

Abstract
Nonstructural protein 15 (Nsp15) of the severe acute respiratory syndrome coronavirus (SARS-CoV) produced in Escherichia coli has endoribonuclease activity that preferentially cleaved 5′ of uridylates of RNAs. Blocking either the 5′ or 3′ terminus did not affect cleavage. Double- and single-stranded RNAs were both substrates for Nsp15 but with different kinetics for cleavage. Mn2+ at 2 to 10 mM was needed for optimal endoribonuclease activity, but Mg2+ and several other divalent metals were capable of supporting only a low level of activity. Concentrations of Mn2+ needed for endoribonuclease activity induced significant conformation change(s) in the protein, as measured by changes in tryptophan fluorescence. A similar endoribonucleolytic activity was detected for the orthologous protein from another coronavirus, demonstrating that the endoribonuclease activity of Nsp15 may be common to coronaviruses. This work presents an initial biochemical characterization of a novel coronavirus endoribonuclease.
The severe acute respiratory syndrome (SARS) epidemic of 2002 to 2003 eventually spread from China to 30 countries, with more than 8,000 confirmed cases and an overall mortality rate of about 10% (31). Due to the lack of more specific antiviral agents, one treatment regimen used consisted of corticoid steroids and ribavirin (15, 16). This regimen was of unknown efficacy and had severe side effects. Thus, there is a need for better therapeutic strategies and the development of potential targets in the etiological agent of SARS.
SARS is caused by a coronavirus (SARS-CoV), which has a positive-strand RNA genome of ∼30 kb in length with a 5′ cap and 3′ poly(A) tail (22). For general reviews of coronaviruses, see Lai and Cavanagh (13) and Spaan et al. (26). Of the three coronavirus phylogenetic groups, groups 1 and 2 infect mammals, causing respiratory, central nervous system, enteric, and even systemic infections, while group 3 infects birds. Phylogenetic analysis of the SARS-CoV RNA-dependent RNA polymerase sequence has placed SARS-CoV in group 2 (25). Analysis of the complete genomes of more than a dozen SARS-CoV isolates from around the world indicate that SARS-CoV is not a mutant or recombinant of known coronaviruses, but a novel coronavirus that acquired the ability to infect humans. A nearly identical coronavirus was isolated from civet cats, raccoon dogs, and badgers, an observation that raises speculations that this virus jumped species to humans (4).
Coronavirus produces two large polyproteins that are proteolytically processed to yield replication-associated proteins and several structural and infection-associated proteins that are translated from subgenomic RNAs (Fig. 1). The coronavirus proteases that are required for processing of the polyproteins have been characterized extensively (1, 32). In addition, the structure of SARS-CoV nonstructural protein 9 (Nsp9) was solved recently and is a novel RNA binding protein (27). Nsp13 is a helicase with the ability to unwind both DNA and RNA duplexes. Nsp13 is also reported to have dNTPase and RNA 5′-triphosphatase activities (6).
FIG. 1.

Schematic of the SARS-CoV genome and the sequence of Nsp15. The approximate locations of polyproteins 1a (pp1a) and 1ab (pp1ab) in the SARS-CoV genome are indicated. Proteins made from subgenomic messages encoded by the 3′ portion of the SARS-CoV genome are shown. For clarity, the individual coding sequences are not shown. The Nsp15 sequence is from the Urbani isolate and is represented by the standard one-letter code. Comparable sequences from MHV and IBV were aligned with the CLUSTAL program (28). Residues that have identical side chains (asterisks), functionally similar side chains (colons), and somewhat similar side chains (periods) are indicated under the sequences. Gaps introduced to maximize alignment are indicated by dashes. The central portion of the MHV protein is more divergent than the comparable sequence from the SARS-CoV and IBV proteins. To help demonstrate the possible relevance of the alignments, the PHD program (21) was used to predict the positions of secondary structures predicted within each polypeptide. The residues putatively involved in the formation of β-strands or α-helices are underlined or shaded, respectively. Sequences under the thick black lines represent the region with similarities to the putative XendoU active site, as described by Snijder et al. (25).
In general, the biochemical characterizations of the coronavirus replication-associated proteins are at an early stage. Snijder and colleagues have proposed that Nsp15 has similarities to the endoribonuclease XendoU (25), a Mn2+-dependent endoribonuclease required for the biogenesis of U16 small nucleolar RNA (3, 14). The predicted secondary structures of the two proteins have few similarities (C. C. Kao, unpublished observations), but the SARS-CoV Nsp15 is rich in acidic residues (46 of 346 total residues are aspartates or glutamates) that could coordinate metal cofactor(s). These observations prompted us to examine whether Nsp15 is indeed an endoribonuclease. Furthermore, Nsp15 orthologs of coronaviruses are well conserved at the N and C termini but are more divergent in the central portion (Fig. 1), raising the question of whether orthologs in different coronaviruses have a common enzymatic activity. In this work, we report the production of a recombinant version of the SARS-CoV Nsp15 protein and provide biochemical evidence that it is indeed an Mn2+-dependent endoribonuclease that cleaves 5′ of uridylates.
MATERIALS AND METHODS
Protein expression and purification.
The SARS-CoV Nsp15 cDNA was amplified by reverse transcription-PCR (RT-PCR) using RNAs extracted from the Urbani isolate of SARS-CoV using oligonucleotides D23.3 and D29.2 (Table 1). RT-PCR was performed using a kit provided by Invitrogen following the protocol suggested by the manufacturer. The PCR product was cloned into the vector pET15b to provide an N-terminal six-histidine tag to facilitate purification of the SARS-CoV Nsp15. The DNA sequence was determined in its entirety to confirm its identity to the one reported by Rota et al. (22) (accession number AY278741). Similar procedures were used to amplify the cDNAs coding for the orthologs of mouse hepatitis virus (MHV) (A59 strain) and infectious bronchitis virus (IBV) (Beaudette strain).
TABLE 1.
Oligonucleotides and RNAs used in this study
| Oligonucleotide or RNA | Orientation | Use | Sequence |
|---|---|---|---|
| DNA oligonucleotides | |||
| D23.3 | 5′ | PCR-SARS-CoV | 5′ CATATGAGTTTAGAAAATGTGGC 3′ |
| D29.2 | 3′ | RT-PCR, SARS-CoV | 5′ GGATCCTTATTGTAGTTTTGGGTAGAAGG 3′ |
| D27.2 | 5′ | PCR, IBV | 5′ ACATATGTCTATCGACAATATTGCTTA 3′ |
| D27.3 | 3′ | RT-PCR, IBV | 5′ ACTCGAGTTGAAGCTGTGGATAACATG 3′ |
| D27.4 | 5′ | PCR, MHV | 5′ ACATATGAGTTTAGAAAATGTAGTGTA 3′ |
| D27.5 | 3′ | RT-PCR, MHV | 5′ ACTCGAGCTGCAAACGAGGATAGAAAG 3′ |
| Chemically synthesized RNAs | |||
| R10.2 | Nsp15 substrate | 5′ UGGGAGUAUA 3′ | |
| R12 | Nsp15 substrate | 5′ CCCCCGCCCCCC 3′ | |
| SK21 | Nsp15 substrate | 5′ GACGCUGAAUUAGGACAUAGA 3′ | |
| LE19P | Nsp15 substrate | 5′ UGUUAUAAUUAUUGUAUAC-puromycin 3′ | |
| F16.2 | Nsp15 substrate | 5′ fluorescein-GGUCAGGAUACCUGGC 3′ | |
| LE21 | Nsp15 substrate | 5′ UGUUAUAAUUAUUGUAUAUAC 3′ | |
| T7 RNA polymerase transcript SL13 | Nsp15 substrate | 5′ GGUGCAUAGCACC 3′ | |
| Double-stranded RNA ds34 | Nsp15 substrate | 5′ UGUUAUAAUUAUUGUAUAUACAAUAAUUAUAACU 3′ | |
| 3′ ACAAUAUUAAUAACAUAUAUGUUAUUAAUAUUGA 5′ |
Bacterial cultures were grown at 37°C in standard Luria-Bertani medium supplemented with 50 μg of ampicillin per ml until the culture reached an optical density at 600 nm of 1.0. The culture temperature was then lowered to 16°C, and expression was induced for 36 h with 1 mM isopropyl-thiogalactoside. Cells were harvested after centrifugation, and recombinant proteins were purified in buffer lacking divalent metals by passage through a Talon nickel affinity column (Invitrogen, Inc.), followed by gel filtration through Superdex 75 (Pharmacia, Inc.) or an ion-exchange (Mono Q) column (Pharmacia, Inc.). The protein was adjusted to 0.1 mg/ml after quantification by staining with Coomassie blue using bovine serum albumin as a concentration control and stored in buffer containing 50 mM Tris (pH 7.5) and 50% glycerol.
Preparation of endoribonuclease substrates.
DNA oligonucleotide D23.2 was synthesized by GeneSys (Sigma, Inc., St. Louis, Mo.). Oligoribonucleotides were either chemically synthesized by Dharmacon, Inc. (Boulder, Colo.), or transcribed by use of the T7 RNA polymerase as described previously (8). Unless stated otherwise, substrates used for cleavage assays were labeled at the 5′ end with T4 polynucleotide kinase and [γ-32P]ATP. Prior to labeling reactions, all RNAs were purified from denaturing 7.5 M urea-20% polyacrylamide gels (20:1 acrylamide to bisacrylamide) by the method of Ranjith-Kumar et al. (18). Double-stranded RNA ds34 was made by extending from two partially annealed molecules of RNA LE21 using the hepatitis C virus RNA-dependent RNA polymerase by the method of Ranjith-Kumar et al. (17).
Endoribonuclease assays.
The standard RNA cleavage assay used 10,000 cpm of radiolabeled RNA substrate (final RNA concentration of ∼20 μM) and 0.26 μM Nsp15 in a buffer consisting of 50 mM Tris (pH 7.5), 100 mM KCl, 1 mM dithiothreitol, and 5 mM MnCl2. The reaction mixture was incubated at 30°C for 30 min unless stated otherwise. The reactions were terminated by the addition of the gel-loading buffer that contained 7.5 M urea. Products were usually separated by electrophoresis in denaturing gels composed of 7.5 M urea and polyacrylamide. Gels were wrapped in plastic and exposed to a PhosphorImager screen. Each result shown was reproduced in at least one and usually two independent experiments. Quantification of radiolabeled bands was performed using a PhosphorImager (Molecular Dynamics).
Intrinsic fluorescence measurements.
All fluorescence measurements were made using Perkin-Elmer luminescence spectrometer LS55 and cuvette (Perkin-Elmer) with an optical path length of 0.4 cm at room temperature (22 to 23°C). Nsp15 protein was at 0.5 μM in a buffer containing 50 mM Tris (pH 7.5) and 100 mM KCl. When divalent metal was added during titration, the final volume did not exceed 10% of the initial sample volume. The samples were excited with light at a wavelength of 295 nm, and the emission was scanned with light at a wavelength from 310 to 450 nm (slit widths of excitation and emission were 5 and 10 nm, respectively). The emission scan was repeated three times, and all results were averaged. All data were corrected for the background intensity of the buffer and for dilution. Binding data were analyzed by nonlinear least-squares fitting using KaleidaGraph software (Synergy Software, Reading, Pa.). A simple 1:1 binding model was fitted to the binding isotherm using equation 1: ΔF/F0 = Bmax [ligand]total/(Kd + [ligand]total), where ΔF is the magnitude of the difference between the observed fluorescence intensity (arbitrary unit) at a given metal ion concentration and the fluorescence intensity in the absence of metal ions, F0 is the fluorescence intensity in the absence of metal ions, and Bmax is the value of the maximum relative fluorescence change.
RESULTS
Endoribonuclease activity.
Recombinant SARS-CoV Nsp15 was expressed with six N-terminal histidine residues and purified from Escherichia coli BL21star (deficient in RNase E activity) (Fig. 2A). Expression of the protein was higher in E. coli BL21star strain than in E. coli BL21(DE3) (K. Bhardwaj, data not shown), with approximately 10% of the overexpressed protein in the soluble fraction after centrifugation at 10,000 × g. The recombinant protein had the expected mass and bound to Ni-nitrilotriacetic acid (NTA) resin. The bound sample was washed with a buffer containing 25 mM imidazole before elution with a buffer containing 250 mM imidazole (Fig. 2A).
FIG. 2.

Purification and endoribonucleolytic activity of Nsp15. (A) Sodium dodecyl sulfate-polyacrylamide gel electrophoresis of the recombinant SARS-CoV purified from a Ni-NTA column. Nsp15 was loaded at two concentrations, and the gel was stained with Coomassie brilliant blue. The positions (in kilodaltons) of molecular mass markers (M) are shown to the left of the gel. (B) A gel image demonstrating the cleavage of a radiolabeled 10-nt RNA and nucleotide resolution of the cleavage products. Nsp15 was added to one master reaction mixture, and aliquots of the reaction mixture were removed at the times shown above the gel image for electrophoresis in a 7.5 M urea-20% polyacrylamide gel. The sequence of the RNA substrate, R10.2, is shown above the gel, and the sizes of the bands (in nucleotides) are indicated to the left of the gel. The percentage of the 10-nt R10.2 that remains after each treatment (% FL) is indicated under the gel. The smaller gel at the right shows that R12, which lacks uridylates, is not cleaved to the same extent as R10.2. (C) Copurification of the endoribonucleolytic activity with the recombinant protein. The protein enriched from the metal affinity column was fractionated over a Mono Q column, with Nsp15 eluting primarily in fractions 17 and 18. The amount of endoribonucleolytic activity in the fraction (% Activity) is indicated. The activities in these column fractions were normalized to that of fraction 17 (set at 100%). (D) Sequences of SK21 and potential products that can be generated from a DNA template coding for SK21 by the T7 RNA polymerase when UTP is absent. (E) Cleavage of a radiolabeled RNA SK21. The percentage of the full-length substrate RNA remaining after each treatment (% FL) is indicated under the gel. Lanes where the input RNA was not treated with Nsp15 (φ) are indicated above the gel. (F) Analysis of the cleavage site preferred by Nsp15. The final product generated by Nsp15 from SK21 should be 5 nt long if cleavage occurs 5′ of uridylates. If cleavage occurs 3′ of uridylates, the stable product should be 6 nt long. To examine the length of the stable Nsp15 product, DNA template T7-SK21 was used to transcribe RNA products in the absence of UTP. T7 polymerase should generate an abundant 5-nt RNA product. Other slightly longer products could be accounted for by terminal nucleotide addition and/or nucleotide misincorporation. End-labeled SK21 RNA was treated with Nsp15 for 1 h (+) or left alone (−) (lanes 1 and 2) as described above. To confirm the length of the Nsp15 cleavage product, T7 transcription was performed in the absence of UTP using template T7-SK21 as described above for panel F (lane 3). Samples from lanes 1 and 3 were mixed together and loaded in lane 4 to show that both products are the same length.
To monitor enzymatic activity, we incubated Nsp15 with a 10-nucleotide (nt)-long RNA named R10.2 that was radiolabeled at the 5′ phosphate. This RNA contains three uridylates (Table 1 and Fig. 2B). Should Nsp15 cleave the phosphodiester bond 5′ of the uridylate, radiolabeled products of 8 and 6 nt would result from partial digestion (Table 1). There is also a minor amount of a 9-nt RNA in this preparation that arose from incomplete chemical synthesis of R10.2. We also used a 12-nt RNA named R12 that lacked uridylates. When incubated for up to 40 min in a buffer containing 5 mM Mn2+, a concentration optimal for XendoU (3), the amount of full-length R10.2 decreased over time, and 6- and 8-nt cleavage products accumulated. Cleavage was not observed in reactions lacking divalent metal, and R12 was not cleaved under the same conditions (Fig. 2B and data not shown). These results suggest that Nsp15 is an endoribonuclease that preferentially cleaves 5′ of uridylates and that endoribonucleolytic activity requires divalent metal. These specific requirements will be confirmed and characterized further below.
To demonstrate further that endoribonucleolytic activity is the property of the recombinant Nsp15, we monitored this activity in Nsp15 preparations fractionated by additional chromatography columns. In a Mono Q ion-exchange column, activity copurified with the peak of Nsp15 protein (Fig. 2C). Endoribonucleotytic activity also copurified with Nsp15 fractionated by gel filtration through Superdex 75 (data not shown).
To confirm that Nsp15 specifically cleaves 5′ of uridylates, we examined the digestion pattern for a second RNA, SK21, which contained four uridylates (Fig. 2D). Over a period of 30 min, four fragments were produced (Fig. 2E), as would be expected if Nsp15 were to cleave at uridylates. Complete digestion of SK21 5′ of uridylates should result in a radiolabeled RNA fragment 5 nt long. To determine whether cleavage occurred 5′ or 3′ of the uridylates, we compared the mobility of the smallest fragment generated upon digestion of SK21 with 5-nt RNA made by in vitro transcription of SK21 in the absence of UTP. The two RNAs comigrated in a denaturing gel, demonstrating that Nsp15 cleaved 5′ of a uridylate residue (Fig. 2F). The requirement for divalent metals, the preferred cleavage 5′ of uridylates in RNAs, and the copurification of activity with the protein indicate that Nsp15, and not a contaminating protein, is an endoribonuclease.
Specificity for RNA.
To determine whether Nsp15 cleavage is specific for RNAs, a 5′-end-labeled DNA molecule (D23.2) of 23 nt that contained all four bases was incubated with Nsp15 at standard assay conditions. D23.2 was not cleaved by Nsp15 (Fig. 3A). Furthermore, D23.2 did not inhibit Nsp15-mediated cleavage of R10.2 (Fig. 3B), suggesting that Nsp15 does not stably bind DNA.
FIG. 3.

Interaction of Nsp15 with DNA and heparin. (A) PhosphorImage of 7.5 M urea-20% polyacrylamide gel showing 5′-end-labeled DNA oligonucleotide (D23.2) incubated in the presence (+) or absence (−) of Nsp15 under standard assay conditions. Each treatment was done in triplicate. (B) Effects of heparin and DNA on cleavage of R10.2 RNA by Nsp15. R10.2 digestion was performed in the absence or presence of various concentrations of either heparin or DNA (indicated above the bars). After the RNAs were resolved on urea-polyacrylamide gels, uncleaved R10.2 was quantitated and shown as a percentage [FL (%)]. Substrate RNA incubated in the absence (−) of Nsp15 was set at 100%. Each error bar represents 1 standard deviation from the mean for each treatment.
We sought to identify an inhibitor for Nsp15. Many nucleic acid binding proteins, such as polymerase, are inhibited by heparin, a polysulfonate that mimics nucleic acid (9). When heparin was present in the reaction mixture at 0.01 to 5 μg, Nsp15 cleavage of R10.2 was inhibited approximately two- to threefold (Fig. 3B). This result suggests that heparin can be used for future characterizations of Nsp15-substrate interactions.
Requirements for divalent metals.
We characterized further the role of divalent metal ions in Nsp15 activity. The ability to cleave R10.2 was determined under a range of Mn2+ and Mg2+ concentrations (Fig. 4A). Increasing the concentration of Mn2+ from 0 to 10 mM increased ribonucleolytic activity more than 50-fold. Furthermore, only minimal activity was observed with Mg2+ or several other divalent metals (Fig. 4A and B). The requirement for Mn2+ at concentrations higher than 1 mM and the inability to cleave RNA in the presence of Mg2+ were also observed with other RNAs (data not shown). The SARS-CoV Nsp15 protein thus exhibits a strong preference for Mn2+ as a cofactor for RNA cleavage.
FIG. 4.

Requirements for divalent metal ions for SARS-CoV endoribonucleolytic activity. (A) Mg2+ and Mn2+ concentrations needed for endoribonucleolytic activity of Nsp15. RNA R10.2 was used as the substrate in the titrations. All divalent metals used in this figure were in the form of chloride salts. (B) Examination of the effects of other divalent metals on cleavage of R10.2 by Nsp15. The effects of several divalent metals (at 0.5 or 5 mM) were quantified and are shown below the gel. Only the full-length RNA (FL) is shown. φ indicates no divalent ion. (C) Effects of Mn2+ on the spectroscopic properties of the SARS-CoV Nsp15. Change in intrinsic fluorescence (FU) of Nsp15 in response to increasing Mn2+ concentration. The emission maximum of Nsp15 was determined to be at 337 nm. Scans at increasing concentrations of divalent metal were taken, and values at 337 nm were plotted. (D) Modeling of the binding constant for Mn2+. A 1:1 model was fitted to the binding isotherm to derive Kd using equation 1 (see Materials and Methods).
To determine whether Mn2+ affects the conformation of Nsp15, we measured its intrinsic tryptophan fluorescence. There are three tryptophans in the SARS-CoV Nsp15, two in the N-terminal region and one near the C terminus. A scan performed in the absence of divalent metals revealed that 337 nm was the wavelength for maximal emission. Titration of Mn2+ resulted in a significant decrease in tryptophan signal, indicating that a conformational change(s) is taking place (Fig. 4C). Buffer lacking Nsp15, but containing Mn2+ at concentrations up to 20 mM, contributed minimally to the measurements. In addition, Mg2+ was unable to induce the same level of change in Nsp15 intrinsic fluorescence (Fig. 4C). The degree of conformational change correlated well with the concentrations needed for endoribonuclease activity (Fig. 4A), suggesting that Mn2+ association with Nsp15 resulted in the formation of an enzyme capable of endoribonucleolytic activity.
The rate of change in intrinsic tryptophan fluorescence, corrected for buffer effects, was used to calculate the dissociation constant for Mn2+. A 1:1 binding model was found to fit the data well, generating an R2 value of 0.98. The calculated Kd for Mn2+ was 5.3 mM (Fig. 4D).
Substrate requirements.
The cleavage pattern with RNA R10.2 suggests that Nsp15 is an endoribonuclease. Next, we determined whether an accessible 5′ or 3′ terminus was needed for RNA cleavage using two previously characterized RNAs named F16.2 and LE19P that have fluorescein and puromycin, respectively, covalently attached to the 5′ or 3′ terminus (12, 17). LE19P has an accessible 5′ terminus that was radiolabeled with kinase. In a standard cleavage reaction, the degradation kinetics of LE19P was identical to that of LE19 (Fig. 5A and data not shown). F16.2 cannot be 5′ end labeled due to the fluorescein and so was visualized by staining with toluidine blue. F16.2 was cleaved and with kinetics similar to that of unmodified RNA (Fig. 5A and data not shown). These results indicate that blocked 5′ and 3′ termini do not significantly affect RNA cleavage by the SARS-CoV Nsp15, consistent with Nsp15 being an endoribonuclease.
FIG. 5.
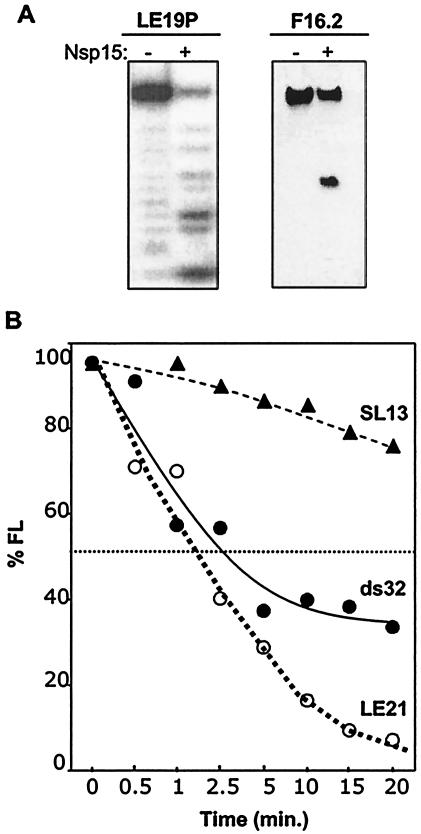
Substrate requirements for endoribonucleolytic activity of SARS-CoV Nsp15. (A) RNA substrates with chemically modified 3′ and 5′ termini. RNA LE19P, with a covalently attached 3′ puromycin is a weakly base-paired RNA that could form a weakly stable intramolecular hairpin or partially base-paired dimer (19). F16.2 is a 16-nt RNA that was synthesized with a 5′ fluorescein. The amount of cleavage was normalized to the percentage of full-length RNA remaining after the treatment (% FL). The RNA used in each experiment and whether Nsp15 was added (+) or not (−) are indicated above the gels. Digestion of LE19P is shown as a PhosphorImage of radiolabeled RNA, while F16.2 was from a toluidine blue-stained gel. (B) Comparison of the rate of cleavage of single- and double-stranded RNAs. ds32 is a double-stranded RNA with complete complementarity in the two strands. LE21, a single-stranded RNA with minimal intramolecular base pairing, was used to generate ds34. The amount of each input RNA was normalized to 100%. SL13, a 13-nt RNA was produced by in vitro transcription by use of T7 polymerase. 5′-end phosphate was replaced with radiolabeled phosphate by sequential treatment with alkaline phosphatase and T4 polynucleotide kinase. Radiolabeled RNA was gel purified before use.
RNA substrates tested thus far do not have extensive stable structures, as predicted by the Mfold computer program (33) and according to previous characterizations of these RNAs (18). To determine whether structures in RNA affect Nsp15 activity, we produced an internally labeled 34-nt double-stranded RNA named ds34 that was generated by primer-dependent extension from two molecules of RNA LE21 (18). The ds34 was cleaved slightly slower than LE21 was (Fig. 5B). Last, we observed that an unusually stable RNA hairpin named SL13 that contains a 5-bp stem and a 3-nt loop with an uridylate (Table 1) (11) was digested by Nsp15 at a significantly reduced rate (Fig. 5B). These results demonstrate that in vitro, Np15 is catalytically active on RNAs that are single or double stranded. However, there are differences in the rates of cleavage that may be significant in the regulation of Nsp15 activity during SARS-CoV infection.
Activity of other coronavirus orthologs.
The SARS-CoV has a number of unique open reading frames (22), and the central region of Nsp15 appears to be more divergent than the orthologs of MHV and IBV (Fig. 1). To address whether endoribonucleolytic activity of Nsp15 is a common property of coronaviruses or is unique to the SARS-CoV, the corresponding proteins from MHV and IBV were engineered with histidine tags and purified by the same protocol as the protein from SARS-CoV. Since the corresponding proteins from different viruses were given different names, we will refer to them as orthologs. When tested for the ability to cleave R10.2, the IBV ortholog did so and with kinetics faster than the protein from the SARS-CoV (Fig. 6A). There was also an increase in the degradation of the 8- and 6-nt cleavage intermediates (Fig. 6A). The IBV ortholog also cleaved SK21 with kinetics similar to that of the SARS-CoV Nsp15 (data not shown). In contrast, two independently constructed orthologs from MHV were much less efficient in this activity (Fig. 6B); cleavage by the MHV ortholog was reproducible and reduced the amount of R10.2 by only 15 to 20%. These results demonstrate that while the endoribonuclease activity of the SARS-CoV Nsp15 is not unique among coronaviruses, the amount of activity could differ in proteins from different viruses, at least in vitro.
FIG. 6.
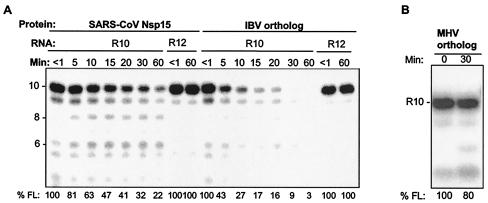
Comparison of endoribonuclease activities of the SARS-CoV Nsp15 and orthologs from other coronaviruses. (A) Activities of the IBV ortholog. An image of the cleavage products from R10.2 and R12 are shown. The proteins and RNA substrates used in each reaction mixture are indicated above the gel. The contents of all lanes were exposed to Nsp15, but an aliquot was removed and placed in the denaturing gel loading buffer within seconds of addition of the enzyme (<1 min). The sizes of the products (in nucleotides) are indicated to the left of the gel. Quantification of endoribonuclease activity, shown below the gel, refers to the percentage of the full-length RNA (% FL) remaining in each lane. (B) Activity of the MHV ortholog.
DISCUSSION
Characterization of the biochemical activities of the proteins in the SARS-CoV is a necessary first step toward identifying molecular targets for antiviral agents. In this work, we have established that the replication-associated Nsp15 protein has endoribonucleolytic activity with properties that resemble those of the Xenopus laevis XendoU (3, 14), as predicted by Snijder and colleagues (25). Nsp15 preferentially cleaved 5′ of uridylates and was insensitive to modifications at both the 5′ and 3′ ends of the RNA substrate. In addition, it can cleave single- and double-stranded RNAs in a manganese-dependent reaction but with notable differences in the rates of cleavage.
The concentrations of Mn2+ needed for catalysis caused a significant change in the tryptophan fluorescence of Nsp15, suggesting that Mn2+ could induce a conformational change in Nsp15, perhaps leading to the catalytically active form. Last, the demonstrated activity was not unique to the SARS-CoV but was also observed in the IBV ortholog. Whether MHV Nsp15 possesses the identical activity requires additional characterizations. RNA endoribonuclease activity is unusual among plus-strand RNA viruses. One other example is found in the negative-strand influenza virus, which encodes a specific endoribonuclease that is involved in cleaving capped cellular transcripts (20). This unique nature of Nsp15 for SARS-CoV suggests that it could be a target for the development of antiviral agents.
A number of Mn2+-dependent endoribonucleases have previously been characterized, including retroviral and cellular RNase H (5), which selectively degrades the RNA strand duplexed to DNA. E. coli RNase H has a single Mg2+ in the active site, while the one from the human immunodeficiency virus uses two Mn2+ that are coordinated by a network of negatively charged amino acid side chains. Nsp15 likely coordinates Mn2+ by a combination of acidic side chains, although the ones participating in metal binding and the residues that participate in nucleophilic attack of the phosphodiester backbone remain to be identified.
Interestingly, E. coli RNase H is active with micromolar concentrations of Mn2+ but requires millimolar levels of Mg2+, leading to the proposal that two metal ions are bound, with the first activating the enzyme and the second inhibiting its activity (10). However, unlike E. coli RNase H, Nsp15 does not appear to be inhibited by high concentrations of Mn2+ and is not particularly active with any concentration of Mg2+. Nonetheless, since the concentration of Mn2+ needed for Nsp15 endoribonucleolytic activity (millimolar) is not physiologically relevant, we cannot rule out additional roles of divalent metals in regulating Nsp15 activity. We note, however, that XendoU also requires millimolar amounts of Mn2+ in vitro (14), again indicating that the two proteins have similar activities. It would not be surprising to learn that the intracellular environment could influence the Kd for divalent metal requirements for Nsp15. Hence, it is possible that physiological concentrations of Mn2+ are sufficient for Nsp15 activity in the SARS virus replication/transcription complex or that the availability of divalent metal(s) provides a means to regulate the activity of this enzyme.
How could an RNA endoribonucleolytic activity function in coronavirus infection? One possibility is that Nsp15 contributes to pathogenesis by affecting cellular RNA processing. Another possibility is that Nsp15 plays a role in coronavirus replication and/or transcription (24, 32). It has been suggested that coronavirus subgenomic RNA synthesis requires nuclease activity (2, 7, 29). Coronaviruses produce between five and eight subgenomic RNAs whose 3′ ends are coterminal with the 3′ end of the plus-strand genomic RNA. Interestingly, despite being of different lengths, the subgenomic RNAs share a common 5′ leader of 65 to 98 nt. How the leader is attached to the body of the mRNA is still under investigation, but two models are generally invoked. The first, the leader-primed model (13), posits that the leader RNA is synthesized from the 3′ end of the genomic minus-strand RNA, released by the viral transcriptase, and primes RNA synthesis at the intergenic sites in the minus-strand template RNA. The precursor leader RNAs tend to be longer than the version fused to the body RNA, suggesting that it is processed prior to the priming reaction (7). Such processing could use Nsp15. In the second model, the discontinuous transcription model, it is hypothesized that the replicating polymerase ternary complex translocates at specific intergenic sequences during minus-strand RNA synthesis and then reattaches to the 5′ leader sequence and resumes RNA synthesis (23). It is also possible that an endoribonuclease activity is required to digest the nascent RNA to untether the ternary complex prior to the translocation of the ternary polymerase complex. How Nsp15 affects the SARS-CoV infection process remains to be determined.
Acknowledgments
We thank J. Leibowitz for providing cDNA to MHV and the SARS-CoV RNA, E. Collisson for providing IBV cDNA, R. Kumar for supplying double-stranded RNA ds34, and Y. C. Kim for help with fluorescence spectroscopy.
We thank the National Science Foundation for an SGER grant to initiate research on the SARS virus.
REFERENCES
- 1.Anand, K., J. Ziebuhr, P. Wadhwani, J. R. Mesters, and R. Hilgenfeld. 2003. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science 300:1763-1767. [DOI] [PubMed] [Google Scholar]
- 2.Baker, S. C., and M. M. Lai. 1990. An in vitro system for the leader-primed transcription of coronavirus mRNAs. EMBO J. 9:4173-4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Caffarelli, E., L. Maggi, A. Fatica, J. Jiricny, and I. Bozzoni. 1997. A novel Mn2+-dependent ribonuclease that functions in U16 SnoRNA processing in X. laevis. Biochem. Biophys. Res. Commun. 233:514-517. [DOI] [PubMed] [Google Scholar]
- 4.Enserink, M. 2003. Clues to the animal origins of SARS. Science 300:1351. [DOI] [PubMed] [Google Scholar]
- 5.Goedken, E. R., and S. Marqusee. 2001. Co-crystal of Escherichia coli RNase HI with Mn2+ ions reveals two divalent metals bound in the active site. J. Biol. Chem. 276:7266-7271. [DOI] [PubMed] [Google Scholar]
- 6.Ivanov, K. A., V. Theil, J. C. Dobbe, Y. van der Meer, E. J. Snijder, and J. Ziebuhr. 2004. Multiple activities associated with severe acute respiratory syndrome coronavirus helicase. J. Virol. 78:5619-5632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Joo, M., and S. Makino. 1992. Mutagenic analysis of the coronavirus intergenic consensus sequence. J. Virol. 66:6330-6337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kao, C., M. Zheng, and S. Rudisser. 1999. A simple and efficient method to reduce nontemplated nucleotide addition at the 3′ terminus of RNAs transcribed by T7 RNA polymerase. RNA 5:1268-1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kao, C. C., and J. H. Sun. 1996. Initiation of minus-strand RNA synthesis by brome mosaic virus RNA-dependent RNA polymerase: use of oligoribonucleotide primers. J. Virol. 70:6826-6830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Keck, J. L., E. R. Goedken, and S. Marqusee. 1998. Activation/attenuation model for RNase H. A one-metal mechanism with second-metal inhibition. J. Biol. Chem. 273:34128-34133. [DOI] [PubMed] [Google Scholar]
- 11.Kim, C. H., C. C. Kao, and I. Tinoco. 2000. RNA motifs that determine specificity between a viral replicase and its promoter. Nat. Struct. Biol. 7:415-423. [DOI] [PubMed] [Google Scholar]
- 12.Kim, Y.-C., G. Roberts, M. Thomson, and C. C. Kao. Spectroscopic analysis of the hepatitis C virus RNA polymerase. Submitted for publication.
- 13.Lai, M. M. C., and D. Cavanagh. 1997. The molecular biology of coronaviruses. Adv. Virus Res. 48:1-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Laneve, P., F. Altieri, M. E. Fiori, A. Scaloni, I. Bozzoni, and E. Caffarelli. 2003. Purification, cloning, and characterization of XendoU, a novel endoribonuclease involved in processing of intron-encoded small nucleolar RNAs in Xenopus laevis. J. Biol. Chem. 278:13026-13032. [DOI] [PubMed] [Google Scholar]
- 15.Peiris, J. S., C. M. Chu, V. C. Cheng, K. S. Chan, I. F. Hung, L. L. Poon, K. I. Law, B. S. Tang, T. Y. Hon, C. S. Chan, K. H. Chan, J. S. Ng, B. J. Zheng, W. L. Ng, R. W. Lai, Y. Guan, and K. Y. Yuen. 2003. Clinical progression and viral load in a community outbreak of coronavirus-associated SARS pneumonia: a prospective study. Lancet 361:1767-1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Poutanen, S. M., D. E. Low, B. Henry, S. Finkelstein, D. Rose, K. Green, R. Tellier, R. Draker, D. Adachi, M. Ayers, A. K. Chan, D. M. Skowronski, I. Salit, A. E. Simor, A. S. Slutsky, P. W. Doyle, M. Krajden, M. Petric, R. C. Brunham, and A. J. McGeer. 2003. Identification of severe acute respiratory syndrome in Canada. N. Engl. J. Med. 348:1995-2005. [DOI] [PubMed] [Google Scholar]
- 17.Ranjith-Kumar, C. T., J. Gajewski, L. Gutshall, D. Maley, R. T. Sarisky, and C. C. Kao. 2001. Terminal nucleotidyl transferase activity of recombinant Flaviviridae RNA-dependent RNA polymerases: implication for viral RNA synthesis. J. Virol. 75:8615-8623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ranjith-Kumar, C. T., Y. C. Kim, L. Gutshall, C. Silverman, S. Khandekar, R. T. Sarisky, and C. C. Kao. 2002. Mechanism of de novo initiation of RNA synthesis by the hepatitis C virus RNA-dependent RNA polymerase: role of divalent metals. J. Virol. 76:12513-12525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ranjith-Kumar, C. T., X. Zhang, and C. C. Kao. 2003. Enhancer-like activity of a brome mosaic virus RNA promoter J. Virol. 77:1830-1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rao, P., W. Yuan, and R. M. Krug. 2003. Crucial role of CA cleavage sites in the cap-snatching mechanism for initiating viral mRNA synthesis. EMBO J. 22:1188-1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rost, B. 1996. PHD: predicting one-dimensional protein structure by profile based neural networks. Methods Enzymol. 266:525-539. [DOI] [PubMed] [Google Scholar]
- 22.Rota, P. A., M. S. Oberste, S. S. Monroe, W. A. Nix, R. Campagnoli, J. P. Icenogle, S. Penaranda, B. Bankamp, K. Maher, M. H. Chen, S. Tong, A. Tamin, L. Lowe, M. Frace, J. L. DeRisi, Q. Chen, D. Wang, D. D. Erdman, T. C. Peret, C. Burns, T. G. Ksiazek, P. E. Rollin, A. Sanchez, S. Liffick, B. Holloway, J. Limor, K. McCaustland, M. Olsen-Rasmussen, R. Fouchier, S. Gunther, A. D. Osterhaus, C. Drosten, M. A. Pallansch, L. J. Anderson, and W. J. Bellini. 2003. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 300:1394-1399. [DOI] [PubMed] [Google Scholar]
- 23.Sawicki, S. G., and D. L. Sawicki. 1990. Coronavirus transcription: subgenomic mouse hepatitis virus replicative intermediates function in RNA synthesis. J. Virol. 64:1050-1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sawicki, S. G., and D. L. Sawicki. 1998. A new model for coronavirus transcription. Adv. Exp. Med. Biol. 440:215-219. [DOI] [PubMed] [Google Scholar]
- 25.Snijder, E. J., P. J. Bredenbeek, J. C. Dobbe, V. Thiel, J. Ziebuhr, L. L. Poon, Y. Guan, M. Rozanov, W. J. Spaan, and A. E. Gorbalenya. 2003. Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. J. Mol. Biol. 331:991-1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Spaan, W., D. Cavanagh, and M. C. Horzinek. 1998. Coronaviruses: structure and genome expression. J. Gen. Virol. 69:2939-2952. [DOI] [PubMed] [Google Scholar]
- 27.Sutton, G., E. Fry, L. Carter, S. Sainsbury, T. Walter, J. Nettleship, N. Berrow, R. Owens, R. Gilbert, A. Davidson, S. Siddell, L. L. Poon, J. Diprose, D. Alderton, M. Walsh, J. M. Grimes, and D. I. Stuart. 2004. The nsp9 replicase protein of SARS-coronavirus, structure and functional insights. Structure 12:341-353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van der Most, R. G., R. J. de Groot, and W. J. Spaan. 1994. Subgenomic RNA synthesis directed by a synthetic defective interfering RNA of mouse hepatitis virus: a study of coronavirus transcription initiation. J. Virol. 68:3656-3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Marle, G., J. C. Dobbe, A. P. Gultyaev, W. Luytjes, W. J. Spaan, and E. J. Snijder. 1999. Arterivirus discontinuous mRNA transcription is guided by base pairing between sense and antisense transcription-regulating sequences. Proc. Natl. Acad. Sci. USA 96:12056-12061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.World Health Organization. 15. August 2003, posting date. Summary table of SARS cases by country, 1 November 2002-7 August 2003. [Online.] http://www.who.int/csr/sars/country/en/country2003_08_15.pdf.
- 32.Ziebuhr, J., E. J. Snijder, and A. E. Gorbalenya. 2002. Virus-encoded proteinases and proteolytic processing in the Nidovirales. J. Gen. Virol. 81:853-879. [DOI] [PubMed] [Google Scholar]
- 33.Zuker, M., D. H. Mathews, and D. H. Turner. 1999. Algorithms and thermodynamics for RNA secondary structure prediction: a practical guide, p. 11-43. In J. Barciszewski and B. F. C. Clark (ed.), RNA biochemistry and biotechnology. NATO ASI Series, Kluwer Academic Publishers, Dordrecht, The Netherlands.