clinical research results with dl1520 (Onyx-015), a replication-selective adenovirus for the treatment of cancer: what have we learned? Gene Ther. 8:89-98. [DOI] [PubMed] [Google Scholar]
- 25.Kleinberger, T. 2000. Induction of apoptosis by adenovirus E4orf4 protein. Apoptosis 5:211-215. [DOI] [PubMed] [Google Scholar]
- 26.Koch, P., J. Gatfield, C. Lober, U. Hobom, C. Lenz-Stoppler, J. Roth, and M. Dobbelstein. 2001. Efficient replication of adenovirus despite the overexpression of active and nondegradable p53. Cancer Res. 61:5941-5947. [PubMed] [Google Scholar]
- 27.Kroemer, G. 1997. The proto-oncogene Bcl-2 and its role in regulating apoptosis. Nat. Med. 3:614-620. [DOI] [PubMed] [Google Scholar]
- 28.Kruyt, F. A. E., and D. T. Curiel. 2002. Towards a new generation of conditionally replicating adenoviruses: pairing tumor selectivity with maximal oncolysis. Hum. Gene Ther. 13:485-495. [DOI] [PubMed] [Google Scholar]
- 29.Lamfers, M. L. M., J. Grill, C. M. F. Dirven, V. W. van Beusechem, B. Geoerger, J. van den Berg, R. Alemany, J. Fueyo, D. T. Curiel, G. Vassal, H. M. Pinedo, W. P. Vandertop, and W. R. Gerritsen. 2002. Potential of the conditionally replicative adenovirus Ad5-Δ24RGD in the treatment of malignant gliomas and its enhanced effect with radiotherapy. Cancer Res. 62:5736-5742. [PubMed] [Google Scholar]
- 30.Lavoie, J. N., M. Nguyen, R. C. Marcellus, P. E. Branton, and G. C. Shore. 1998. E4orf4, a novel adenovirus death factor that induces p53-independent apoptosis by a pathway that is not inhibited by zVAD-fmk. J. Cell Biol. 140:637-645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leist, M., and M. Jaattela. 2001. Four deaths and a funeral: from caspases to alternative mechanisms. Nat. Rev. Mol. Cell Biol. 2:1-10. [DOI] [PubMed] [Google Scholar]
- 32.Lichtenstein, D. L., K. Toth, K. Doronin, A. E. Tollefson, and W. S. Wold. 2004. Functions and mechanisms of action of the adenovirus E3 proteins. Int. Rev. Immunol. 23:75-111. [DOI] [PubMed] [Google Scholar]
- 33.Lichtenstein, D. L., P. Krajcsi, D. J. Esteban, A. E. Tollefson, and W. S. M. Wold. 2002. Adenovirus RIDβ subunit contains a tyrosine residue that is critical for RID-mediated receptor internalization and inhibition of Fas- and TRAIL-induced apoptosis. J. Virol. 76:11329-11342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lomonosova, E., T. Subramanian, and G. Chinnadurai. 2002. Requirement of BAX for efficient adenovirus-induced apoptosis. J. Virol. 76:11283-11290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marcellus, R., J. Teodoro, T. Wu, D. Brough, G. Ketner, G. Shore, and P. Branton. 1996. Adenovirus type 5 early region 4 is responsible for E1A-induced p53- independent apoptosis. J. Virol. 70:6207-6215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martin, M. E., and A. J. Berk. 1998. Adenovirus E1B 55K represses p53 activation in vitro. J. Virol. 72:3146-3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McCormick, F. 2001. Cancer gene therapy: fringe or cutting edge? Nat. Rev. Cancer 1:130-141. [DOI] [PubMed] [Google Scholar]
- 38.McNees, A. L., and L. R. Gooding. 2002. Adenoviral inhibitors of apoptotic cell death. Virus Res. 88:87-101. [DOI] [PubMed] [Google Scholar]
- 39.Mi, J., Z. Y. Li, S. Ni, D. Steinwaerder, and A. Lieber. 2001. Induced apoptosis supports spread of adenovirus vectors in tumors. Hum. Gene Ther. 12:1343-1352. [DOI] [PubMed] [Google Scholar]
- 40.Nemunaitis, J., F. Khuri, I. Ganly, J. Arseneau, M. Posner, E. Vokes, J. Kuhn, J. McCarty, S. Landers, A. Blackburn, L. Romel, B. Randlev, S. Haye, and D. Kirn. 2001. Phase I trial of intratumoral administration of ONYX-015, a replication selective adenovirus, in patients with refractory head and neck cancer. J. Clin. Oncol. 19:289-298. [DOI] [PubMed] [Google Scholar]
- 41.Reed, J. C. 1998. Bcl-2 family proteins. Oncogene 17:3225-3236. [DOI] [PubMed] [Google Scholar]
- 42.Ring, C. J. A. 2002. Cytolytic viruses as potential anti-cancer agents. J. Gen. Virol. 83:491-502. [DOI] [PubMed] [Google Scholar]
- 43.Rothmann, T., A. Hengstermann, N. J. Whitaker, M. Scheffner, and H. zur Hausen. 1998. Replication of ONYX-015, a potential anticancer adenovirus, is independent of p53 status in tumor cells. J. Virol. 72:9470-9478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sauthoff, H., T. Pipiya, S. Heitner, S. Chen, R. G. Norman, W. N. Rom, and J. G. Hay. 2002. Late expression of p53 from a replicating adenovirus improves tumor cell killing and is more tumor cell specific than expression of the adenoviral death protein. Hum. Gene Ther. 13:1859-1871. [DOI] [PubMed] [Google Scholar]
- 45.Sun, X.-M., M. MacFarlane, J. Zhuang, B. B. Wolf, D. R. Green, and G. M. Cohen. 1999. Distinct caspase cascades are initiated in receptor-mediated and chemical-induced apoptosis. J. Biol. Chem. 274:5053-5060. [DOI] [PubMed] [Google Scholar]
- 46.Suzuki, K., R. Alemany, M. Yamamoto, and D. T. Curiel. 2002. The presence of the adenovirus E3 region improves the oncolytic potency of conditionally replicative adenoviruses. Clin. Cancer Res. 8:3348-3359. [PubMed] [Google Scholar]
- 47.Suzuki, K., J. Fueyo, V. Krasnykh, P. N. Reynolds, D. T. Curiel, and R. Alemany. 2001. A conditionally replicative adenovirus with enhanced infectivity shows improved oncolytic potency. Clin. Cancer Res. 7:120-126. [PubMed] [Google Scholar]
- 48.Thornberry, N. A., and Y. Lazebnik. 1998. Caspases: enemies within. Science 281:1312-1316. [DOI] [PubMed] [Google Scholar]
- 49.Tollefson, A. E., T. W. Hermiston, D. L. Lichtenstein, C. F. Colle, R. A. Tripp, T. Dimitrov, K. Toth, C. E. Wells, P. C. Doherty, and W. S. Wold. 1998. Forced degradation of Fas inhibits apoptosis in adenovirus-infected cells. Nature 392:726-730. [DOI] [PubMed] [Google Scholar]
- 50.Tollefson, A. E., A. Scaria, T. W. Hermiston, J. S. Ryerse, L. J. Wold, and W. S. Wold. 1996. The adenovirus death protein (E3-11.6K) is required at very late stages of infection for efficient cell lysis and release of adenovirus from infected cells. J. Virol. 70:2296-2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.van Beusechem, V. W., P. B. van den Doel, J. Grill, H. M. Pinedo, and W. R. Gerritsen. 2002. Conditionally replicative adenovirus expressing p53 exhibits enhanced oncolytic potency. Cancer Res. 62:6165-6171. [PubMed] [Google Scholar]
- 52.Wold, W. S., K. Doronin, K. Toth, M. Kuppuswamy, D. L. Lichtenstein, and A. E. Tollefson. 1999. Immune responses to adenoviruses: viral evasion mechanisms and their implications for the clinic. Curr. Opin. Immunol. 11:380-386. [DOI] [PubMed] [Google Scholar]
- 53.Wu, G. S., and W. S. El-Deiry. 1996. Apoptotic death of tumor cells correlates with chemosensitivity, independent of p53 or bcl-2. Clin. Cancer Res. 2:623-633. [PubMed] [Google Scholar]
- 54.Yang, Y. L., and X. M. Li. 2000. The IAP family: endogenous caspase inhibitors with multiple biological activities. Cell Res. 10:169-177. [DOI] [PubMed] [Google Scholar]