

Abstract
Herpes simplex virus (HSV) enters cells by fusion with target membranes, commonly the plasma membrane. In some cells, including CHO cells expressing the nectin1 or herpesvirus entry mediator receptors, entry occurs through an endocytic route. We report the following results. (i) When expressed in J cells, nectin1 and HVEM mediated a pathway of entry insensitive to endosome acidification inhibitors. (ii) A chimeric nectin1 receptor competent for endosomal uptake by fusion of the nectin1 ectodomain with the transmembrane sequence and cytoplasmic tail of the epidermal growth factor receptor (EGFR1) (nectin1-EGFR1) and chimeric nectin1 sorted to lipid rafts by a glycosylphosphatidylinositol anchor mediated endocytic entry blocked by the early endosome inhibitor wortmannin and by the endosome acidification inhibitors bafilomycin and NH4Cl. (iii) Entry mediated by nectin1-EGFR1 was selectively inhibited by AG1478, a tyrosine phosphorylation inhibitor that targets the EGFR1 cytoplasmic tail and blocks the signaling pathway that culminates in clathrin-dependent uptake of the receptor into endosomes. We draw the following conclusions. (i) The same receptor may initiate different routes of infection, depending on the cell in which it is expressed. Hence, the cell is a determinant that controls whether a given receptor initiates a plasma membrane or an endocytic route of entry. (ii) Receptors whose physiology involves uptake into endosomes or sorting to lipid rafts are suitable to serve as HSV receptors. (iii) Structural features of the receptors are additional determinants that control whether HSV entry occurs at the plasma membrane or at endosomes. These findings are relevant to studies of HSV retargeting to specific receptors.
Herpes simplex virus (HSV) enters cells by fusion of the envelope with target membranes. This event follows virus attachment mediated by the binding of glycoprotein C (gC) to the glycosaminoglycan moieties of heparan sulfate proteoglycan and requires virion envelope gD as the receptor-binding glycoprotein (3, 31). gD interacts with two alternative protein receptors, nectin1, an intercellular adhesion molecule belonging to the immunoglobulin (Ig) superfamily, and herpesvirus entry mediator (HVEM), a member of the tumor necrosis factor receptor family (7, 11, 16, 25). The crystal structure of the gD ectodomain showed that it is made of an Ig-folded core with N- and C-terminal extensions (4). The N terminus is disordered in the crystal of gD alone and forms a hairpin when gD is bound to HVEM, testifying to a conformational modification in gD following receptor binding (4). The interaction of gD with its receptor triggers fusion. According to a current model, the triggering event is a conformational modification that ensues in gD after receptor binding, such that the pro-fusion domain of gD, which is proline rich and located membrane proximally in the ectodomain, can recruit or activate three additional fusion glycoproteins, gH, gL, and gB (5). These glycoproteins are conserved throughout the family Herpesvirinae and execute fusion.
It has been a long-held paradigm that the site of HSV fusion with the cell is the plasma membrane (3, 31). Recently, it was found that entry into HeLa cells, but not into Vero cells, is inhibited by NH4Cl or monensin (28, 28a). NH4Cl acts as a weak base which buffers the pH and causes a generalized increase in the intracellular pH, including that of endosomes. Monensin perturbs the ionic gradient of endocytic and exocytic membranes. Furthermore, bafilomycin A1, a highly selective inhibitor of endosome acidification that blocks endosomal ATPase, inhibits HSV entry into CHO cells ectopically expressing human nectin1 (CHO-nectin1) or HVEM (CHO-HVEM) (28, 28a). Cumulatively, these findings indicate that HSV enters some cells through an endocytic pathway and other cells through the plasma membrane.
Because the receptor actually active in Vero cells was not characterized, the above findings could be accounted for either by differences in the receptors being used in Vero versus CHO-nectin1 and CHO-HVEM cells or by the fact that the cell is a determinant that defines the pathway of HSV entry. To discriminate between these two possibilities and to determine whether the receptors themselves are determinants that control the HSV entry pathway, we expressed nectin1 or HVEM in a cell line other than CHO cells (J cells) and modified nectin1 to make it competent either for endosomal uptake or for sorting to lipid rafts. Specifically, in a nectin1-epidermal growth factor (EGF) receptor 1 (EGFR1) chimera, the ectodomain of nectin1 was fused to the transmembrane (TM) sequence and cytoplasmic tail (cyt-tail) of EGFR1, a typical receptor that is endocytosed following binding to its ligand, EGF (2, 13, 30). In the second construct, nectin1-glycosylphosphatidylinisitol (GPI), the nectin1 ectodomain was anchored to the membrane through the GPI lipid moiety (27). We report the following. (i) When ectopically expressed in receptor-negative J cells, wild-type nectin1 mediated a route of entry insensitive to endosome acidification inhibitors. (ii) Both nectin1-EGFR1 and nectin1-GPI were competent to mediate HSV entry. (iii) In contrast to wild-type nectin1, the retargeted receptors mediated a pathway of entry through acidic endosomes when expressed in J cells. Thus, not only the cell but also the structural features of the receptors are key determinants that control the pathway of entry.
MATERIALS AND METHODS
Cells and viruses.
Cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 5% fetal calf serum. The J1.1-2 cell line (J cells), a derivative of BHKtk− cells and highly resistant to HSV infection, was described previously (7). The HSV type 1 (HSV-1) recombinant R8102, expressing β-galactosidase (β-Gal), was described previously (21). Virus infectivity was detected as β-Gal activity by light microscopy observation of β-Gal-expressing cells stained with 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal) or by reading the optical density at 405 nm after the addition of o-nitrophenyl-β-d-galactopyranoside (7, 25).
Plasmids.
pCF18 carries nectin1β cDNA cloned into BstXI and NotI of pcDNA3.1(+) (Invitrogen) (7). Expression plasmids for HSV-1 gD, gB, gH, and gL under the control of the cytomegalovirus immediate-early promoter were described elsewhere; pEA99 carries the gD gene cloned in pcDNA3.1(−); the gB, gH, and gL genes were cloned in MTS-1, a vector derived from pAcSG2 (Pharmingen), by the insertion of the cytomegalovirus immediate-early promoter (23, 34).
Mutagenesis.
pCF18 was mutagenized with the oligonucleotide N1b_990HpaNotKsp (5′-GGT GGA GGT TAA CAT CGC GGC CGC GCC GCG GCC CCA GAG GGG-3′) in order to introduce HpaI (silent), NotI, and KspI (silent) restriction sites as described previously (21). The resulting plasmid, pCF18HNK, contained nectin1β with the substitutions TEK334-336AAA due to the introduction of the NotI restriction site and was sequenced for accuracy.
Generation of chimeric receptors and cell lines.
To construct nectin1-GPI, the sequence encoding the last 37 residues (311 to 347) of human CD55 (decay-accelerating factor [DAF]) (8, 20) was generated by 25 cycles of annealing and extension of an equimolar mixture of synthetic oligonucleotides DAF_5′P_Hpa_f (5′-P-AAC CCA AAT AAA GGA TCC GGA ACC ACT TCA GGT ACT ACC CGT CTT CTA TCT GGG CAC ACG TGT TTC ACG TTG ACA GG-3′; sense; previously phosphorylated at the 5′ end) and DAF_3′Xho_r (5′-CCG CTC GAG CTA AGT CAG CAA GCC CAT GGT TAC TAG CGT CCC AAG CAA ACC TGT CAA CGT GAA ACA CGT GTG CCC-3′; antisense). The DAF_5′P_Hpa_f oligonucleotide was designed with a silent BamHI site for ease of screening. The XhoI-digested 131-bp product containing a blunt phosphorylated HpaI end was ligated to pCF18HNK previously digested with HpaI and XhoI. The resulting plasmid, pNec1-GPI, contained the sequence encoding nectin1 residues 1 to 332 fused to residues 311 to 347 of human CD55. Plasmid p163A2 contains the EGFR1 cDNA as a SacII-XhoI fragment with a unique NaeI site 32 amino acids upstream of the TM sequence (13, 30, 32). The NaeI-XhoI restriction fragment from p163A2 was ligated to pCF18HNK digested with HpaI and XhoI to produce plasmid pNec1-EGFR1. Plasmids encoding the chimeric receptors were transfected into J cells; after selection with neomycin (G418), the cells were cloned by limiting dilution. Clones stably expressing the receptors were designated J-nectin1-EGFR1 and J-nectin1-GPI.
IFA.
For the indirect immunofluorescence assay (IFA), cells were grown on glass coverslips, fixed with 4% paraformaldehyde for 10 min at room temperature or methanol for 10 min at −20°C, incubated with monoclonal antibody (MAb) R1.302 (17) at 1:100 in 20% newborn calf serum in phosphate-buffered saline (PBS) for 1 h at room temperature, and incubated with fluorescein isothiocyanate (FITC)-conjugated anti-mouse immunoglobulin G (IgG) antibody (Jackson Immunoresearch) for 45 min at room temperature. Samples were observed with a Zeiss microscope, and micrographs were taken with a Kodak DC290 digital camera.
Digestion with PI-PLC.
Cells grown on glass coverslips (2 cm2) were incubated with phosphatidylinositol-specific phospholipase C (PI-PLC) obtained from Bacillus cereus (Molecular Probes) and diluted in 25 μl of PBS in amounts ranging from 0.02 to 2 U/coverslip for 20 min at 4°C according to the manufacturer's instructions. Cells were rinsed twice with PBS, fixed with 4% paraformaldehyde in PBS, and assayed for the expression of nectin1 or the chimeric receptors by IFA. For infectivity assays, cells were grown in 1-cm2 wells, treated with PI-PLC diluted in 100 μl of PBS at 0.1 to 0.01 U/well for 20 min at 4°C, washed twice with ice-cold PBS, and infected with R8102 at 10 PFU/cell. β-Gal expression was measured at 6 h after infection (7, 25).
Lipid raft flotation on sucrose gradients.
Membrane fractions containing lipid rafts were prepared essentially as described previously (9). Confluent J cells expressing nectin 1 (J-nectin1 cells) and J-nectin1-GPI cells were trypsinized, washed with PBS, and counted. A total of 107 cells were centrifuged, suspended in 1 ml of TNE buffer (10 mM Tris-HCl [pH 7.5], 150 mM NaCl, 5 mM EDTA) containing 1% Triton X-100 (Sigma Aldrich, Milan, Italy) and 0.036 mg/ml each of the protease inhibitors Nα-p-tosyl-l-lysine chloromethyl ketone (TLCK; Sigma Aldrich) and N-tosyl-l-phenylalanine chloromethyl ketone (TPCK; Sigma Aldrich), and incubated on ice for 1 h. The lysate was homogenized by 15 strokes with a 200-μl pipette and centrifuged at 900 × g for 10 min. The supernatant was diluted 1:1 with 85% sucrose in TNE buffer containing protease inhibitors and was placed at the bottom of an ultracentrifuge tube. A discontinuous sucrose gradient was prepared by the sequential overlay of 6 ml of 35% sucrose and 3.5 ml of 5% sucrose (both in TNE buffer containing protease inhibitors). The gradient was centrifuged at 200,000 × g for 20 h at 4°C in an SW41 swing-out rotor. Eleven 1-ml fractions were collected from the top. The pellet was suspended in fraction 12 (500 μl) at the bottom. The proteins were precipitated with a final trichloroacetic acid concentration of 15% on ice for 2 h, followed by centrifugation at 14,000 × g for 15 min at 4°C in a microcentrifuge. The pellet was washed with cold acetone and air dried.
Western blotting.
The protein samples obtained from the sucrose gradient fractions were resuspended in sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) loading buffer, boiled, resolved by SDS-PAGE, and transferred to Hybond enhanced chemiluminescence (ECL) nitrocellulose membranes (Amersham). The membranes were blocked overnight with 5% nonfat dry milk in PBS at 4°C, washed, and reacted with IgG purified from MAb CK6, directed to the nectin1 ectodomain (Santa Cruz) (15), followed by peroxidase-conjugated anti-mouse IgG antibody (Sigma Aldrich). The blots were developed by ECL (Amersham) according to the manufacturer's instructions.
Cell-cell fusion assay.
BHK cells were seeded at a density of 2 × 104 cells/2-cm2 well and transfected on the following day with a mixture of plasmids encoding gD, gB, gH, and gL (80 ng of each plasmid/well). At 24 h after transfection, effector BHK cells were stained with 10 μM CellTracker Orange CMTMR [5- (and 6-) (((4-chloromethyl)benzoyl)amino)tetramethylrhodamine] (Molecular Probes) in DMEM for 30 min at 37°C. Target cells (J, J-nectin1, J-nectin1-GPI, and J-nectin1-EGFR1) grown in 25-cm2 flasks were stained with 15 μM CellTracker Green CMFDA (5-chloromethylfluorescein diacetate) (Molecular Probes) in DMEM for 30 min at 37°C, trypsinized, and used to seed transfected effector BHK cells at a density of 0.7 × 105 to 1.4 × 105 cells/well. After 24 h, cells were rinsed with PBS, fixed with 4% paraformaldehyde for 10 min at room temperature, and observed with a Zeiss fluorescence microscope. Micrographs were taken with a Kodak DC290 digital camera.
Treatment with endosome inhibitors.
The stock solution of NH4Cl (1.5 M) was prepared immediately prior to use. The stocks solutions of bafilomycin (0.16 M), AG1478 (0.1 M), and wortmannin (2 M) (all from Sigma Aldrich) were prepared in dimethyl sulfoxide, adjusted to pH 7.4, and stored at −20 or 4°C. Cells were exposed to the inhibitors for 30 min at 37°C and then infected with R8102 at 25 PFU/cell for 90 min at 37°C in the presence of the inhibitors. The viral inoculum was removed, and the cells were overlaid with medium containing the inhibitors for 6 h.
RESULTS
HSV entry into J-nectin1 or J-HVEM cells is not inhibited by bafilomycin and NH4Cl.
CHO-nectin1 and CHO-HVEM cells mediate an endocytic pathway of HSV entry (28, 28a). They were therefore not suitable for investigating whether the retargeting of nectin1 to endosomes or its sorting to lipid rafts modifies the pathway of HSV entry. In order to determine whether the route of HSV entry into J-nectin1 cells or J cells expressing HVEM (J-HVEM cells) occurs through acidic endosomes, we tested whether HSV infection of these cells was inhibited by bafilomycin or NH4Cl. We included in the assay CHO-nectin1 and CHO-HVEM cells as controls and a number of cell lines commonly used in HSV research, such as BHK, 143tk−, COS, and HEp-2, that were not characterized in the previous studies (28, 28a). Cells were pretreated with bafilomycin or NH4Cl for 30 min and then infected with R8102, an HSV recombinant that expresses β-Gal under the control of the α27 promoter. The extent of entry was quantified as β-Gal activity. The results shown in Fig. 1 indicate that entry into J-nectin1 or J-HVEM cells was not decreased by either of the two inhibitors. These results contrast with those for entry into CHO-nectin1 or CHO-HVEM cells, which was almost abolished by the inhibitors. Entry into HeLa cells was inhibited by NH4Cl and only moderately decreased by bafilomycin, testifying to the differential effects of these two inhibitors in these cells. All other cell lines mediated a pathway of entry not affected by these two inhibitors, confirming the notion that in the majority of cells, HSV entry is initiated at the plasma membrane. We conclude that the pathway of entry into J-nectin1 and J-HVEM cells differed from that into CHO-nectin1 and CHO-HVEM cells and that J cells were a suitable cellular system for investigating the effects of receptor retargeting.
FIG. 1.
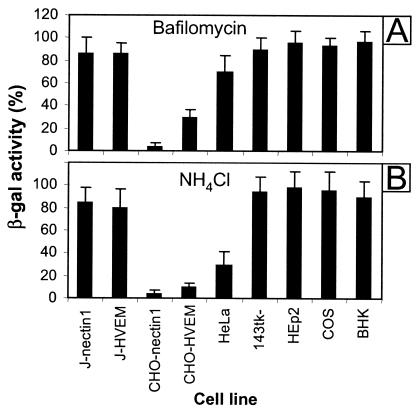
Effects of bafilomycin and NH4Cl on R8102 entry into J-nectin1, J-HVEM, CHO-nectin1, CHO-HVEM, HeLa, 143tk−, HEp-2, COS, and BHK cells. Monolayers of the indicated cell lines were treated with bafilomycin (200 nM) (A) or NH4Cl (45 mM) (B) for 30 min prior to infection with R8102 at 25 PFU/cell for 90 min in the presence of the inhibitors. Unadsorbed virus then was removed, and the cells were overlaid with medium containing the inhibitors for 6 h. The efficiency of virus entry was measured as β-Gal activity and expressed as a percentage of the activity in untreated infected cultures. Each cell line was tested in three independent experiments. The results of a typical experiment are shown. Data represent the mean of triplicates and the standard error.
Construction of the nectin1-EGFR1 chimera.
To construct the nectin1 chimeras, endonuclease restriction sites for HpaI, NotI, and KspI were introduced into nectin1 cDNA by site-directed mutagenesis at residues 332 to 339, i.e., at the site of alternative splicing (amino acid 335) that generates the three natural isoforms of nectin1, α, β, and γ. In the resulting plasmid, pCF18HNK (Fig. 2A, line d), the mutations that introduced the HpaI and KspI sites were silent, while those that introduced the NotI site caused the TEK334-336AAA substitutions. These mutations did not alter the expression of nectin1 at the plasma membrane or its ability to serve as a receptor for HSV entry (data not shown). The nectin1-EGFR1 chimera (Fig. 2A, line f) was generated by ligation to HpaI-XhoI-digested pCF18HNK of an EGFR1 fragment that encompasses 32 amino acids upstream of the TM sequence, the TM sequence itself, and the cyt-tail of EGFR1 (Fig. 2A, line e). The resulting plasmid was designated pNec1-EGFR1. The predicted structure of the nectin1-EGFR1 chimera is schematically represented in Fig. 2B.
FIG. 2.
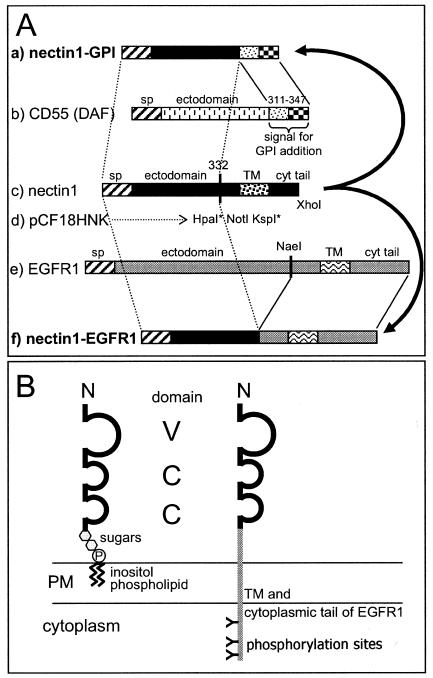
(A) Schematic diagram of construction of chimeric receptors. Numbers indicate amino acid residues. Line c shows a schematic linear map of nectin1. Different patterns indicate the signal peptide (sp) and the TM segment. The sequence encoding residues 332 to 339 was mutagenized to introduce restriction sites HpaI (at amino acids 332 to 333), NotI (at amino acids 334 to 336), and KspI (at amino acids 338 to 339), thus generating pCF18HNK (line d). Asterisks denote silent substitutions. The fragment encoding the TM and cyt-tail regions of nectin1 plus 13 amino acids upstream of the TM sequence was removed by digestion with HpaI and XhoI. The sequence encoding residues 311 to 347 of human CD55 DAF (line b), which enables the addition of the GPI anchor, was synthesized and used to replace the HpaI-XhoI fragment deleted from nectin1. This procedure generated nectin1-GPI (line a). The NaeI-XhoI fragment from EGFR1 (line e), encompassing 32 amino acids upstream of the TM sequence, the TM sequence, and the cyt-tail of EGFR1, was used to replace the HpaI-XhoI fragment deleted from nectin1. This procedure generated nectin1-EGFR1 (line f). Bars are not drawn to scale. (B) Schematic representation of the structures of nectin1-GPI and nectin1-EGFR1 when inserted in the plasma membrane (PM). The nectin1 ectodomain is depicted as containing three Ig domains (loops), one V type and two C type. The sugar moiety of the GPI anchor is represented by hexagons. N, amino terminus. P, phosphate of the GPI anchor. The letter “Y” on its side indicates phosphorylation sites in the cyt-tail of EGFR1. Structures are not drawn to scale.
Construction of the nectin1-GPI chimera.
In order to generate a form of nectin1 anchored to the plasma membrane by means of GPI, we ligated to HpaI-XhoI-digested pCF18HNK a synthetic oligonucleotide encoding the C-terminal 37 amino acids of human CD55 DAF (Fig. 2A, line b). The resulting plasmid, designated pNec1-GPI, encodes a chimeric protein made of nectin1 residues 1 to 332 fused to residues 311 to 347 of human CD55 (Fig. 2A, line a). In the endoplasmic reticulum, the engineered CD55 DAF sequence is cleaved from nectin1 by a GPI-transamidase complex, which thereafter catalyzes the attachment of the resulting new C terminus to the amino group of an ethanolamine residue in the GPI precursor. The final structure of the nectin1-GPI chimera as inserted in cellular membranes is schematically represented in Fig. 2B.
Nectin1-EGFR1 and nectin1-GPI are expressed in J cells and are localized at plasma membranes.
The objective of these experiments was to determine whether transfection of the chimeric receptors into J cells would lead to their expression and plasma membrane localization. J cells were transfected with plasmids pNec1-GPI and pNec1-EGFR1, and cell lines expressing the two receptors were established by neomycin (G418) selection and cloning. The expression of the chimeric receptors was detected by IFA with MAb R1.302 to nectin1. Figure 3a and b show that J-nectin1-GPI and J-nectin1-EGFR1 were expressed in J cells. The pattern of staining indicated that the receptors were localized in part to the plasma membrane.
FIG. 3.
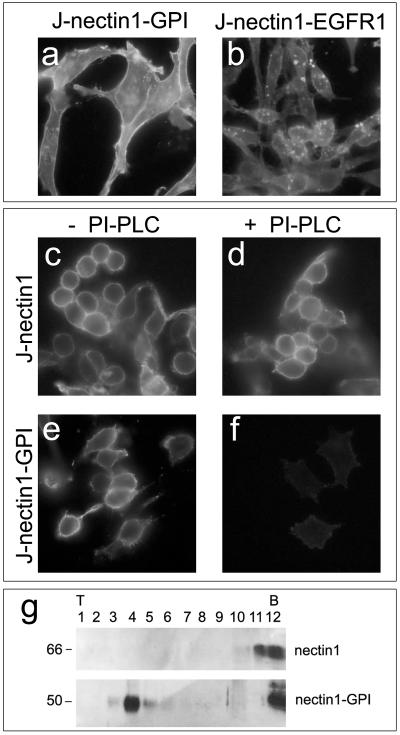
Expression of nectin1-GPI and nectin-EGFR1. Nectin1-GPI is anchored to the plasma membrane through the GPI lipid moiety and is sorted to lipid rafts. Images show J-nectin1-GPI cells (a, e, and f), J-nectin1-EGFR1 cells (b), and J-nectin1 cells (c and d). (a and b) Cells were fixed with methanol and reacted with MAb R1.302 to nectin1, followed by secondary FITC-conjugated antibody. Fluorescence localized at the level of the plasma membrane and intracellularly. (c to f) Cleavage of nectin1 from nectin1-GPI with PI-PLC. J-nectin1 cells (c and d) and J-nectin1-GPI cells (e and f) were mock treated (c and e) or treated with PI-PLC (d and f). Cells then were fixed with paraformaldehyde and reacted with MAb R1.302, followed by secondary FITC-conjugated antibody. (g) Flotation of nectin1-GPI in raft-containing low-density sucrose fractions. J-nectin1-GPI and J-nectin1 cells were solubilized in Triton X-100 and fractionated on sucrose gradients. The proteins were separated by SDS-PAGE, and nectin1 was visualized by Western blotting with MAb CK6 directed to the nectin1 ectodomain, followed by peroxidase-conjugated anti-mouse antibody and ECL. Numbers above panels indicate fractions collected from top (T) to bottom (B); numbers at left indicate the apparent molecular weight.
Next, we determined whether nectin1-GPI was indeed anchored to the plasma membrane through the GPI lipid moiety and whether it was sorted to lipid rafts. J-nectin1-GPI cells were treated with PI-PLC, which specifically cleaves the GPI anchor into myo-inositol-1,2-cyclic phosphate, conjugated to the nectin1 ectodomain, and lipid-soluble diacylglycerol. Figure 3f shows that exposure of J-nectin1-GPI cells to PI-PLC abolished the reactivity to MAb R1.302, demonstrating that the nectin1 portion was cleaved off. To determine the sorting of nectin1-GPI to lipid rafts, lysates of J-nectin1-GPI and J-nectin1 cells made in Triton X-100 were subjected to flotation in discontinuous sucrose gradients. Fractions were analyzed for the presence of nectin1 by Western blotting. Figure 3g shows that in the gradient from J-nectin1-GPI cells, nectin1 was partitioned in the low-density fractions where lipid rafts float; in contrast, in the gradient from J-nectin1 cells, nectin1 did not float and remained in the high-density fractions.
Nectin1-EGFR1 and nectin1-GPI serve as receptors for HSV entry and cell-cell fusion.
To ascertain whether nectin1-EGFR1 and nectin1-GPI are functional receptors for HSV-mediated fusion, we performed a cell-cell fusion assay. Effector BHK cells were cotransfected with expression plasmids encoding the four HSV fusion glycoproteins, gD, gB, gH, and gL. At 24 h after transfection, they were stained with the dye CellTracker CMTMR and overlaid with CellTracker CMFDA-stained target cells, J-nectin1-EGFR1 or J-nectin1-GPI. Syncytium formation led to mixing of the cytoplasm of effector cells and the cytoplasm of target cells, visible as a yellow stain after merging of digital pictures taken with red and green filters. Both J-nectin1-GPI cells (Fig. 4k) and J-nectin1-EGFR1 cells (Fig. 4l) fused with effector BHK cells and could not be differentiated from J cells expressing wild-type nectin1 (Fig. 4j). The results imply that the retargeted receptors were displayed on the cell surface and that their abilities to interact with gD and to trigger fusion were not hampered.
FIG. 4.
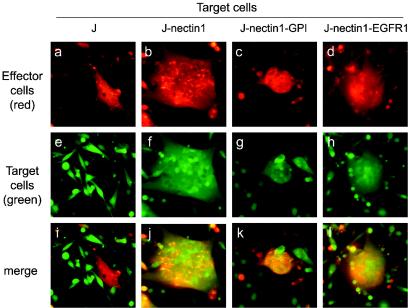
Fusion of J-nectin1, J-nectin1-GPI, and J-nectin1-EGFR1 cells with BHK cells expressing gB, gD, gH, and gL. Effector BHK cells cotransfected with plasmids encoding gD, gB, gH, and gL were stained with CellTracker Orange CMTMR and overlaid with the following target cells stained with CellTracker Green CMFDA: receptor-negative J cells (a, e, and i), J-nectin1 cells (b, f, and j), J-nectin1-GPI cells (c, g, and k), and J-nectin1-EGFR1 cells (d, h, and l). Micrographs were taken 24 h after the target cells were overlaid with red and green filters to visualize effector and target cells, respectively. Cell-cell fusion resulted in mixing of the cytoplasm of effector cells with that of target cells and can be visualized as yellow color in the merged images (i to l). Panels a and e, panels b and f, panels c and g, and panels d and h represent images of a same field taken with the red filter (a to d) and with the green filter (e to h).
Next, we determined whether J-nectin1-EGFR1 and J-nectin1-GPI cells could be infected by HSV. We made use of the R8102 recombinant, which expresses β-Gal under the control of the α27 promoter (22) and is readily quantifiable. Both J-nectin1-EGFR1 cells (Fig. 5a) and J-nectin1-GPI cells (Fig. 5b) were indeed infected by R8102. Furthermore, cleavage of nectin1 from nectin1-GPI by means of PI-PLC significantly reduced infection (Fig. 5h). In addition, we performed a single-step growth curve for R8102 in J cells expressing the retargeted receptors or wild-type nectin1. Figure 5i shows that the yields of R8102 in the three types of cells were approximately the same, indicating that the cells had no defect in productive infection. Altogether, the results show that the retargeted receptors were competent to mediate HSV entry and cell-cell fusion.
FIG. 5.
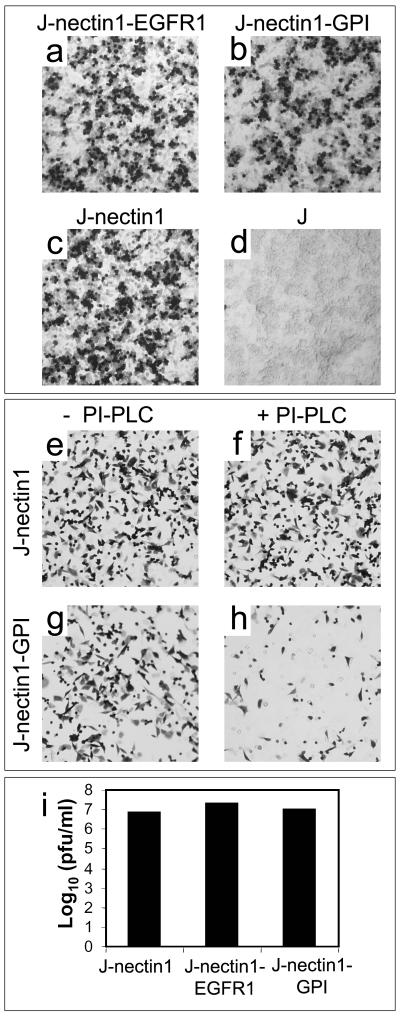
(a to d) The nectin1-EGFR1 and nectin1-GPI chimeras serve as HSV receptors. Micrographs show J-nectin1-EGFR1 cells (a), J-nectin1-GPI cells (b), J-nectin1 cells (c), and receptor-negative J cells (d) infected with R8102 and stained with X-Gal. Note that J cells expressing the chimeric receptors are susceptible to HSV infection, while J cells are not. (e to h) Treatment of J-nectin1-GPI cells with PI-PLC reduces HSV infectivity. J-nectin1 cells (e and f) and J-nectin1-GPI cells (g and h) were mock treated (e and g) or treated with PI-PLC (f and h), infected with R8102, and stained with X-Gal at 6 h after infection. The treatment specifically reduced infectivity in J-nectin1-GPI cells but not in J-nectin1 cells. (i) One-step growth of R8102 in J-nectin1, J-nectin1-EGFR1, and J-nectin1-GPI cells. Cells were infected at 25 PFU/cell. Progeny virus was harvested at 24 h after infection and titrated on Vero cells.
HSV entry into J-nectin1-EGFR1 and J-nectin1-GPI cells is inhibited by bafilomycin, NH4Cl, and wortmannin.
We next examined whether the pathway of HSV entry into J-nectin1-EGFR1 and J-nectin1-GPI cells differed from that into J-nectin1 cells and determined the sensitivity to two types of inhibitors: wortmannin, an inhibitor of early endosomes, and inhibitors of endosome acidification. Soon after early endosomes are formed, they may be sorted to different routes, including maturation to multivesicular bodies and late endosomes and recycling to the cell surface. This sorting is regulated by an effector domain consisting of the active GTP-bound Rab5 and phosphatidylnositol 3-phosphate-binding proteins, including phosphatidylinositol 3-kinases (12). The latter are blocked by wortmannin. J-nectin1-EGFR1 and J-nectin1-GPI cells were pretreated with increasing concentrations of the inhibitors and then infected with R8102. J-nectin1 cells, HeLa cells, CHO-nectin1 cells, and CHO-HVEM cells were included in the assay. The efficiency of entry was quantified as β-Gal activity. As shown in Fig. 6A, entry into J-nectin1-EGFR1 and J-nectin1-GPI cells was decreased in a dose-dependent fashion by wortmannin which, at the highest concentration, abolished entry. The curve differed considerably from that obtained with J-nectin1 cells, where no significant inhibition was observed. Of note, CHO-nectin1 cells exhibited a different dose-response curve and an overall lower extent of inhibition relative to J-nectin1-EGFR and J-nectin1-GPI cells. HeLa cells, which displayed low levels of sensitivity to NH4Cl and particularly to bafilomycin (Fig. 1), behaved in an intermediate fashion between CHO-nectin1 cells and J-nectin1-EGFR1 and J-nectin1-GPI cells (Fig. 6A). The bafilomycin and NH4Cl dose-response curves for J-nectin1-EGFR1 and J-nectin1-GPI cells are shown in Fig. 6B and C and indicate almost complete inhibition with both compounds. These results are particularly significant given that bafilomycin is a highly specific inhibitor of endosome ATPase. Cumulatively, the results indicate that the pathway of entry into J cells mediated by the retargeted nectin1-EGFR1 and nectin1-GPI receptors differed considerably from the pathway of entry mediated by wild-type nectin1 and that entry was decreased by both the early endosome inhibitor wortmannin and inhibitors of endosome acidification: Thus, entry mediated by the retargeted receptors involves an acidic endosome compartment, whereas entry mediated by the wild-type receptor does not.
FIG. 6.
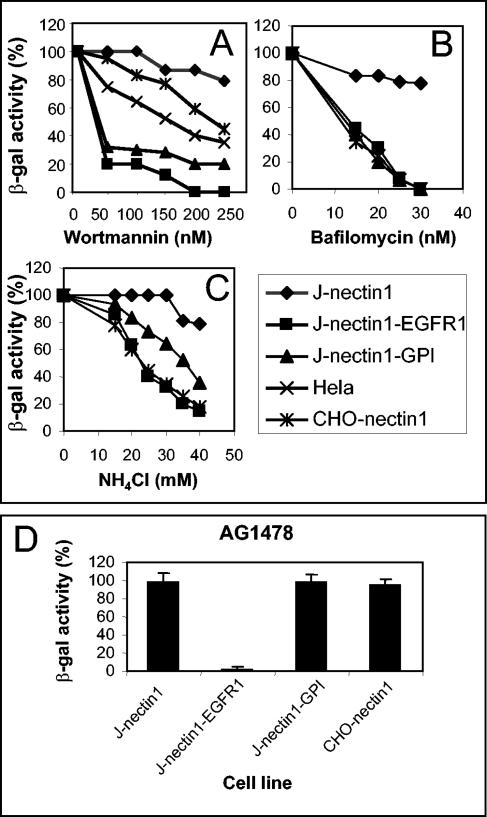
Effect of wortmannin, bafilomycin, NH4Cl, and AG1478 on HSV-1 infectivity in J-nectin1, J-nectin1-EGFR1, J-nectin1-GPI, and CHO-nectin1 cells. (A to C) Cells grown in 96 wells were exposed to increasing concentrations of wortmannin (A), bafilomycin (B), or NH4Cl (C) for 30 min, infected with R8102 at 25 PFU/cell for 90 min in the same medium, and overlaid with medium containing the inhibitors for 6 h. Virus entry was measured as β-Gal activity and was expressed as a percentage of the value obtained in the absence of the inhibitors. Two to four independent experiments were performed. A typical experiment is shown. Data represent the mean of triplicates. (D) Cells grown in 96 wells were exposed to 100 mM AG1478 for 30 min, infected with R8102 at 25 PFU/cell for 90 min in the same medium, and overlaid with medium containing AG1478 for 6 h. Virus entry was measured and expressed as described for panels A to C. Bars represent standard errors.
HSV entry into J-nectin1-EGFR1 cells but not into J-nectin1-GPI or J-nectin1 cells is inhibited by the tyrosine kinase inhibitor AG1478.
The binding of EGF to its receptor induces the autophosphorylation of tyrosine residues located in the EGFR1 cyt-tail. This signaling activity results in the interaction of the EGFR1 cyt-tail with the clathrin-associated AP-2 protein complex and is responsible for initiating endocytosis of the receptor (2, 30). Here we wanted to determine whether the endocytic pathway of HSV entry into J-nectin1-EGFR1 cells involves autophosphorylation of the EGFR1 cyt-tail, likely induced by the binding of virion gD to nectin1. AG1478 is a highly specific inhibitor of the protein tyrosine kinases which catalyze the phosphorylation of tyrosine residues. J-nectin1-EGFR1 cells and, for comparison, J-nectin1-GPI, J-nectin1, and CHO-nectin1 cells were pretreated with AG1478 and then infected with R8102. Entry was quantified as β-Gal activity. Figure 6D shows that AG1478 selectively inhibited HSV infection of J-nectin1-EGFR1 cells but not of J-nectin1-GPI, J-nectin1, and CHO-nectin1 cells. These results indicate that the nectin1-EGFR1 chimera is able to respond to virion binding through tyrosine phosphorylation and that this autophosphorylation is critical for initiation of the acidic endosome pathway of HSV entry mediated by nectin1-EGFR1.
DISCUSSION
The site of HSV virion fusion with target membranes is the plasma membrane in some cells and an endosomal compartment in other cells (3, 28, 28a, 31). The significance of this difference is unclear at present and may reflect the need for fusion factors provided by the cell or differences in the properties of the receptors used in different cells. Of note, the two well-characterized human receptors, nectin1 and HVEM, when expressed in CHO cells, initiate an endocytic pathway of entry that is sensitive to inhibitors of endosome acidification (28). Surprisingly, when nectin1 and HVEM were expressed in receptor-negative J cells, the pathway of entry was not sensitive to inhibitors of endosome acidification (bafilomycin and NH4Cl) or to wortmannin, an inhibitor of phosphatidylinositol 3-kinases that regulate a number of processes; these processes include the differentiation of early endosomes to multivesicular bodies and late endosomes and plasma membrane recycling (12, 19). The actual site of HSV entry into J-nectin1 and J-HVEM cells remains to be identified and may be either the plasma membrane or a nonacidic endocytic compartment. Irrespective of this, our results clearly indicate that the routes of entry differed when the same receptors were expressed in the context of CHO or J cells. We infer that the cell is a determinant that controls whether the same receptor will mediate entry at the plasma membrane or through an endocytic pathway.
Endocytosis is a very complex and broad phenomenon that includes several different processes, such as pynocytosis, receptor-mediated endocytosis, and fluid-phase internalization, and that involves different endocytic compartments, such as clathrin-coated vesicles that mature into early endosomes and caveolae (19). The rationale of our approach was to modify nectin1 and create two receptors with different predicted properties. EGFR1 is a typical receptor that, upon binding to EGF, is taken up by clathrin-coated vesicles that mature into early endosomes (2, 13, 30); the rationale for the construction of nectin1-EGFR1 was to render nectin1 competent for acidic endosomal uptake. For the construction of nectin1-GPI, the rationale was to make nectin1 competent for sorting to lipid rafts, which serve as platforms for receptor clustering and enable endocytosis of the receptors into caveolae; these may be caveolin positive or sometimes caveolin negative (27). A previous report showed that neither nectin1 nor gD locates to lipid rafts when HSV enters cells, whereas gB does (1). Both retargeted receptors were capable of carrying out cell-cell fusion, indicating that they were properly exposed on the plasma membrane and functional. More importantly, when expressed in J cells, they were competent to mediate HSV entry, confirming and extending the notion that the TM region and the cyt-tail of nectin1 do not play a critical role and that they can be readily exchanged with heterologous regions or with a lipid anchor (6, 10).
The key result of this investigation was that entry into J cells mediated by nectin1-EGFR1 or nectin1-GPI was blocked by wortmannin, an inhibitor of early endosomes, and by endosome acidification inhibitors, demonstrating that the retargeting was successful and that entry occurred through acidified endosomes. We infer that receptors that have the inherent ability to be taken up by acidic endosomes or to be sorted to lipid rafts may serve as HSV receptors. In addition, we infer that the structural characteristics of receptors are additional determinants that control the choice between different pathways of HSV entry.
Viruses that enter through endosomes are equipped with fusion glycoproteins that require a low pH for conformational modifications and conversion to a fusion-active state. HSV differs in that the viral glycoproteins are equally able to carry out fusion at the plasma membrane and in an acidic endosomal compartment, with two obvious implications: they do not require low-pH-induced modifications to trigger fusion, and low pH is not detrimental to their activity. Cumulatively, these considerations further strengthen the conclusion that some of the determinants that control whether HSV entry occurs at the plasma membrane or in an endosomal compartment are not the viral glycoproteins but are intrinsic properties of the receptors. Furthermore, the retargeted receptors were able to carry out both endocytic entry and cell-cell fusion; hence, they were equally active in acidic endosomes and at the plasma membrane. The question then arises as to why, in the presence of wortmannin or of endosome acidification inhibitors, entry did not take place at the level of the primary endosome. A likely explanation is that the membrane of the primary endosome is not suitable for fusion, possibly because of its association with proteins such as clathrin, AP-2, or other connectors.
Our results further suggest that the two retargeted receptors mediated HSV uptake into different endocytic compartments. The findings that entry into nectin1-EGFR1 cells but not entry into nectin1-GPI cells was inhibited by the protein tyrosine kinase inhibitor AG1478 and that the NH4Cl dose-response curves with the two receptors differed strongly argue that nectin1-EGFR1 and nectin1-GPI induce an initial internalization of HSV into different types of vesicles, both of which eventually mature into acidified endosomes.
As noted by Nicola et al., HSV entry into HeLa cells is reduced by endosome inhibitors (28). We further report here that entry is reduced by wortmannin. However, in our determinations, HeLa cells were less sensitive to all of the inhibitors than nectin1-EGFR1 and nectin1-GP cells. The reason for this result is unclear at present. Given that the only functional HSV receptor in HeLa cells is nectin1 (7), this lower sensitivity cannot be accounted for by the receptor that is used and may reflect strain differences among HeLa cells that result in a lower efficiency of uptake of the inhibitors, their degradation, and so forth. Nevertheless, the finding that HSV entry into a human epithelial cell line involves an endocytic compartment implies that this route of entry may be relevant for virus infection of the human host.
There are some notable analogies and differences between HSV and Epstein-Barr virus (EBV) routes of entry. Like HSV, EBV is able to exploit different routes of entry, namely, the plasma membrane in epithelial cells and the endocytic compartment in B lymphocytes (24, 26, 33). In contrast to the routes of entry used by HSV, for which the quartet of gD, gB, gH, and gL and one of the two gD receptors are sufficient to enter any cell line tested (3, 14, 28a, 31), the two different routes of entry used by EBV involve different entities. Thus, gB, gH, and gL are required for infection of any cell line; however, gp42 and its HLA class II cognate receptor are required for infection of B cells but not of epithelial cells. Irrespective of the route of entry, the pH of the involved compartment is irrelevant, a further difference between EBV and HSV (24, 26, 33).
Understanding how HSV enters the cell is important not only for elucidation of the molecular basis of this process but also for application of HSV as a vector or as a therapeutic agent. Thus, HSV is a promising virus for use as an oncolytic agent in the therapy of glioblastoma and as a lytic agent in the therapy of restenosis (18, 29). Some applications are under phase I clinical experimentation. HSV might also be used as a vector for heterologous vaccination. All of these applications require that HSV be retargeted to the specific cells that need to be infected and not to the cells normally infected by the virus. A strategy for retargeting HSV to a specific receptor was described recently (35). Our study has relevant implications in the design of HSV-based therapeutic agents and viral vectors, as it establishes that receptors whose physiology involves acidic endosomes or sorting to lipid rafts may be functional for HSV entry and can be considered suitable candidates for HSV retargeting.
Acknowledgments
We thank L. Beguinot (DIBIT, San Raffaele University, Milan, Italy) for the gift of plasmid p163A2. We are grateful to Elisabetta Romagnoli for invaluable help with cell cultures.
These studies were supported by FIRB autonomous project, FIRB coordinated project, Cofin-MIUR 40% 2002 and 2003, CNR-Functional Genomics, University of Bologna 60%, and Fondo Pallotti.
REFERENCES
- 1.Bender, F. C., J. C. Whitbeck, M. Ponce de Leon, H. Lou, R. J. Eisenberg, and G. H. Cohen. 2003. Specific association of glycoprotein B with lipid rafts during herpes simplex virus entry. J. Virol. 77:9542-9552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bishayee, A., L. Beguinot, and S. Bishayee. 1999. Phosphorylation of tyrosine 992, 1068, and 1086 is required for conformational change of the human epidermal growth factor receptor c-terminal tail. Mol. Biol. Cell 10:525-536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Campadelli-Fiume, G., F. Cocchi, L. Menotti, and M. Lopez. 2000. The novel receptors that mediate the entry of herpes simplex viruses and animal alphaherpesviruses into cells. Rev. Med. Virol. 10:305-319. [DOI] [PubMed] [Google Scholar]
- 4.Carfi, A., S. H. Willis, J. C. Whitbeck, C. Krummenacher, G. H. Cohen, R. J. Eisenberg, and D. C. Wiley. 2001. Herpes simplex virus glycoprotein D bound to the human receptor HveA. Mol. Cell 8:169-179. [DOI] [PubMed] [Google Scholar]
- 5.Cocchi, F., D. Fusco, L. Menotti, T. Gianni, R. J. Eisenberg, G. H. Cohen, and G. Campadelli-Fiume. The soluble ectodomain of herpes simplex virus gD contains a membrane-proximal pro-fusion domain and suffices to mediate virus entry. Proc. Natl. Acad. Sci. USA 101:7445-7450. [DOI] [PMC free article] [PubMed]
- 6.Cocchi, F., M. Lopez, P. Dubreuil, G. Campadelli-Fiume, and L. Menotti. 2001. Chimeric nectin1-poliovirus receptor molecules identify a nectin1 region functional in herpes simplex virus entry. J. Virol. 75:7987-7994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cocchi, F., L. Menotti, P. Mirandola, M. Lopez, and G. Campadelli-Fiume. 1998. The ectodomain of a novel member of the immunoglobulin superfamily related to the poliovirus receptor has the attributes of a bona fide receptor for herpes simplex viruses 1 and 2 in human cells. J. Virol. 72:9992-10002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davitz, M. A., M. G. Low, and V. Nussenzweig. 1986. Release of decay-accelerating factor (DAF) from the cell membrane by phosphatidylinositol-specific phospholipase C (PIPLC). Selective modification of a complement regulatory protein. J. Exp. Med. 163:1150-1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dykstra, M. L., R. Longnecker, and S. K. Pierce. 2001. Epstein-Barr virus coopts lipid rafts to block the signaling and antigen transport functions of the BCR. Immunity 14:57-67. [DOI] [PubMed] [Google Scholar]
- 10.Geraghty, R. J., A. Fridberg, C. Krummenacher, G. H. Cohen, R. J. Eisenberg, and P. G. Spear. 2001. Use of chimeric nectin-1 (HveC)-related receptors to demonstrate that ability to bind alphaherpesvirus gD is not necessarily sufficient for viral entry. Virology 285:366-375. [DOI] [PubMed] [Google Scholar]
- 11.Geraghty, R. J., C. Krummenacher, G. H. Cohen, R. J. Eisenberg, and P. G. Spear. 1998. Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science 280:1618-1620. [DOI] [PubMed] [Google Scholar]
- 12.Gruenberg, J. 2003. Lipids in endocytic membrane transport and sorting. Curr. Opin. Cell Biol. 15:382-388. [DOI] [PubMed] [Google Scholar]
- 13.Helin, K., and L. Beguinot. 1991. Internalization and down-regulation of the human epidermal growth factor receptor are regulated by the carboxyl-terminal tyrosines. J. Biol. Chem. 266:8363-8368. [PubMed] [Google Scholar]
- 14.Krummenacher, C., F. Baribaud, M. Ponce De Leon, I. Baribaud, J. C. Whitbeck, R. Xu, G. H. Cohen, and R. J. Eisenberg. 2004. Comparative usage of herpesvirus entry mediator A and nectin-1 by laboratory strains and clinical isolates of herpes simplex virus. Virology 322:286-299. [DOI] [PubMed] [Google Scholar]
- 15.Krummenacher, C., I. Baribaud, M. Ponce De Leon, J. C. Whitbeck, H. Lou, G. H. Cohen, and R. J. Eisenberg. 2000. Localization of a binding site for herpes simplex virus glycoprotein D on herpesvirus entry mediator C by using antireceptor monoclonal antibodies. J. Virol. 74:10863-10872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lopez, M., F. Eberlé, M. G. Mattei, J. Gabert, F. Birg, F. Bardin, C. Maroc, and P. Dubreuil. 1995. Complementary DNA characterization and chromosomal localization of a human gene related to the poliovirus receptor-encoding gene. Gene 155:261-265. [DOI] [PubMed] [Google Scholar]
- 17.Lopez, M., F. Jordier, F. Bardin, L. Coulombel, C. Chabannon, and P. Dubreuil. 1997. Identification of a new class of IgG superfamily antigens expressed in hemopoiesis, p. 1081-1083. In T. Kishimoto, H. Kikutani, A. E. G. von dem Borne, S. M. Goyert, D. Y. Mason, M. Miyasaka, A. Moretta, K. Okumura, S. Shaw, T. A. Springer, K. Sugamura, and H. Zola (ed.), Leukocyte typing VI: white cell differentiation antigens. Garland Publishing, Inc., New York, N.Y.
- 18.Markert, J. M., G. Y. Gillespie, R. R. Weichselbaum, B. Roizman, and R. J. Whitley. 2000. Genetically engineered HSV in the treatment of glioma: a review. Rev. Med. Virol. 10:17-30. [DOI] [PubMed] [Google Scholar]
- 19.Maxfield, F. R., and T. E. McGraw. 2004. Endocytic recycling. Nat. Rev. Mol. Cell. Biol. 5:121-132. [DOI] [PubMed] [Google Scholar]
- 20.Medof, M. E., E. I. Walter, W. L. Roberts, R. Haas, and T. L. Rosenberry. 1986. Decay accelerating factor of complement is anchored to cells by a C-terminal glycolipid. Biochemistry 25:6740-6747. [DOI] [PubMed] [Google Scholar]
- 21.Menotti, L., R. Casadio, C. Bertucci, M. Lopez, and G. Campadelli-Fiume. 2002. Substitution in the murine nectin1 receptor of a single conserved amino acid at a position distal from the herpes simplex virus gD binding site confers high-affinity binding to gD. J. Virol. 76:5463-5471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Menotti, L., F. Cocchi, and G. Campadelli-Fiume. 2002. Critical residues in the CC′ ridge of the human nectin1 receptor V domain enable herpes simplex virus entry into the cell and act synergistically with the downstream region. Virology 301:6-12. [DOI] [PubMed] [Google Scholar]
- 23.Menotti, L., M. Lopez, E. Avitabile, A. Stefan, F. Cocchi, J. Adelaide, E. Lecocq, P. Dubreuil, and G. Campadelli-Fiume. 2000. The murine homolog of human Nectin1delta serves as a species nonspecific mediator for entry of human and animal alpha herpesviruses in a pathway independent of a detectable binding to gD. Proc. Natl. Acad. Sci. USA 97:4867-4872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miller, N., and L. M. Hutt-Fletcher. 1992. Epstein-Barr virus enters B cells and epithelial cells by different routes. J. Virol. 66:3409-3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Montgomery, R. I., M. S. Warner, B. J. Lum, and P. G. Spear. 1996. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell 87:427-436. [DOI] [PubMed] [Google Scholar]
- 26.Mullen, M. M., K. M. Haan, R. Longnecker, and T. S. Jardetzky. 2002. Structure of the Epstein-Barr virus gp42 protein bound to the MHC class II receptor HLA-DR1. Mol. Cell 9:375-385. [DOI] [PubMed] [Google Scholar]
- 27.Nabi, I. R., and P. U. Le. 2003. Caveolae/raft-dependent endocytosis. J. Cell Biol. 161:673-677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nicola, A. V., A. M. McEvoy, and S. E. Straus. 2003. Roles for endocytosis and low pH in herpes simplex virus entry into HeLa and Chinese hamster ovary cells. J. Virol. 77:5324-5332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28a.Nicola, A. V., and S. E. Straus. 2004. Cellular and viral requirements for rapid endocytic entry of herpes simplex virus. J. Virol. 78:7508-7517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Skelly, C. L., M. A. Curi, S. L. Meyerson, D. H. Woo, D. Hari, J. E. Vosicky, S. J. Advani, H. J. Mauceri, S. Glagov, B. Roizman, R. R. Weichselbaum, and L. B. Schwartz. 2001. Prevention of restenosis by a herpes simplex virus mutant capable of controlled long-term expression in vascular tissue in vivo. Gene Ther. 8:1840-1846. [DOI] [PubMed] [Google Scholar]
- 30.Sorkin, A., M. Mazzotti, T. Sorkina, L. Scotto, and L. Beguinot. 1996. Epidermal growth factor receptor interaction with clathrin adaptors is mediated by the Tyr974-containing internalization motif. J. Biol. Chem. 271:13377-13384. [DOI] [PubMed] [Google Scholar]
- 31.Spear, P. G., R. J. Eisenberg, and G. H. Cohen. 2000. Three classes of cell surface receptors for alphaherpesvirus entry. Virology 275:1-8. [DOI] [PubMed] [Google Scholar]
- 32.Ullrich, A., L. Coussens, J. S. Hayflick, T. J. Dull, A. Gray, A. W. Tam, J. Lee, Y. Yarden, T. A. Libermann, J. Schlessinger, et al. 1984. Human epidermal growth factor receptor cDNA sequence and aberrant expression of the amplified gene in A431 epidermoid carcinoma cells. Nature 309:418-425. [DOI] [PubMed] [Google Scholar]
- 33.Wang, X., W. J. Kenyon, Q. Li, J. Mullberg, and L. M. Hutt-Fletcher. 1998. Epstein-Barr virus uses different complexes of glycoproteins gH and gL to infect B lymphocytes and epithelial cells. J. Virol. 72:5552-5558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou, G., E. Avitabile, G. Campadelli-Fiume, and B. Roizman. 2003. The domains of glycoprotein D required to block apoptosis induced by herpes simplex virus 1 are largely distinct from those involved in cell-cell fusion and binding to nectin1. J. Virol. 77:3759-3767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou, G., G. Ye, W. Debinski, and B. Roizman. 2002. Engineered herpes simplex virus 1 is dependent on IL13Ralpha 2 receptor for cell entry and independent of glycoprotein D receptor interaction. Proc. Natl. Acad. Sci. USA 99:15124-15129. [DOI] [PMC free article] [PubMed] [Google Scholar]