

Abstract
The pathway of entry of polyomavirus (Py) has been investigated with glycolipid-deficient C6 cells and added ganglioside GD1a as a specific virus receptor. Unsupplemented C6 cells show a low basal level of infection but become highly infectable by Py following preincubation with the sialic acid-containing ganglioside GD1a (38). Addition of GD1a has no effect on the overall level of virus binding but mediates the internalization and transit of virus to the endoplasmic reticulum (ER). This pathway of entry is cholesterol and caveola dependent and requires intact microtubules as well as a dynamic state of the microfilament system. In contrast to vesicular transport of other cargo via glycolipids, Py particles do not appear to pass through the Golgi apparatus. Colcemid and brefeldin A block transport of the virus to the ER in GD1a-supplemented cells and lead to accumulation of virus in a caveolin-1-containing environment. Several features distinguish the efficient GD1a-mediated pathway of virus uptake from the less-efficient pathway of basal infection in C6 cells.
Murine polyomavirus (Py) is a small nonenveloped DNA virus that induces tumors in a wide variety of tissues in the mouse and efficiently transforms cells in culture. The Py virion is composed of 360 copies of the major capsid protein VP1, which assemble into 72 pentamers to form the outer shell of the virus and bind to cell surface sialic acid (8), and 72 copies of the minor proteins VP2-VP3, which form an inner scaffold (2). The ability of the virus to bind to oligosaccharides with terminal α-2,3-linked sialic acid enables the virus to infect multiple cell types in its natural host (3, 6, 7). The crystal structure of VP1 complexed with an oligosaccharide receptor fragment indicates that changes in the contact residues within the binding pocket profoundly affect the ability of sialic acid variants to bind (36, 37). This discrimination between different sialic acid linkages determines the ability of different Py strains to infect cells (14) and to spread in the animal host (3).
The mechanism by which Py is internalized is not fully understood. Early electron microscopic studies indicated that virus particles are taken up into small, uncoated vesicles presumably destined for the endoplasmic reticulum (ER) (13, 18-20, 27, 28). The exact nature of these vesicles has proved to be elusive, however. Previous work indicated that Py is endocytosed by nonclathrin, non-caveola-derived vesicles in a dynamin-independent manner (10, 11), whereas work from other labs implicated a caveola-based vesicle uptake pathway (19, 27). Given the ubiquity of α-2,3-linked sialic acid residues on the cell surface and the broad range of cell types the virus can infect, the possibility exists that Py may attach to multiple receptors and be taken up through more than one endocytic pathway. The effectiveness of different pathways could vary among cell types, depending on the efficiency of delivery to a compartment, presumably the ER, where Py can penetrate the host membrane regardless of the exact pathway of delivery.
Recent efforts in a number of laboratories have focused on the identification of specific molecules bearing sialic acid as Py receptors and on the subsequent pathway of virus internalization and entry in the nucleus. Efforts to identify a specific Py receptor by screening for protective monoclonal antibodies were unsuccessful (3). However, α4β1 integrin, which carries sialic acid moieties, has been reported to function at the postattachment level as a possible secondary receptor (4, 5). C6, a rat glioma cell line that is deficient in the formation of complex gangliosides (34) is poorly infectable by both Py and simian virus 40 (SV40). These cells can be rendered infectable by preincubation with specific gangliosides, GD1a for Py and GM1 for SV40 (38). The present study focuses on the C6 cell system as a model for entry of the whole virus mediated by ganglioside GD1a as a specific receptor.
MATERIALS AND METHODS
Cells and viruses.
C6 cells (rat glioma cells) that are deficient in complex ganglioside production were a gift of B. Tsai. Cells were maintained in F-12K medium (American Type Culture Collection, Manassas, Va.), 10% heat-inactivated fetal calf serum (Invitrogen; Carlsbad, Calif.), 2.5% heat-inactivated horse serum (Invitrogen), 100 IU of penicillin per ml, and 100 IU of streptomycin (Invitrogen) per ml in a 5% CO2, humidified incubator at 37°C. Prior to infectivity studies, cells were supplemented or not with 30 μm GD1a or GM1 (conditions designated C6+GD1a or C6+GM1) (Matreya, Inc., State College, Pa.), as indicated.
The Py RA strain (small plaque strain) used in this study was propagated on baby mouse kidney cells. For infectivity studies, a crude virus lysate (titered by plaque assay) prepared by freezing-thawing and centrifugation of cellular debris was used. CsCl-purified Py was labeled as described previously (10) with the FluoReporter Texas red protein labeling kit (Molecular Probes, Eugene, Oreg.) and designated TRPy. By plaque assay titration and optical density measurements, the infectious particle-to-physical particle ratio of TRPy was determined to be ∼1 to 12.5.
Antibodies and reagents.
4,6′-Diamidino-2-phenylindole (DAPI), paclitaxel (Taxol), methyl-β-cyclodextrin (MBCD), and the monoclonal antibodies to β-actin (clone AC-74), α-tubulin (clone B-5-1-2) and the Golgi 58-kDa protein (clone 58K-9) were purchased from Sigma Chemical Company (St. Louis, Mo.). Brefeldin A (BFA), Colcemid, jasplakinolide, latrunculin A, α-2,3,6,8 neuraminidase, and nystatin were purchased from Calbiochem (San Diego, Calif.). The rabbit polyclonal antibody to caveolin-1 (cav-1) and the mouse monoclonal antibodies to GM130 (clone 35) and GS28 (clone 1) were purchased from Transduction Laboratories (Lexington, Ky.). The polyclonal antibody to β-COPII was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, Calif.). The rabbit polyclonal antibodies to β-COPI and to GRP78 (BiP) were purchased from Affinity BioReagents (Golden, Colo.). Rat antibody to Py large T antigen (Py LTAg) was generated within the laboratory. Oregon green-conjugated goat anti-rabbit, anti-mouse, or anti-rat immunoglobulin G was purchased from Molecular Probes. Streptavidin-horseradish peroxidase was purchased from Pierce Chemical Company (Rockford, Ill.).
Transfection.
The RA strain genome was excised from pBluescriptKS+ (Stratagene, La Jolla, Calif.), gel purified, and transfected into C6 or C6+GD1a cells with Lipofectamine Plus (Invitrogen). Cells were cotransfected with pEGFPN1 (Clontech, Palo Alto, Calif.) as a marker for transfection-positive cells.
Indirect immunofluorescence assay (IFA).
After the time of desired incubation, cells were fixed in 4% paraformaldehyde (Electron Microscopy Sciences, Ft. Washington, Pa.). Samples were permeabilized by treatment with either 0.1% Triton X-100 in phosphate-buffered saline containing 1% calf serum (Atlanta Biologicals, Norcross, Ga.) for examination of all antigens except LTAg or β-actin, which were permeabilized by incubation in ethanol/acetic acid (2:1) or methanol, respectively. Samples were incubated with primary antibody for 1 h at room temperature. Samples then were incubated with Oregon green-labeled secondary antibodies and DAPI and incubated for 1 h at room temperature. The washed coverslips were mounted with Mowiol, sealed with nail polish, and examined by standard fluorescence microscopy with a Nikon Eclipse TE300 microscope with an apochromatic Plan 60X/1.4 oil objective (magnification, 60×). For deconvolution microscopy, six random fields were selected per time point, and data were collected with a Nikon Eclipse TE200 microscope with a an apochromatic Plan 60X/1.4 oil objective equipped with a DeltaVision optical sectioning system employing SoftWoRx software (Applied Precision, Inc., Issaquah, Wash.) with 0.2-μm-diameter step Z-sections. Deconvolved Z sections were examined for colocalization of TRPy and epitopes of interest with the SoftWoRx program. The selected images were saved as TIFFs and then imported and prepared in Adobe Photoshop 6.0. Enlargements were ×50.
Binding.
C6, C6+GM1, or C6+GD1a cells were treated or not for 1 h at 37°C with neuraminidase, which removes both protein and lipid-linked sialic acid residues. Washed cells were chilled on ice and then incubated with ∼2 μg of biotinylated Py for 1 h at 4°C. Samples were lysed in either 0.1% Triton X-100 (Sigma) in phosphate-buffered saline, and the debris was cleared and processed for sodium dodecyl sulfate-polyacrylamide gel electrophoresis and detection of biotinylated proteins as described by Gilbert et al. (12).
Infectivity assay.
For analysis of Py infectivity, cells were plated on 12-mm glass coverslips; 3 h after plating, cells were supplemented or not with gangliosides and grown to approximately 80% confluency at 37°C in a CO2 incubator for a minimum of 27 h. Cells were washed extensively prior to infection with virus to remove any unincorporated gangliosides. Cells were pretreated (or not, as indicated) with various compounds, as described in the text. The virus was diluted in F-12K medium containing the additional compounds, as indicated, and infection was carried out at a multiplicity of infection (MOI) of approximately 500 PFU/cell. Cells and virus were incubated from 1 h at 37°C in a CO2 incubator, and then the virus was removed by aspiration. The virus was allowed to replicate for 32 h at 37°C. Successful entry was assessed by nuclear expression of Py LTAg by IFA, as described above. Data are presented as the percentages of nuclei that were LTAg positive in the treated sample relative to the percentages of nuclei that were LTAg positive in the untreated control, normalized to 100%. Unsupplemented C6 cells showed an actual infection rate of roughly 10% LTAg-positive cells under these conditions. Values are presented as the averages of triplicate samples where approximately 500 nuclei were counted per sample.
Uptake assay.
To assess uptake of labeled Py into cells, TRPy was added to cells on ice after any indicated pretreatments. The virus was allowed to bind for 60 min at 4°C. Unbound virus was removed by aspiration, and cells were washed and then incubated at 37°C in a CO2 incubator. Samples were then fixed in 4% paraformaldehyde and processed for IFA as described above, at the indicated times. Time points were in duplicate, and experiments were performed twice with similar results.
RESULTS
Preincubation of C6 cells with GD1a enhances infectability at an early postadsorption step.
Addition of the sialic acid-containing ganglioside GD1a prior to infection increases infectability of C6 cells, as judged by nuclear expression of the viral LTAg (38). This effect of GD1a is specific for cells infected by virus as opposed to transfection with viral DNA. Thus, only 9% of untreated C6 cells become LTAg positive after infection at a high MOI. In contrast, C6 cells that were supplemented with GD1a prior to infection showed a five- to sixfold enhancement in infectability, regardless of MOI. This increase was not seen in C6 cells transfected with Py DNA, which showed equivalent numbers of LTAg-positive cells with or without preincubation with GD1a (Table 1).
TABLE 1.
Effect of GD1a on infection versus transfection of C6 cells
| Treatment | No. (%) of Py LT Ag-positive cellsc
|
|
|---|---|---|
| −GD1a | +GD1a | |
| Infectiona | 100 (9) | 542 (49) |
| Transfectionb | 100 (80) | 105 (84) |
For infected cells, the values indicate the percentages of cells that were Py LTAg positive out of the total number of cells counted.
For transfected cells, the values indicate the percentages of cells that were Py LTAg positive out of the total number of transfected (green fluorescent protein-positive) cells.
All values are normalized to C6 cells without GD1a control as 100%.
The effect of GD1a on viral infection as opposed to DNA transfection suggests that the added glycolipid may provide sites for virus adsorption. A direct binding experiment with biotinylated virus was carried out to test this possibility. Addition of GD1a had no discernible effect on the overall level of virus binding. C6 cells bound the same amount of virus with or without supplementation with GD1a or the related ganglioside GM1 (Fig. 1). Presumably, sialoglycoproteins on the cell surface provide abundant binding sites for the virus. Treatment with neuraminidase decreased the amount of Py binding to the same degree, independently of ganglioside addition. This indicates that the enhancement of infectivity by GD1a is not due simply to an increase in overall cell surface binding but also to an effect on uptake of virus from the surface and penetration of the cell.
FIG. 1.
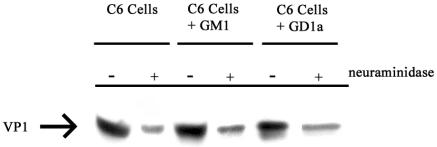
Binding of Py to C6 Cells. C6, C6+GM1, or C6+GD1a cells were treated with neuraminidase or not as indicated. Cells were then allowed to bind biotinylated Py for 1 h at 4°C. Samples were washed and processed for detection of biotinylated proteins with streptavidin-horseradish peroxidase.
Uptake of Py via GD1a is cholesterol and caveola dependent.
Gangliosides can be found associated with lipid rafts on the cell surface, and these cholesterol-rich microdomains are thought to play a role in the uptake and trafficking of lipids within the cell (9, 17, 30, 32). To determine whether GD1a-facilitated infection by Py is cholesterol dependent, the effect of cholesterol depletion and sequestration of drugs on Py infectivity was examined. Incubation of C6 cells with MBCD or nystatin beginning 1 h prior to infection had little or no effect on the low basal level of infectivity in unsupplemented cells (Table 2). In contrast, the enhancement of infectivity in GD1a-supplemented C6 cells was totally reversed by treatment with the drugs. These results indicate that GD1a-facilitated virus entry requires cholesterol and occurs via lipid rafts or caveolae.
TABLE 2.
Effect of cholesterol-blocking drugs on virus infection
| Treatment | % Py LTAg-positive cellsa
|
|
|---|---|---|
| −GD1a | +GD1a | |
| None | 100 ± 8 | 468 ± 20 |
| MBCD | 104 ± 9 | 85 ± 3 |
| Nystatin | 92 ± 6 | 78 ± 8 |
All values are normalized to C6 cells without GD1a control as 100%.
Fluorescence microscopy was used to determine if caveolae are important for GD1a-dependent uptake. TRPy was used to infect C6 cells with or without GD1a supplementation. Cells were fixed at various times postinfection and examined for colocalization with cav-1 by indirect immunofluorescence (IFA) and TRPy. By fluorescence microscopy and deconvolution analysis, cells that had been preincubated with GD1a, but not untreated control cultures, showed colocalization with cav-1 (Fig. 2A). The kinetics of association between TRPy and cav-1 indicated an increase as early as 15 min postinfection, peaking at 30 min postinfection and then tapering off after 2 h (Fig. 2B). These kinetics correlate well with the uptake of the virus from the cell surface as defined by the escape of virus from antibody neutralization (data not shown). The involvement of cholesterol and colocalization with cav-1 clearly indicate a role for caveolae in the uptake of virus from the cell surface in GD1a-supplemented C6 cells.
FIG. 2.
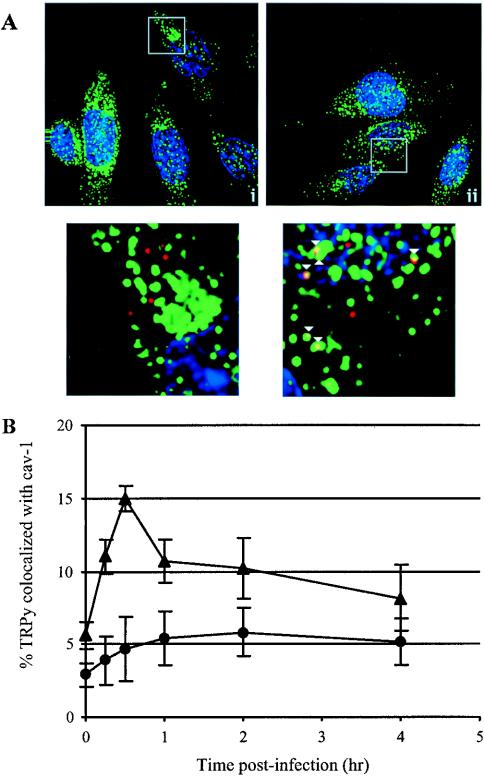
Colocalization of TRPy and cav-1. (A) Immunofluorescence. C6 cells (i) or C6+GD1a cells (ii) were infected with TRPy (in red). Cells were fixed, processed for immunodetection of cav-1 (in green), imaged, and subjected to deconvolution. Z sections were examined, and representative ones showing cells that were incubated for 0.5 h at 37°C prior to fixation are presented (magnification, ×60). Enlargements are shown below (enlargement, ×50). Colocalized particles are indicated in the enlargements by white arrowheads. (B) Quantitation and time course of colocalization. C6 cells (circles) or C6+GD1a cells (triangles) were infected with TRPy. Cells were fixed at the indicated times, processed for immunodetection of cav-1, imaged, and subjected to deconvolution. Z sections (0.2 μm thick) were examined for total number of virus particles and the number of particles colocalized with cav-1.
Participation of actin filaments in the endocytosis of caveolar vesicles has been suggested (24, 26). To investigate whether actin is involved in GD1a-mediated infection, GD1a-supplemented C6 cells were treated with either the g-actin sequestering compound latrunculin A or the f-actin stabilizing agent jasplakinolide. Infection was reduced two- to threefold in these treated cells (Table 3). Actin and TRPy were colocalized in GD1a-supplemented cells (Fig. 3A) but not in unsupplemented cells (not shown). The kinetics of virus colocalization with actin filaments in GD1a-treated C6 cells were roughly the same as found for cav-1 (Fig. 3B). The uptake of Py into GD1a-supplemented C6 cells thus appears to utilize caveolae and is dependent upon actin and a dynamic state of microfilaments, similar to what has been described for the uptake of SV40 into CV-1 cells (26).
TABLE 3.
Effect of cytoskeletal binding drugs on infection of GD1a-supplemented C6 cells
| Treatment | % Py LTAg-positive cells |
|---|---|
| +GD1aa | |
| None | 100 ± 5 |
| Latrunculin A | 47 ± 4 |
| Jasplakinolide | 36 ± 7 |
| Colcemid | 12 ± 6 |
| Taxol | 107 ± 10 |
All values are normalized to C6 cells without GD1a control as 100%.
FIG. 3.
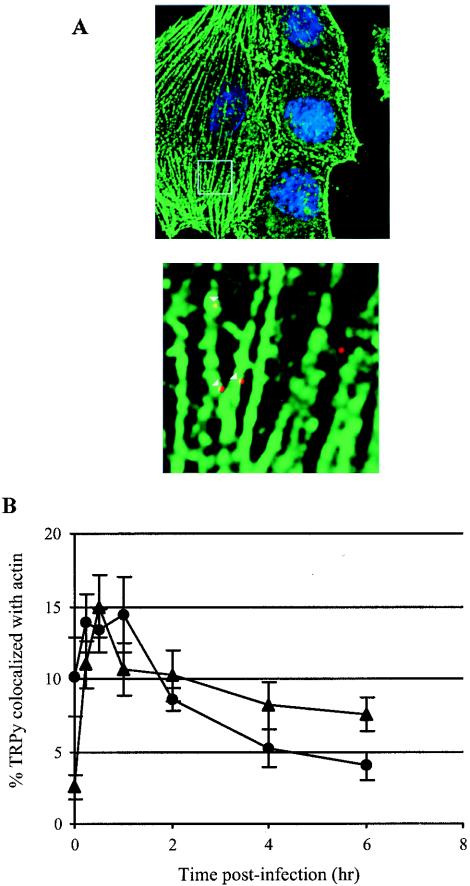
Colocalization of TRPy and actin. (A) Immunofluorescence. C6+GD1a cells were infected with TRPy (in red). Cells were fixed, processed for immunodetection of β-actin (in green), imaged, and subjected to deconvolution. Z sections were examined, and representative ones showing cells that were incubated for 0.5 h at 37°C prior to fixation are presented. Enlargement is shown below. Colocalized particles are indicated in the enlargement by white arrowheads. (B) Quantitation and time course. C6+GD1a cells were infected with TRPy. Cells were fixed at the indicated times, processed for immunodetection of β-actin (circles) or cav-1 (triangles), imaged, and subjected to deconvolution. Z sections were examined for the total number of viral particles and the number of particles colocalized with either actin or cav-1.
Lack of evidence for trafficking of Py via the Golgi in GD1a-treated cells.
Endocytosis and trafficking of most nonclathrin-coated vesicles are destined for, or pass through, the Golgi complex, though exceptions have been described (17, 21, 25, 30). Fluorescence microscopy was used to determine whether Py particles colocalized with any Golgi associated protein markers in GD1a-supplemented C6 cells. TRPy was not seen adjacent to β-COPI, β-COPII, or GM130 (Fig. 4A) at any of the time points examined (Fig. 4B). Two other Golgi markers were also examined (the 58-kDa Golgi protein and GS28) with no evidence of colocalization with virus (data not shown). Although it is thought that caveola-derived vesicles traffic through the Golgi complex and travel in a retrograde pathway to the ER, the apparent lack of virus association with Golgi may indicate that a certain subset of cav-1 vesicles are diverted into a non-Golgi-destined pathway (17, 22, 24). Similar findings have been reported for SV40 uptake into CV-1 cells (25).
FIG. 4.
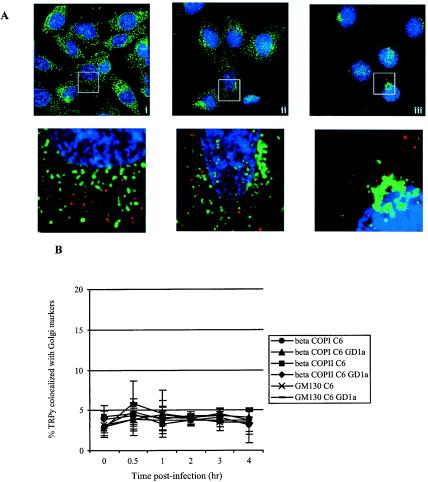
Absence of colocalization of TRPy and Golgi markers. (A) Immunofluorescence. C6+GD1a cells were infected with TRPy (in red). Cells were fixed; processed for immunodetection of β-COPI (i), β-COPII (ii), or GM130 (iii) (in green); imaged; and subjected to deconvolution. Z sections were examined, and representative ones showing cells that were incubated for 1 h at 37°C prior to fixation are presented. Enlargements are shown below. (B) Quantitation and time course. C6 cells or C6+GD1a cells were infected with TRPy. Cells were fixed at the indicated times, processed for immunodetection of β-COPI (circles and triangles for C6 and C6+GD1a cells, respectively), β-COPII (squares and diamonds for C6 and C6+GD1a cells, respectively), GM130 (x's and dashes for C6 and C6+GD1a cells, respectively), imaged, and subjected to deconvolution. Z sections were examined for total number of viral particles and the number of particles colocalized with each Golgi marker.
Interestingly, the GD1a-mediated uptake pathway of Py in C6 cells is sensitive to BFA (Fig. 5). BFA, an inhibitor of the Arf1 guanine nucleotide exchange factor (GEF), is required for β-COPI association with Golgi membranes and inhibits β-COPI-dependent transport between the Golgi and the ER. Pretreatment of GD1a-supplemented C6 cells with BFA and infection in the continuous presence of BFA strongly inhibits the ability of Py to infect. Addition of BFA to cells at various times postinfection indicates that GD1a-mediated infection becomes largely insensitive to BFA between 2 and 4 h postinfection (Fig. 5). The acquisition of BFA resistance occurs after cav-1 colocalization decreases. The fact that C6 cells remain infectable at these later times indicates that BFA treatment is not cytotoxic under the conditions used.
FIG. 5.
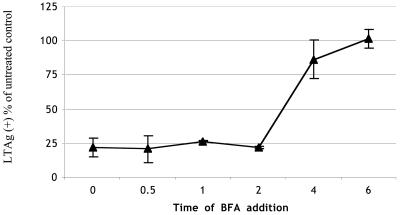
Effect of BFA on Py infection. C6+GD1a cells were infected with Py, and BFA was added at the indicated times postinfection and left in until cells were processed for Py LTAg staining. Infected cells were analyzed for Py LTAg as described in the legend to Table 1.
Trafficking of Py to the ER via GD1a requires microtubules.
Many gangliosides function as receptors for toxins that must be transported from the plasma membrane to the ER, possibly through the Golgi, for intoxication to occur (9, 29, 39). Work on Py and other polyomaviruses has indicated that these viruses are also transported to the ER (15, 18, 19, 23, 25, 27, 38; our unpublished data). TRPy particles taken up into GD1a-pretreated but not untreated C6 cells were found in the ER colocalizing with BiP (GRP78) as a marker (Fig. 6A) (38). Virus particles reach the ER and are seen adjacent to BiP as early as 1 to 2 h postinfection. Colocalization with the ER marker reaches a maximum at around 4 h and decreases thereafter (Fig. 6B). The kinetics of Py trafficking via GD1a to the ER in these cells are comparable to what has been reported for the transport of Py to the ER in previous studies with other cells (19, 27).
FIG. 6.
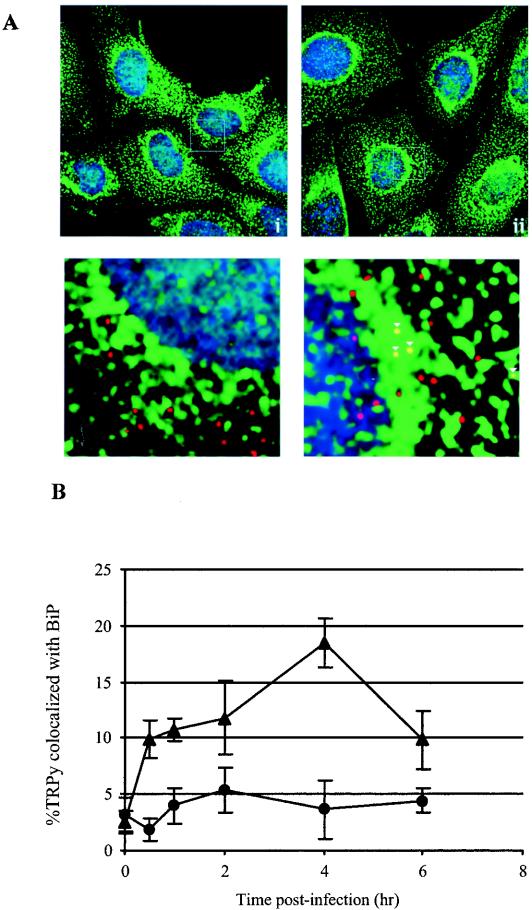
Colocalization of TRPy and BiP. (A) Immunofluorescence. C6 cells (i) or C6+GD1a cells (ii) were infected with TRPy (in red). Cells were fixed, processed for immunodetection of BiP (in green), imaged and subjected to deconvolution. Z sections were examined, and representative sections showing cells that were incubated for 4 h at 37°C prior to fixation are presented. Enlargements are shown below. Colocalized particles are indicated in the enlargements by white arrowheads. (B) Quantitation and time course. C6 cells (circles) or C6+GD1a cells (triangles) were infected with TRPy. Cells were fixed at the indicated times, processed for immunodetection of BiP, imaged, and subjected to deconvolution. Z sections were examined for the total number of viral particles and the number of particles colocalized with BiP.
A requirement for microtubules in the routing of Py to the ER has been indicated in several studies (11, 16, 25, 27, 31). The requirement for microtubules in GD1a-mediated uptake in C6 cells was examined by treating GD1a-supplemented C6 cells with either Colcemid to disrupt intact microtubules or with Taxol to stabilize microtubules. Cells were infected with Py, and the extent of infection in treated cells was compared to that of control untreated cells. Similar to what was seen with native host NIH 3T3 and BMK cells (11), microtubule disruption but not stabilization strongly inhibited Py infection (Table 3). Microscopic examination of TRPy and microtubules by IFA (Fig. 7A) indicated that the interaction of virus particles and microtubules occurred between 1 and 4 h, peaking at 2 h postinfection (Fig. 7B). The interaction between TRPy and microtubules appears to occur just prior to TRPy particles arriving in the ER (Fig. 7B), suggesting that virus particles still in vesicles are transported along microtubules to the ER.
FIG. 7.
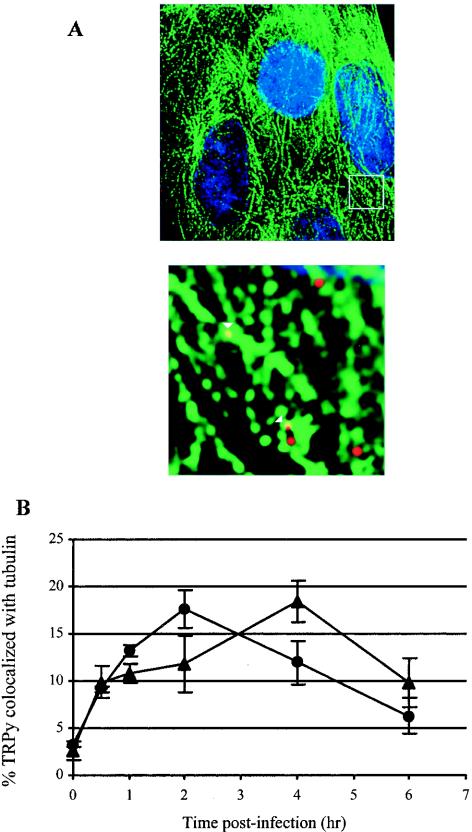
Colocalization of TRPy and microtubules. (A) Immunofluorescence. C6+GD1a cells were infected with TRPy (in red). Cells were fixed, processed for immunodetection of α-tubulin (in green), imaged, and subjected to deconvolution. Z sections were examined, and representative sections showing cells that were incubated for 2 h at 37°C prior to fixation are presented. Enlargement is shown below. Colocalized particles are indicated in the enlargement by white arrowheads. (B) Quantitation and time course. C6+GD1a cells were infected with TRPy. Cells were fixed at the indicated times, processed for immunodetection of either α-tubulin (circles) or BiP (triangles), imaged, and subjected to deconvolution. Z sections were examined for total number of viral particles and the number of particles colocalized with either tubulin or BiP.
Disruption of GD1a-mediated Py infection by Colcemid or brefeldin A strands virus in a cav-1-containing compartment.
It appears that transport of Py to the ER with GD1a as the virus receptor occurs via caveolae and depends on intact microtubules as well as on some as-yet-undefined vesicular transport step that is inhibitable by BFA. To further test whether microtubules and a BFA-sensitive GEF are important for transport of Py to the ER, GD1a-supplemented C6 cells were treated with Colcemid or BFA, and uptake of TRPy was compared with untreated C6 cells supplemented with GD1a as a control. Localization of the particles by fluorescence microscopy was determined at various times postinfection in relation to BiP. No TRPy particles were seen to colocalize with BiP during the 6-h period examined either when the microtubules were disrupted or when BFA-sensitive trafficking was impeded (Fig. 8A).
FIG. 8.
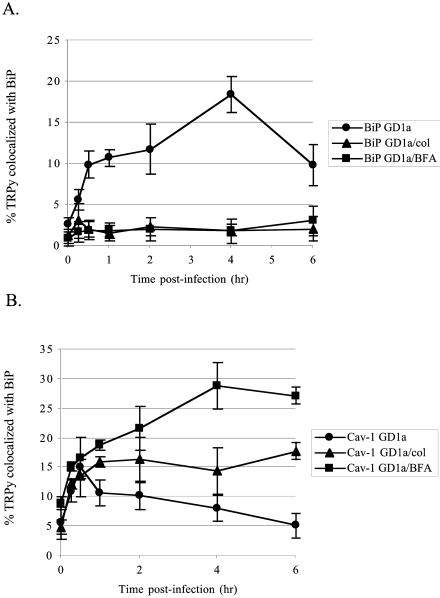
Quantitation of colocalization of TRPy with BiP and cav-1 in Colcemid- and BFA-disrupted cells. (A) Colocalization with BiP. C6+GD1a cells were either pretreated or not (circles) with either Colcemid (triangles) or BFA (squares) for 1 h at 37°C and then infected with TRPy. Cells were fixed, processed for immunodetection of BiP, imaged, and subjected to deconvolution. Z sections were examined for the total number of viral particles and the number of particles colocalized with BiP. (B) Colocalization with Cav-1. C6+GD1a cells were either pretreated or not (circles) with either Colcemid (triangles) or BFA (squares) for 1 h at 37°C and then infected with TRPy. Cells were fixed, processed for immunodetection of cav-1 imaged and subjected to deconvolution. Z sections were examined for total number of viral particles and the number of particles colocalized with cav-1.
Neither Colcemid nor BFA was able to block the association of virus with cav-1 in GD1a-supplemented cells (Fig. 8B). Virus colocalization with cav-1 increased at early times in a manner unaffected by either drug. However, beginning at about 30 min, the association with cav-1 decreased in the untreated cells but continued to increase and remained high throughout the 6-h period in the continued presence of Colcemid or BFA. Attempts to determine if TRPy could be colocalized with Golgi markers (β-COPI or GM130) in BFA- or Colcemid-treated C6 cells were negative. These data implicate a cav-1-containing intracellular location that TRPy reaches prior to traveling to the ER. Escape from this compartment depends upon both intact microtubules and GEFs. This intracellular location has similarities to the caveosome described by Pelkmans et al. (25).
DISCUSSION
Py may utilize different receptors and follow different pathways of internalization, depending in part on the target host cell. In some primary and established murine cells, Py enters in a noncaveolar, nonclathrin-dependent manner (10, 11). Other studies, using different cells, showed that Py infection was dependent on functional caveolae (27). Py was able to infect certain cells under conditions of cholesterol depletion in which infection of the same cells by SV40, known to depend on caveolae for uptake (1, 23, 25, 35), was blocked (11).
Apart from bearing sialic acid as an essential component, the nature and identity of cell receptors for Py remain to be clarified. Tests for binding of Py to a variety of gangliosides bearing sialic acid showed that GD1a and GT1b are able to serve as receptors (38). Structural modeling indicated that it is the sialic acid in α-2,3 linkage to the galactose moiety on the longer branches of GD1a and GT1b that makes appropriate contacts with residues in the binding pocket of Py VP1, while the shorter branch bearing α-2,3 sialic acid in GM1 is unable to bind because of interference with ceramide (36-38). In contrast, Py-like particles made from bacterially expressed VP1 appear to be able to utilize GM1 in different cells for uptake (33). Studies of the C6 rat glioma cells deficient in the expression of complex gangliosides showed that the efficiency of infection by Py is facilitated by addition of the ganglioside GD1a (Fig. 1) (38). Addition of GD1a to C6 cells prior to exposure to virus leads to a five- to sixfold increase in the number of cells infected. The increase occurs in the absence of a discernible effect on the overall level of virus binding to the cells. These findings indicate that, in addition to presenting an appropriate binding site for the virus, the ganglioside provides an efficient pathway of internalization leading to infection.
The addition of ganglioside GM1 to C6 cells renders them susceptible to SV40 (38), presumably enabling a pathway similar to that of Py in GD1a-supplemented C6 cells and comparable to the pathway in monkey cells that is cholesterol and caveola dependent (23, 25). GD1a-Py complexes are routed via caveolae from the plasma membrane to the ER, apparently bypassing the Golgi. While colocalization of Py with caveolea and ER markers was apparent, no colocalization of virus with the Golgi was detected with a variety of markers. When trafficking to the ER was disrupted by either Colcemid or BFA, Py particles were seen to accumulate and become trapped in an intracellular location that contains cav-1- but not BiP- or Golgi-related markers. These observations parallel those of SV40, which also appears to bypass the Golgi while passing through an intermediate vesicular compartment termed the caveosome on the way to the ER (25). Despite the lack of evidence for Golgi involvement in the pathway of Py entry, infection of GD1a-supplemented C6 cells was blocked by BFA, a known inhibitor of the GEF Arf1 involved in trafficking of vesicles between the Golgi and ER. These results could be explained by an effect of BFA on some unidentified GEF or other target that functions in a different pathway. It remains possible that Py passes rapidly through the Golgi and escapes detection in our experiments; however, blocking infection with BFA in this case might be expected to give rise to an association of virus with Golgi markers, and this was not observed.
A schematic representation of the uptake pathway of Py in GD1a-supplemented C6 cells as currently understood is shown in Fig. 9. The binding of virus to GD1a in supplemented C6 cells clearly initiates an efficient entry pathway for Py. This retrograde pathway occurs via caveolae and trafficking to the caveosome but bypassing the Golgi on the way to the ER. Critical but as-yet-undefined steps of disassembly must occur in the ER prior to escape into the cytoplasm and entry into the nucleus. It is expected that penetration of the ER as a site of disassembly, as described for SV40 (23), will be an essential feature of the infectious pathway for Py.
FIG. 9.
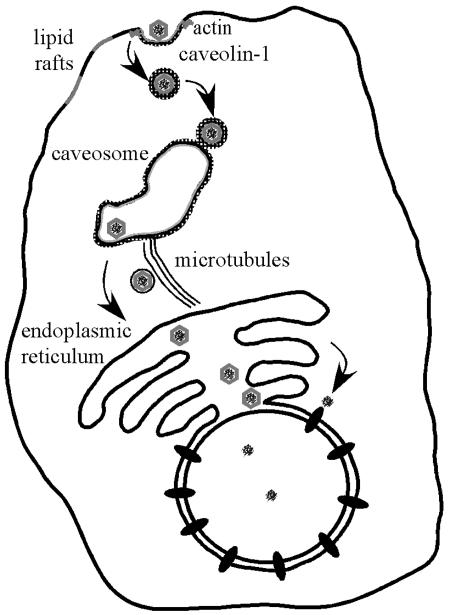
GD1a-mediated uptake pathway. Py virus binds to GD1a that is associated with lipid rafts and/or caveolae. Vesicles are endocytosed and routed to the caveosome. Movement from the caveosome to the ER is dependent upon microtubules. From the ER, the virus exits and presumably employs the nuclear pore to reach the nucleus, the site of viral replication.
Unsupplemented C6 cells have a low but reproducible level of infectability by Py. The basal and GD1a-supplemented routes of infection in C6 cells are most likely the same beyond the initial steps of internalization, converging on a pathway requiring intact microtubules before arriving at the ER as a common destination. The basal pathway can be distinguished from the GD1a-enhanced pathway in the initial steps of virus binding and internalization. The two pathways are ostensibly based on different classes of receptor molecules. Virus internalization in basal infection is not blocked by cholesterol disrupting agents, suggesting a process of internalization that is independent of lipid rafts or caveolae. This contrasts with infection of GD1a-supplemented C6 cells that is cholesterol dependent, consistent with evidence that trafficking of gangliosides is dependent upon lipid rafts and/or caveolae (9, 29, 39). It is possible that the receptor(s) for the two pathways is the same but is located in different domains of the plasma membrane. For example, unsupplemented C6 cells may express a low level of GD1a (or GT1b) that is not present in lipid rafts or in association with caveolae, and these gangliosides may mediate virus internalization by a different and perhaps less-efficient endocytic process. Alternatively, the basal pathway may be mediated by a sialoglycoprotein receptor(s). A recent study focusing on cells derived from the mouse as the natural host and selected for resistance to Py virus has shown that, like C6 cells, GD1a restores infectability (unpublished data) pointing to the importance and generality of the glycolipid-mediated entry pathway.
Acknowledgments
This work has been supported by grant RO1CA082395 from the National Cancer Institute. T.B. is a Virginia and D. K. Ludwig Professor of Cancer Research and Teaching.
We acknowledge fruitful discussions with T. Rapoport and B. Tsai and the technical assistance of J. You.
REFERENCES
- 1.Anderson, H. A., Y. Chen, and L. C. Norkin. 1996. Bound simian virus 40 translocates to caveolin-enriched membrane domains and its entry is inhibited by drugs that selectively disrupt caveolae. Mol. Biol. Cell 7:1825-1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barouch, D. H., and S. C. Harrison. 1994. Interactions among the major and minor coat proteins of polyomavirus. J. Virol. 68:3982-3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bauer, P. H., C. Cui, T. Stehle, S. C. Harrison, J. A. DeCaprio, and T. L. Benjamin. 1999. Discrimination between sialic acid-containing receptors and pseudoreceptors regulates polyomavirus spread in the mouse. J. Virol. 73:5826-5832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Caruso, M., L. Belloni, O. Sthandier, P. Amati, and M. I. Garcia. 2003. α4β1 Integrin acts as a cell receptor for murine polyomavirus at the postattachment level. J. Virol. 77:3913-3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Caruso, M., M. Cavaldesi, M. Gentile, O. Sthandier, P. Amati, and M-I. Garcia. 2003. Role of sialic acid-containing molecules and the α4β1 integrin receptor in the early steps of polyomavirus infection. J. Gen. Virol. 84:2927-2936. [DOI] [PubMed] [Google Scholar]
- 6.Chen, M., and T. Benjamin. 1997. Roles of N-glycans with α-2,6 as well as α-2,3 linked sialic acid in infection by polyoma virus. Virology 233:400-442. [DOI] [PubMed] [Google Scholar]
- 7.Dawe, C. J., R. Freund, G. Mandel, K. Ballmer-Hofer, D. A. Talmage, and T. L. Benjamin. 1987. Variations in polyoma virus genotype in relation to tumor induction in mice. Characterization of wild type strains with widely differing tumor profiles. Am. J. Pathol. 127:243-261. [PMC free article] [PubMed] [Google Scholar]
- 8.Fried, H., L. D. Cahan, and J. C. Paulson. 1981. Polyoma virus recognizes specific sialyloligosaccharide receptors on host cells. Virology 109:188-192. [DOI] [PubMed] [Google Scholar]
- 9.Fujinaga, Y., A. A. Wolf, C. Rodighiero, H. Wheeler, B. Tsai, L. Allen, M. G. Jobling, T. Rapoport, R. K. Holmes, and W. I. Lencer. 2003. Gangliosides that associate with lipid rafts mediate transport of cholera and related toxins from the plasma membrane to ER. Mol. Biol. Cell 14:4783-4793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gilbert, J. M., and T. L. Benjamin. 2000. Early steps of polyomavirus entry into cells. J. Virol. 74:8582-8588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gilbert, J. M., I. G. Goldberg, and T. L. Benjamin. 2003. Cell penetration and trafficking of polyomavirus. J. Virol. 77:2615-2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gilbert, J. M., L. D. Hernandez, T. Chernov-Rogan, and J. M. White. 1993. Generation of a water soluble oligomeric ectodomain of the Rous sarcoma virus envelope glycoprotein. J. Virol. 67:6889-6892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Griffith, G. R., S. J. Marriott, D. A. Rintoul, and R. A. Consigli. 1988. Early events in polyomavirus infection: fusion of monopinocytotic vesicles containing virions with mouse kidney cell nuclei. Virus Res. 10:41-52. [DOI] [PubMed] [Google Scholar]
- 14.Herrmann, M., C. Wilhelm von der Lieth, P. Stehling, W. Reutter, and M. Pawlita. 1997. Consequences of a subtle sialic acid modification on the murine polyomavirus receptor. J. Virol. 71:5922-5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kartenbeck, J., H. Stukenbrok, and A. Helenius. 1989. Endocytosis of simian virus 40 into the endoplasmic reticulum. J. Cell Biol. 109:2721-2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krauzewicz, N., J. Stokrova, C. Jenkinds, M. Elliott, C. F. Higgins, and B. E. Griffin. 2000. Virus-like gene transfer into cells mediated by polyoma virus pseudocapsids. Gene Ther. 7:2122-2131. [DOI] [PubMed] [Google Scholar]
- 17.Le, P. U., and I. R. Nabi. 2003. Distinct caveolae-mediated endocytic pathways target the Golgi apparatus and the endoplasmic reticulum. J. Cell Sci. 116:1059-1071. [DOI] [PubMed] [Google Scholar]
- 18.MacKay, R. L., and R. A. Consigli. 1976. Early events in polyoma virus infection: attachment, penetration, and nuclear entry. J. Virol. 19:620-636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mannová, P., and J. Forstová. 2003. Mouse polyomavirus utilizes recycling endosomes for a traffic pathway independent of COPI vesicle transport. J. Virol. 77:1672-1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mattern, C. F. T., K. K. Takemoto, and W. A. Daniel. 1966. Replication of polyoma virus in mouse embryo cells: electron microscopic observations. Virology 30:242-256. [DOI] [PubMed] [Google Scholar]
- 21.Nabi, I. R., and P. U. Le. 2003. Caveolae/raft-dependent endocytosis. J. Cell Biol. 161:673-677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nichols, B. J. 2002. A distinct class of endosome mediated clathrin-independent endocytosis to the Golgi complex. Nat. Cell Biol. 4:374-378. [DOI] [PubMed] [Google Scholar]
- 23.Norkin, L. C., H. A. Anderson, S. A. Wolfrom, and A. Oppenheim. 2002. Caveolar endocytosis of simian virus 40 is followed by brefeldin A-sensitive transport to the endoplasmic reticulum, where the virus disassembles. J. Virol. 76:5156-5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pelkmans, L., and A. Helenius. 2002. Endocytosis via caveolae. Traffic 3:311-320. [DOI] [PubMed] [Google Scholar]
- 25.Pelkmans, L., J. Kartenbeck, and A. Helenius. 2001. Caveolar endocytosis of simian virus 40 reveals a new two-step vesicular-transport pathway to the ER. Nat. Cell Biol. 3:473-483. [DOI] [PubMed] [Google Scholar]
- 26.Pelkmans, L., D. Puntener, and A. Helenius. 2002. Local actin polymerization and dynamin recruitment in SV40-induced internalization of caveolae. Science 296:535-539. [DOI] [PubMed] [Google Scholar]
- 27.Richterová, Z., D. Liebl, M. Horák, Z. Palková, J. Stokrová, P. Hozák, J. Korb, and J. Forstová. 2001. Caveolae are involved in the trafficking of mouse polyomavirus virions and artificial VP1 pseudocapsids toward cell nuclei. J. Virol. 75:10880-10891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sahli, R., R. Freund, D. T., R. Garcea, R. Bronson, and T. Benjamin. 1993. Defect in entry and altered pathogenicity of a polyoma virus mutant blocked in VP2 myristylation. Virology 192:142-153. [DOI] [PubMed] [Google Scholar]
- 29.Sandvig, K., S. Grimmer, S. U. Lauvrak, M. L. Torgersen, G. Skretting, B. van Deurs, and T. G. Iversen. 2002. Pathways followed by ricin and Shiga toxin into cells. Histochem. Cell Biol. 117:131-141. [DOI] [PubMed] [Google Scholar]
- 30.Sharma, D. K., A. Choudhury, R. D. Singh, C. L. Wheatley, D. L. Marks, and R. E. Pagano. 2003. Glycosphingolipids internalized via caveolar-related endocytosis rapidly merge with the clathrin pathway in early endosomes and form microdomains for recycling. J. Biol. Chem. 278:7564-7572. [DOI] [PubMed] [Google Scholar]
- 31.Shimura, H., Y. Umeno, and G. Kimura. 1987. Effects of inhibitors of the cytoplasmic structure and functions on the early phase of infection of cultured cells with simian virus 40. Virology 158:34-43. [DOI] [PubMed] [Google Scholar]
- 32.Singh, R. D., V. Puri, J. T. Valiyaveettil, D. L. Marks, R. Bittman, and R. E. Pagano. 2003. Selective caveolin-1 dependent endocytosis of glycosphingolipids. Mol. Biol. Cell 14:3254-3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith, A. E., H. Lilie, and A. Helenius. 2003. Ganglioside-dependent cell attachment and endocytosis of murine polyomavirus-like particles. FEBS Lett. 555:199-203. [DOI] [PubMed] [Google Scholar]
- 34.Sottocornola, E., I. Colombo, B. Vergani, G. Taraboletti, and B. Berra. 1999. Increased tumorigenicity and invasiveness of C6 rat glioma cells transfected with the human α-2,8 sialyltransferase cDNA. Invasion Metastasis 18:142-154. [DOI] [PubMed] [Google Scholar]
- 35.Stang, E., J. Kartenbeck, and R. G. Parton. 1997. Major histocompatibility complex class I molecules mediate association of SV40 with caveolae. Mol. Biol. Cell 8:47-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stehle, T., and S. C. Harrison. 1996. Crystal structures of murine polyomavirus in complex with straight-chain and branched-chain sialyloligosaccharide receptor fragments. Structure 4:183-194. [DOI] [PubMed] [Google Scholar]
- 37.Stehle, T., and S. C. Harrison. 1997. High-resolution structure of a polyomavirus VP1-oligoaccharide complex: implications for assembly and receptor binding. EMBO J. 16:5139-5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsai, B., J. M. Gilbert, T. Stehle, W. Lencer, T. L. Benjamin, and T. Rapoport. 2003. Gangliosides are receptors for murine polyoma virus and SV40. EMBO J. 22:4346-4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wolf, A. A., Y. Fujinaga, and W. I. Lencer. 2002. Uncoupling of the cholera toxin-GM1 ganglioside receptor complex from endocytosis, retrograde Golgi trafficking, and downstream signal transduction by depletion of membrane cholesterol. J. Biol. Chem. 277:16249-16256. [DOI] [PubMed] [Google Scholar]