

Abstract
The HERV-W family contains hundreds of loci diversely expressed in several physiological and pathological contexts. A unique locus termed ERVWE1 encodes an envelope glycoprotein (syncytin) involved in hominoid placental physiology. Here we show that syncytin expression is regulated by a bipartite element consisting of a cyclic AMP (cAMP)-inducible long terminal repeat (LTR) retroviral promoter adjacent to a cellular enhancer conferring a high level of expression and placental tropism. Deletion mutant analysis showed that the ERVWE1 5′ LTR contains binding sites essential for basal placental activity in the region from positions +1 to +125. The region from positions +125 to +310 represents a cAMP-responsive core HERV-W promoter active in all cell types. Site-directed mutagenesis analysis highlighted the complexity of U3 regulation. ERVWE1 placenta-specific positive (e.g., T240) and negative (e.g., G71) regulatory sites were identified, as were essential sites required for basic activity (e.g., A247). The flanking sequences of the ERVWE1 provirus contain several putative regulatory elements. The upstream HERV-H and HERV-P LTRs were found to be inactive. Conversely, the 436-bp region located between the HERV-P LTR and ERVWE1 was shown to be an upstream regulatory element (URE) which is significantly active in placenta cells. This URE acts as a tissue-specific enhancer. Genetic and functional analyses of hominoid UREs revealed large differences between UREs of members of the Hominidae and the Hylobatidae. These data allowed the identification of a positive regulatory region from positions −436 to −128, a mammalian apparent LTR retrotransposon negative regulatory region from positions −128 to −67, and a trophoblast-specific enhancer (TSE) from positions −67 to −35. Putative AP-2, Sp-1, and GCMa binding sites are essential constituents of the 33-bp TSE.
The HERV-W family (5) was initially characterized by screening a placental cDNA library with a polymerase probe derived from a retroviral sequence, multiple sclerosis-associated retrovirus (38), isolated from biological samples from multiple sclerosis patients. The human genome contains about 311 copies of complete and partial HERV-W proviruses, processed pseudogenes of HERV-W generated by long interspersed nuclear elements, and an additional 343 solitary HERV-W long terminal repeats (LTRs) (36). The highest level of expression of the HERV-W multicopy family was detected in the placenta (5, 34). Nevertheless, a lower but highly variable level of mRNA expression was reported in several other physiological and pathological contexts. More precisely, this family was found to be expressed in testis by Northern blotting (34), in fetal liver-spleen by analysis of expressed sequence tags (5), and in many other healthy tissues, including breast, colon, kidneys, skin, spleen, and thyroid, by real-time quantitative PCR (11). Moreover, reverse transcription-PCR allowed the detection of expression in pathological contexts such as multiple sclerosis (38), rheumatoid arthritis (17), schizophrenia (21), and breast carcinoma cells (44). Thus, although HERV-W expression was detected in several contexts, no direct link between mRNA expression and (re)activation of a specific HERV-W locus (or a subset of loci) has been demonstrated to date (15, 20). This simple statement suggests the existence of complex regulatory mechanisms within the HERV-W family.
A notable exception is the ERVWE1 locus (Online Mendelian Inheritance in Man accession number 604659), which produces most of the HERV-W transcripts detected in the placenta (5). The ERVWE1 locus consists of a complete provirus (human chromosome 7q21.2; GenBank accession number AC000064; positions 28068 to 38289) containing gag and pol pseudogenes and an env gene flanked by two LTRs. In addition, a 2-kb intron is located just downstream of the 5′ LTR (5). This locus was shown to be the only member of the whole family which retained a complete env open reading frame (48). The ERVWE1 Env protein was detected in vivo in the syncytiotrophoblast layer of the placenta (6, 14). ERVWE1 Env, also dubbed syncytin, was proven to be a highly fusogenic protein in vitro (6, 34) and is functionally preserved among hominoids (32). This protein has been shown to be involved in human trophoblastic cell fusion and differentiation (14, 34). A correlation between HERV-W expression in the placenta and the ERVWE1 locus is supported by two main arguments (5). First, most of the isolated HERV-W cDNA clones derived from the placenta exhibit 99 to 100% similarity to the ERVWE1 locus. Second, the simple pattern of mRNA expression observed by Northern blot analysis was compatible with (i) 5′ LTR U3 region-driven transcription, (ii) splice sites identified in the ERVWE1 locus, and (iii) splice junctions identified in HERV-W placental cDNA clones.
The U3 region of the ERVWE1 5′ LTR contains putative transcription factor binding sites, including a CAAT box 43 bp distant from a TATA box located 23 bp upstream from the R CAP site (5). U3 was shown to be a functional promoter in placental BeWo choriocarcinoma cells (32). Similar maintenance of active U3 promoters in other HERV families has also been reported. Thus, the retroviral Rec regulatory protein expressed in teratocarcinoma cells was controlled by its own HERV-K 5′ LTR (30, 31). In addition, several HERV-E LTRs were found to promote adjacent cellular gene expression in the placenta; these LTRs included those for pleiotrophin (45), the endothelin B receptor (33), and the MID-1 gene (25). The promoter role of the ERVWE1 5′ LTR was strengthened by the functional preservation demonstrated in the human population and in orthologous loci isolated from chimpanzee, gorilla, orangutan, and gibbon (32). Interestingly, a comparison of the human ERVWE1 5′ LTR with other HERV-W LTRs revealed some specific signatures, apparently correlated with promoter efficiency and tropism (32). These data were supported by the strict sequence conservation of the U3 regions among 48 human alleles (32). Nevertheless, the poor conservation of these signatures through hominoid evolution and the fact that the experimentally tested U3 regions contained flanking sequences raised the question of the putative influence of integration context on placental tropism (32). In keeping with these data, an HERV-H element located 7,082 to 1,001 bp upstream from the ERVWE1 locus (39) represents a candidate regulatory element considering the high level of expression of HERVs in the placenta. In addition, two recent reports supported a transcriptional role for the ERVWE1 upstream region. First, the placenta-specific GCMa transcription factor was shown to regulate syncytin gene expression via two GCMa binding sites located 2,530 and 42 bp upstream from the ERVWE1 5′ LTR (52). Second, Cheng et al. (9) recently reported that upstream regions spanning nucleotides (nt) −1343 to −808 upstream from the retroviral LTR and nt −119 to +28 within U3 were required for maximal expression in normal trophoblastic cells.
We performed transcription analyses with several cell types in order to compare the efficiencies of a set of LTR mutants, i.e., by swapping distinct HERV-W LTR mutants, ERVWE1 5′ LTR deletion mutants, and ERVWE1 5′ LTR mutants bordered by up to 7 kb of upstream and 2.5 kb of downstream flanking sequences. We showed that the syncytin promoter is composed of a bipartite element consisting of a cyclic AMP (cAMP)-inducible retroviral promoter adjacent to a cellular enhancer conferring a high level of expression and placental tropism. Analysis of LTR mutants in various cell lines confirmed the presence of placenta-specific signatures and exemplified general features of the regulation of HERV-W elements. Analysis of hominoid cellular enhancers and site-directed mutants allowed us to identify putative AP-2, Sp-1, and GCMa binding sites as essential constituents of a 33-bp trophoblast-specific enhancer (TSE).
MATERIALS AND METHODS
Isolation of LTRs by PCR.
Human (Homo sapiens) (Promega) and chimpanzee (Pan troglodytes), gorilla (Gorilla gorilla), orangutan (Pongo pygmaeus), and gibbon (Hylobates pileatus) (Quantum Biotechnologies Inc, Canada) genomic DNAs were used. All HERV-W LTRs were obtained by PCR amplification from the above DNAs with the oligonucleotides listed in Table 1. Each reaction tube contained a maximum of 250 ng of DNA, 330 nM each primer, 1.5 mM MgCl2, 250 μM deoxynucleoside triphosphate, and 2 U of DyNAzyme II (Finnzymes OY, Helsinki, Finland) in 50 μl of PCR buffer (DyNAzyme EXT Mg-free 10× buffer; Finnzymes OY). PCR cycling conditions were 5 min at 94°C; 30 cycles of 1 min at 94°C, 1 min at 52°C, and elongation at 72°C for a time depending of the length of the PCR product (1 min/kb); and a final extension at 72°C for 5 min. Amplifications were performed on an Applied Biosystems 9700 apparatus. PCR products were analyzed on a 1% agarose gel. Short and long PCR fragments were cloned into pCR2.1-TOPO and pCR-XL-TOPO (InVitrogen), respectively. Vectors were transformed into Escherichia coli TOP10 cells, and DNA was extracted from different clones and sequenced.
TABLE 1.
Oligonucleotides used to amplify, clone, and mutate HERV-W transcriptional elementsa
| Oligonucleotide | Sequence (5′→3′) | Description |
|---|---|---|
| F6460 | TTG-GTA-CCC-AAA-ACG-CCT-GGA-GAT-ACA-GCA-ATT-ATC | HW5′ −35 KpnI |
| F26 | CGG-TAC-CAG-GTT-TTC-CTG-TTG-AGA-T | HW3′ −25 KpnI |
| F265 | TTG-GTA-CCT-GAG-AGA-CAG-GAC-TAG-CT | HW5′ +1 KpnI |
| F49 | TTG-GTA-CCA-GAA-TCC-CTA-AGC-CTA-GCT-GGG | HW5′ +37 KpnI |
| F6077 | CGG-TAC-CTT-GCA-ACT-TAG-CTC-ACA-CC | HW5′ +90 KpnI |
| F217 | AAG-GTA-CCA-GGG-TCC-TTG-GTC-TCA-CGC-TGA-TTT-AG | HW5′ −7125 KpnI |
| F219 | AAG-GTA-CCT-TAA-TAT-AAG-AAG-ACA-GCA-ATG-TCA-GGC | HW5′ −1421 KpnI |
| F415 | AAG-GTA-CCT-ATG-TAT-TAC-CAT-AAT-TAG-GCA-GC | HW5′ −436 KpnI |
| R6864 | AAA-GAT-CTA-ATA-GAG-TGA-AAT-AGC-ATG-AAA-ACA-GC | HW5′ +310 BglII |
| R24 | AAG-ATC-TTT-AAT-AGA-GTG-AAA-TAG-CAT-G | HW3′ +314 BglII |
| R94 | AAA-GAT-CTC-AAG-ATT-TAA-TAG-AGT-GAA-ATA-GC | Primates +310 BglII |
| R6865 | AAA-GAT-CTT-CGT-GGT-TGC-CAA-AAT-GTT-ACC-G | HW5′ +796 BglII |
| R218 | AAA-CGC-GTT-CTG-AAA-AAA-GAA-CAT-AGG-GAT-GCC-AG | HW5′ +2888 MluI |
| R220 | AAA-CGC-GTT-CTG-AAA-AAA-GAA-CAT-AGG-GAT-GC | HW5′ +2888 MluI |
| QC39 | GGA-AGG-TGA-CCA-CAT-CCA-CCT-TTA-AAC | HW5′ mut71 |
| QC41 | GCT-AAT-TAG-GCA-AAA-ACA-GGA-GGT-AAA-G | HW5′ mut151 |
| QC43 | CAG-CAG-GAG-GGA-CAA-TGA-TCG-GGA-TAT-AAA-CC | HW5′ mut210-211 |
| QC193 | CCC-AGG-CAT-TCG-AGC-CGG-CAA-CAG-CAG-CCC-CC | HW5′ mut240 |
| QC411 | CAT-TCG-AGC-TGG-CAA-CGG-CAG-CCC-CCC-TTT-GGG | HW5′ mut247 |
| QC187 | GGA-AGG-TGA-CCA-CGT-CCA-CCT-TTA-AAC | HW3′ mut71 |
| QC189 | GCT-AAT-TAG-GCA-AAG-ACA-GGA-GGT-AAA-G | HW3′ mut151 |
| QC191 | CAG-CAG-GAG-GGA-CAA-CAA-TCG-GGA-TAT-AAA-CC | HW3′ mut210-211 |
| QC195 | CCC-AAG-TCT-TCG-AGC-TGG-CAA-CGG-CAA-CCC-C | HW3′ mut240 |
| QC413 | CTT-CGA-GCC-GGC-AAC-AGC-AAC-CCC-CTT-TGG-G | HW3′ mut247 |
| QC242 | GGG-ACG-GGG-GTG-ACA-GGT-ACC-CGG-ACA-CAC-ATG-GAG | HW5′ −1000 KpnI |
| QC269 | GCC-TAA-TTA-TGG-TAA-TAC-AAT-GGT-ACC-TAA-GAA-ATA-GTT-TTT-TGG-CAG | HW5′ −436 KpnI |
| QC244 | CAT-GCT-ATT-TCA-CTC-TAT-TAC-GCG-TTG-CAA-CTG-CAC-TCT-TCT-GG | HW5′ −1421 KpnI |
| QC585 | GAG-AGT-GAA-TTA-CTG-AGT-CGG-TAC-CTC-TTC-ACT-GCA-GTC-ATT-TG | HW5′ −129 KpnI |
| QC587 | GGA-CAG-TGA-ACA-TAG-ACG-GTA-CCC-CCT-GGG-GCG-GGC-TTC-C | HW5′ −67 KpnI |
| QC328 | GGT-GCC-AGA-ACA-TTT-CTC-TAT-CGA-TAG-GTA-CCG-GCA-TGA-GGG-CAA-AAC-GCC-TGG-AGA-TAC-AGC-AAT-TAT-CTT-GC | HW5′ −44 KpnI |
| QC629 | CAT-AGA-CAG-AAG-TCC-CTA-GGG-TGG-GCT-TCC-TTT-CTG-GG | UREHu mutGi |
| QC631 | CAT-AGA-CAG-AAG-TCC-CTG-GGG-CGG-GCT-TCC-TTT-CTG-GG | UREGi mutHu |
| QC627 | CAG-TGA-ACA-TAG-ACA-GAA-GTT-TTT-GGG-GCG-GGC-TTC-CTT-TCT-GG | MutAP-2 |
| QC625 | CAT-AGA-CAG-AAG-TCC-CTA-AAA-CGG-GCT-TCC-TTT-CTG-GG | MutAP2/Sp1 |
| QC634 | GGC-TTC-CTT-TCT-GGG-ATG-ATA-GCA-AAA-CGC-CTG-GAG-ATA-C | MutGCMa |
Forward (F) and reverse (R) primers are described by their position (+1 corresponds to the beginning of the ERVWE1 5′ LTR) and by the restriction enzyme site (bold type) added at their 5′ ends. Expected mutations (bold type) are presented for each QuikChange (QC) primer. In the “Description” column, the nature of the ERVWE1 LTR (5′ [HW5′], 3′ [HW3′], or primates) is followed by the position of the primer (+1 corresponds to the beginning of the ERVWE1 5′ LTR) and the name of the restriction site added in the 5′ end. Alternatively, the nature of the LTR is followed by position of the mutation introduced (e.g., mut151).
Plasmid pGL3-LTR construction.
LTR fragments isolated from the pCR2.1-TOPO (or pCR-XL-TOPO) vector after digestion were purified by using a Nucleospin extract kit (Macherey-Nagel) and subcloned into the KpnI/BglII sites or the KpnI/MluI sites of the pGL3-basic luciferase plasmid or the pGL3-SV40 enhancer vector (Promega) according to the restriction enzyme sites present in the primers used (Table 1). Deletion mutants were generated either by nested PCR with the corresponding primers (Table 1) and cloned into the pGL3-basic vector as described above or by insertion of a restriction site by using a QuikChange site-directed mutagenesis kit (Stratagene), followed by deletion of the sequence between the corresponding restriction sites (KpnI for 5′ deletion; BglII or MluI for 3′ deletion). After deletion, plasmids were purified on agarose with the Nucleospin extract kit and ligated. In construct HW125/310, the sequence between the KpnI restriction site and the natural SacI site (located at position 125) was deleted. Purification and religation were performed as described above. Point mutants were generated by in vitro PCR-directed mutagenesis by using the QuikChange site-directed mutagenesis kit as recommended by the manufacturer. The oligonucleotides used for mutagenesis are listed in Table 1.
Sequence analysis.
PCR products were sequenced by using DyeDeoxy terminator reaction chemistry (BigDye Terminator V2.0; Applied Biosystems), and automatic sequence analysis was performed with automatic sequencers (373 Long gel DNA sequencer, ABI Prism 377 Long gel DNA sequencer, and ABI prism 3100 genetic analyzer; Applied Biosystems). The fragment data were assembled by using Seqman (DNAstar, Inc.) and ABI Prism AutoAssembler V1.4.0 (Applied Biosystems). Nucleotide sequences were compared with the sequences in the GenBank and EMBL databases by using the FASTA program. Alignments were performed with ClustalW, and putative transcription factor binding sites were identified by computer analysis with the Transcription Element Search System of the University of Pennsylvania (http://cbil.upenn.edu/tess/index.html) and Alibaba version 2.1 (http://www.alibaba2.com).
Cell culture.
BeWo b30 human choriocarcinoma cells were cultured in F-12-K medium (Life Technologies) supplemented with 10% fetal calf serum (Gibco), antibiotics (100 IU of penicillin/ml and 100 μg of streptomycin/ml) (Gibco), and amphotericin B (Fungizone; Gibco). Jeg-3 human choriocarcinoma cells and TCL-1 immortalized human trophoblasts were cultured in RPMI 1640 medium containing 2 mM l-glutamine, 10% fetal calf serum, antibiotics, and amphotericin B. LC5 human lung fibroblasts, U373 human astrocytoma cells, HeLa human cervix epithelial cells, N-Tera-2 human testicular carcinoma cells, HBL-100 human breast epithelial cells, and TelCeB6 human rhabdomyosarcoma cells were maintained in Dulbecco's modified Eagle's medium without sodium pyruvate (Life Technologies) supplemented with 10% fetal calf serum (FCS) (5% FCS for TelCeB6 cells), antibiotics, and amphotericin B. Finally, MCF-7 and T-47D breast cancer cells were grown in Dulbecco's modified Eagle's medium with nonessential amino acids (Eurobio), 100 mM sodium pyruvate (Eurobio), 10 μg of bovine insulin (Calbiochem)/ml, 10% FCS, antibiotics, and amphotericin B.
Transient transfections and luciferase assays.
At 24 h prior to transfection, cells at 60 to 70% confluence were used to seed 12-well plates. Cells were cotransfected with 0.7 μg of pGL3-LTR firefly luciferase plasmid and 35 ng of pRL-TK renilla luciferase plasmid (Promega) for internal standardization by using Lipofectamine Plus (Life Technologies) as recommended by the manufacturer. All transfection experiments were performed in triplicate. A total of 50 μM forskolin (Calbiochem) was added after 12 h to induce BeWo cell differentiation and fusion. To study the role of a protein kinase A (PKA) inhibitor, transfected cells were preincubated with 20 μM H89 (Calbiochem) for 30 min before treatment with forskolin. Cells were lysed 24 h after transfection with passive lysis buffer (Promega). Firefly and renilla luciferase activities were measured by using a dual-reporter assay system (Promega) according to the manufacturer's instructions. Promoter activities were normalized to the activity of the internal renilla luciferase control, i.e., (firefly/renilla) × 100.
RESULTS
Mapping of the ERVWE1 5′ LTR.
Previous experiments with HERV-W LTR luciferase reporter constructs tested in human trophoblastic choriocarcinoma cells (BeWo cells) and lung fibroblasts (LC5 cells) showed that the ERVWE1 5′ LTR is a functional promoter in placental cells (32). A comparison with other HERV-W LTRs suggested the presence of specific signatures which might be involved in promoter efficiency and tropism. In particular, ERVWE1 5′ and 3′ LTRs which display significant differences in promoter activities differ by only 10 nt within their U3 regions (Fig. 1a).
FIG. 1.
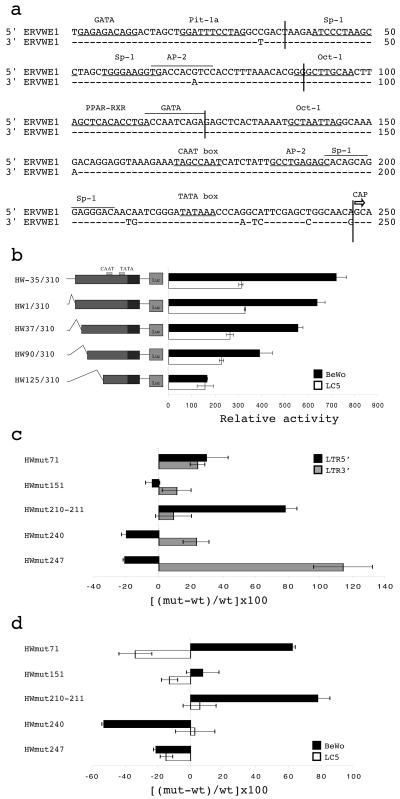
ERVWE1 LTR analysis. (a) ERVWE1 5′ and 3′ LTR alignment. Nucleotide numbering starts from the first position in the U3 region. Putative transcription factor binding sites, the TATA box, and the CAAT box are indicated. The CAP transcription initiation site at the 5′ end of the R region is represented by an arrow. (b) Relative activities of 5′ LTR deletion mutants. Constructs are schematically represented on the left. Their promoter activities were measured in BeWo choriocarcinoma cells (black bars) and in LC5 lung fibroblasts (white bars). Firefly luciferase activities were normalized to the activity of the pRL-TK renilla luciferase plasmid. The mean and standard deviation from at least three independent experiments are shown. (c) Relative activity variations of ERVWE1 5′ and 3′ LTR mutants. Point mutants were generated by PCR-directed mutagenesis, allowing the swap between precise ERVWE1 5′ and 3′ nucleotides: G71A (HWmut71), G151A (HWmut151), C210T and A211G (HWmut210-211), T240C (HWmut240), and A247G (HWmut247). ERVWE1 5′ (black bars) and 3′ (gray bars) LTR mutants were used to transiently transfect BeWo cells. Luciferase activities were normalized and expressed as a percentage of the relative activity increase or decrease compared to the activity of the wild-type reference sequence (shown on the x axis): [(mutant − wild type)/wild type] × 100. The mean and standard deviation from at least three independent experiments are shown. (d) Relative activity variations of ERVWE1 5′ LTR mutants shown in panel c in BeWo (black bars) and LC5 (white bars) cells. The mean and standard deviation from at least three independent experiments are shown.
In order to identify regions conferring high promoter activity in placental cells, sequential 5′ deletions (Fig. 1b) and point mutations (Fig. 1c and d) were constructed. Four 5′ deletion mutants were generated, eliminating successively the flanking cellular host genomic region (positions −35 to −1) upstream from the LTR U3 domain (HW1/310) and U3 putative transcription factor binding sites, including GATA and Pit-1 (HW37/310), Sp-1 and AP-2 (HW90/310), and Oct-1, peroxisome proliferator-activated receptor gamma (PPAR-γ)/retinoid X receptor (RXR), and GATA (HW125/310). These LTR mutants were cloned into the reporter plasmid pGL3-basic and used to transfect BeWo and LC5 cells, and luciferase activities were compared to that of the HW-35/310 reference construct. Progressive deletion of the 5′ region induced a corresponding decrease in relative activity, irrespective of the cell type (Fig. 1b). Nevertheless, all constructs exhibited higher activity in BeWo cells than in LC5 cells, except for the HW125/310 mutant, which contained a large deletion. These results suggested that the region from positions +1 to +125 contains binding sites essential for basal placental activity and that the region from positions +125 to +310 represents the core HERV-W promoter active in all cell types. The presence of the CAAT and TATA boxes toward the 3′ end of the U3 region precluded the use of 3′ deletion experiments. The functional roles of the CAAT box (positions 168 to 175) and the TATA box (positions 219 to 224) were indeed confirmed by analysis of the activities of altered LTRs isolated from other HERV-W loci. An LTR with an altered CAAT box (C171T mutation and A175 insertion) was 8.8 times less active than the ERVWE1 5′ LTR, and a natural double mutant with changes in CAAT (A174G mutation) and TATA (T221C and A224G mutations) did not show any detectable activity (data not shown).
A point mutation analysis was performed by making use of the nucleotide differences between the ERVWE1 5′ LTR and both ERVWE1 3′ and HERV-W8 LTRs, which exhibited lower activity in BeWo cells (32). The discriminating positions in the ERVWE1 5′ LTR versus the ERVWE1 3′ and HERV-W8 LTRs corresponded to nt 71, 151, 210-211, 240, and 247 (Fig. 1a). Swap mutations made between closely related ERVWE1 5′ and 3′ LTRs were generated by PCR-directed mutagenesis. The variations in the promoter efficiencies of the 5′ and 3′ LTR mutants were determined by measuring the relative activities of the reporter genes in BeWo cells (Fig. 1c). The promoter efficiencies of ERVWE1 5′ LTR mutants were also compared in BeWo and LC5 cells in order to estimate the influence of cell type (Fig. 1d). Each mutant exhibited a unique phenotype. Swapping of residue 71 (HWmut71) resulted in the most complex situation. This change induced increases in both 5′ and 3′ LTR activities in BeWo cells. The 5′ LTR G71A mutation caused a 33.7% decrease in activity in LC5 fibroblasts. These results indicated that 5′ LTR nucleotide G71 behaves as a negative regulatory base in BeWo cells (but as a positive regulatory base in LC5 cells). Mutation of residue 151 (HWmut151) was neutral. Mutation of tandem residues 210 and 211 (HWmut210-211) produced a 78% increase in 5′ LTR activity in BeWo cells but did not affect 3′ LTR activity. In LC5 cells, 5′ LTR activity was not altered. These results indicated that 5′ LTR nucleotides C210 and A211 act specifically as negative regulatory sequences only in BeWo cells. Swapping of residue 240 generated a 20% decrease in 5′ LTR activity and a 23% increase in 3′ LTR activity in BeWo cells. Together with the absence of variations in LC5 cells, these results indicated that the 5′ LTR nucleotide T240 belongs to a placenta-specific positive regulatory element. Similarly, mutation of the last U3 nucleotide induced a 21% decrease in 5′ LTR activity associated with a 114% increase in 3′ LTR activity in BeWo cells. In addition, the 5′ LTR activity of the A247G mutant was decreased 14.6% in LC5 cells. Thus, A247, the only base conserved throughout hominoid evolution, is involved in positive transcriptional regulation, independent of the LTR context and the cell type.
The ERVWE1 upstream host DNA sequence displays a high level of transcription.
Analysis of the 7q21.2 chromosomal region surrounding the ERVWE1 locus revealed the presence of two HERV-H LTRs (39) and one HERV-P LTR (Fig. 2a) which could be candidates for placental putative regulatory elements, considering the high levels of expression of HERVs in this tissue. In addition, although no function was assigned to the 2-kb intron located between the ERVWE1 5′ LTR and the gag pseudogene (5), the conservation of this sequence among ERVWE1 hominoid loci (32) deserves attention. Thus, a 10-kb fragment containing all HERV-H LTRs, the HERV-P LTR, the ERVWE1 5′ LTR, and the 2-kb intron was cloned, and the resulting HW-7125/2888-expressing luciferase plasmid was used to transfect BeWo cells and LC5 control cells (Fig. 2b). This construct displayed remarkably higher activity than the HW-35/310 reference construct in BeWo cells, as a 6.7-fold increase in relative activity was detected. To further delineate regions responsible for this activity, sequential 5′ and 3′ deletions were constructed. Deletion of the HERV-H 5′ LTR (HW-1421/2888), the HERV-H 3′ LTR (HW-1000/2888), and the HERV-P LTR (HW-436/2888) did not significantly affect the high level of transcriptional activity observed with the 10-kb fragment. The relative activity remained 4.5- to 5.8-fold higher than that of the HW-35/310 reference construct. Conversely, deletion of the 436-bp region located between the HERV-P LTR and the ERVWE1 5′ LTR (HW-35/2888) drastically reduced the transcriptional activity to a level similar to that of the HW-35/310 reference construct. Hence, this 436-bp upstream regulatory element (URE) was fully responsible for the high level of expression detected in BeWo cells. As all of these constructs exhibited similarly low transcriptional activities in LC5 cells, this URE seems to confer placental specificity.
FIG. 2.
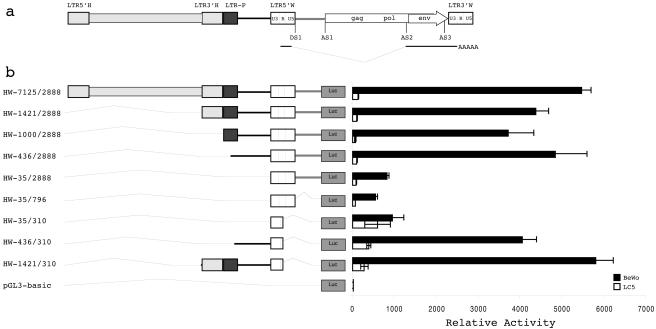
Identification of a URE. (a) Schematic representation of the ERVWE1 locus and its integration context. Retroviral sequences are depicted by rectangles, whereas host nonretroviral DNA is represented by a line. The U3, R, and U5 regions of the ERVWE1 5′ and 3′ LTRs are delineated. The subgenomic env transcript is indicated below the ERVWE1 provirus. Donor (DS) and acceptor (AS) splice sites are represented, as is the 2-kb intron (stippled line). The ERVWE1 upstream flanking sequence consists of a complete HERV-H provirus (light gray), a truncated HERV-P LTR (dark gray), and a host nonretroviral region (black line). (b) Relative activities of the 5′ LTR within its environment. HW-7125/2888 and the corresponding 5′ deletion mutants (HW-1421/2888, HW-1000/2888, HW-436/2888, and HW-35/2888) were used to transiently transfect BeWo (black bars) and LC5 (white bars) cells. 3′ Deletion mutants that eliminated the 2-kb intron (HW-35/796) and the U5 region (HW-35/310, HW-436/310, and HW-1421/310) were also tested. The promoterless pGL3-basic vector was used as a negative control. The mean and standard deviation from at least three independent experiments are shown.
Conversely, deletion of the 2-kb intron (HW-35/796) and the LTR U5 region (HW-35/310) did not affect the transcriptional activity observed in BeWo cells relative to that seen with the HW-35/310 reference construct. Interestingly, deletion of the LTR U5 region (HW-35/310), even in the presence of different URE contexts (HW-436/310 and HW-1421/310), led to a 3.2- to 6.9-fold increase in the relative activity of the LTR in LC5 cells. This result suggested that the U5 region spanning nt 310 to 796 contains negative regulatory elements recognized by LC5 but not BeWo cellular factors. The increase in the relative activity of the U5 deletion mutant was also observed in TelCeB6 (muscle), N-Tera-2 (testis), and U373 (brain) cells (data not shown). These results confirmed the U5 silencer activity in nonplacental cell lines.
The host URE is a placenta-restricted enhancer.
The dichotomy displayed by the URE-LTR and the LTR with regard to their activities in BeWo cells versus their similarly low transcriptional activities in LC5 cells suggested URE involvement in cellular tropism. Moreover, as HERV-W family expression was detected in several physiological and pathological contexts, URE-LTR and LTR activities were compared in 11 human cell types (Fig. 3a). These cells included three cell types of trophoblastic origin, BeWo, Jeg-3, and TCL-1; a testicular carcinoma cell line, N-Tera-2; a breast epithelial cell line, HBL-100; two breast cancer cell lines, MCF-7 and T-47D; a rhabdomyosarcoma cell line, TelCeB6; a cervix epithelial cell line, HeLa; an astrocytoma cell line, U373; and a lung fibroblastic cell line, LC5.
FIG. 3.
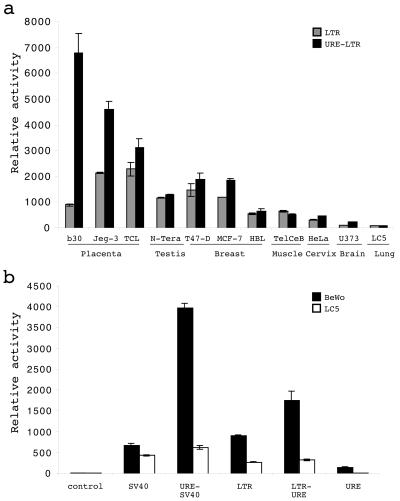
TSE role of the URE. (a) Analysis of URE-LTR relative activities in various cell types. The LTR (gray bars) and the URE-LTR (black bars) were used to transfect 11 human cell types (BeWo [b30], Jeg-3, TCL-1, N-Tera-2, T-47D, MCF-7, HBL-100, TelCeB6, HeLa, U373, and LC5) corresponding to seven organs. Luciferase relative activities from at least three independent experiments (mean and standard deviation) are shown. (b) URE relative activities in heterologous contexts. The URE (positions −436 to +1) was cloned upstream from a heterologous SV40 promoter controlling the firefly luciferase gene (URE-SV40), in a reverse orientation far downstream from the firefly luciferase gene controlled by the ERVWE1 5′ LTR (LTR-URE), and upstream from the firefly luciferase gene (URE). The pGL3-basic (control) and pGL3-SV40 (SV40) vectors were used as negative controls. Promoter activities were measured in BeWo (black bars) and LC5 (white bars) cells. Luciferase relative activities from at least three independent experiments (mean and standard deviation) are shown. LTR means the HW-35/310 construction and corresponds to the ERVWE1 5′ LTR cloned upstream of the luciferase gene from position −35 to position 310.
The highest LTR activity was detected in Jeg-3 and TCL-1 placental cells. Surprisingly, the LTR activity was slightly higher in testicular and tumoral breast cells than in placental BeWo cells. Similar lower activities were detected in normal breast cells and in muscle and cervix epithelial cells. Almost no activity was observed in brain and lung cells. This partitioning was not strictly conserved in the presence of the URE. Thus, the highest activity, shared by the three placental cell lines, was detected in BeWo cells. This relative activity was 3.6-fold higher than the highest activity detected in nonplacental cells, that detected in the breast cancer cell lines. A comparison of the LTR and URE-LTR activities showed that the largest increases in activity conferred by the URE were 7.4- and 2.1-fold in BeWo and Jeg-3 cells, respectively. It is notable that the lowest URE-dependent increase (1.3-fold), that detected in placental TCL-1 cells, correlated with the highest intrinsic LTR activity. A small increase due to the URE was also observed in breast cancer cells. No significant effect of the URE was noted for all other cell types. These data clearly demonstrated that the URE plays a role in placental tropism and may modulate basal transcription by providing binding sites for trophoblastic positively acting transcription factors.
The enhancer capacity of the URE was assessed by using two reporter systems evaluated in BeWo placental cells and LC5 control cells (Fig. 3b). The URE was inserted (i) upstream from the heterologous simian virus 40 (SV40) promoter controlling the luciferase gene (URE-SV40) and (ii) in a reverse orientation far downstream from the reporter gene controlled by the ERVWE1 5′ LTR (LTR-URE). The URE produced 5.8- and 2-fold increases in SV40 and LTR promoter activities, respectively. It should be noted that the URE did not exhibit any transcriptional promoter activity by itself, either in BeWo cells or in LC5 cells. Hence, the position-independent and promoter-independent URE-driven placental expression confirms that the URE is a tissue-specific enhancer.
Characterization of the URE in hominoids.
Positional amplification and sequencing of both alleles of the URE in nonhuman hominoids indicated that the orangutan and gibbon UREs were homozygous. The chimpanzee and gorilla UREs each had a single variable site, G/A at −233 and T/G at −271, respectively. The percentages of similarities observed by pairwise alignments of the human URE and the chimpanzee, gorilla, orangutan, and gibbon UREs were 99.1, 99.5, 97.0, and 96.1%, respectively. The relative placental activities of the LTR and of the URE-LTR were compared for the five orthologous sequences in human BeWo cells and human LC5 cells as a negative control. As observed for the human URE, juxtaposition of the UREs of the Hominidae with their related LTRs induced a drastic increase in activity in BeWo cells but not in LC5 cells (Fig. 4). Thus, 14-, 5.8-, and 4-fold increases were contributed by the chimpanzee, gorilla, and orangutan UREs, respectively. Conversely, not only was this property not conserved for the gibbon URE but also positioning of the URE upstream from the LTR induced a slight decrease in activity. The LTRs of the Hylobatidae also exhibited biased behavior compared to the LTRs of the Hominidae. Gibbon basal LTR activities were 2.6- and 2.4-fold higher than the mean activity of all four LTRs of the Hominidae in BeWo cells and LC5 cells, respectively. Nevertheless, the gibbon LTR remained 2.6-fold more active in BeWo cells than in LC5 cells.
FIG. 4.
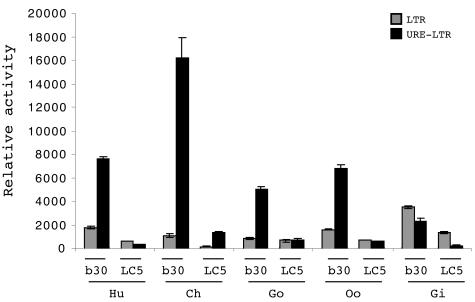
Comparison of hominoid UREs. Relative activities of the LTR (gray bars) and the URE-LTR (black bars) from human (Hu), chimpanzee (Ch), gorilla (Go), orangutan (Oo), and gibbon (Gi) are shown. Promoter activities were measured in BeWo (b30) and LC5 (LC5) cells. The mean and standard deviation from at least three independent experiments are shown.
Identification of the critical domains of the URE conferring TSE activity.
The antagonist activity of the UREs of the Hominidae and the Hylobatidae suggests that the gibbon sequence may lack specific binding sites dedicated to transcription factors present in human trophoblastic cells. Thus, alignment of all five UREs showed that only 11 nt distinguished the gibbon URE from the four other UREs (Fig. 5a). In addition, biocomputational analysis of the URE sequences revealed the presence of (i) putative transcription factor binding sites previously found to be involved in promoter placental regulation (AP-2, Sp-1, PPAR-γ, GATA, c-Myb, and GCMa) and (ii) a 63-bp truncated nonautonomous mammalian apparent LTR retrotransposon (MaLR) located −67 bp upstream from the cellular DNA-LTR junction (Fig. 5a).
FIG. 5.
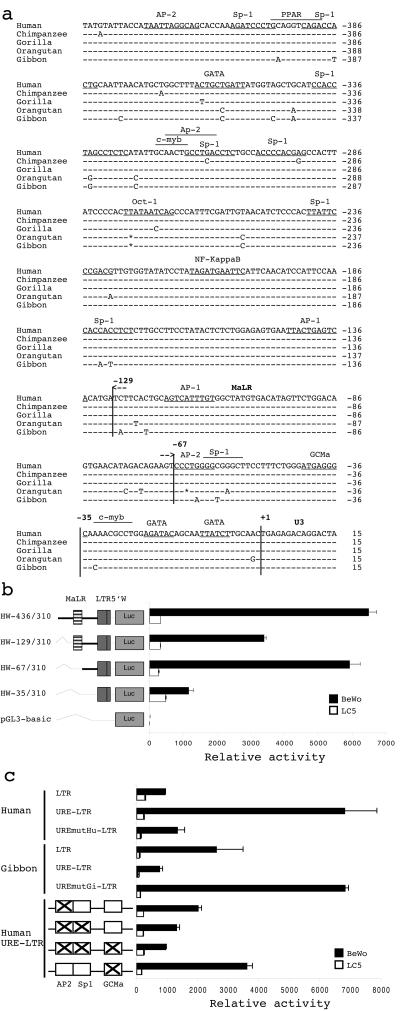
Characterization of TSE constituents. (a) Alignment of orthologous UREs of human, chimpanzee, gorilla, orangutan, and gibbon. Putative transcription factor binding sites are indicated. The truncated MaLR is delineated between positions −129 and −67. The +1 position corresponds to the 5′ start site of the LTR U3 region. (b) Dissection of the human URE. Sequential 5′ deletions of the URE are schematically represented on the left. Constructs HW-129/310 and HW-67/310 started, respectively, from MaLR (hatched box) and after MaLR. The promoterless pGL3-basic vector was used as a negative control. Promoter activities were measured in BeWo (black bars) and LC5 (white bars) cells. The mean and standard deviation from at least three independent experiments are shown. LTR5′W corresponds to the U3 region and the beginning of the R region of the ERVWE1 5′ LTR from position +1 to positioin 310. (c) Relative activities of URE mutants. Point mutations were generated by PCR-directed mutagenesis with the primers listed in Table 1, allowing the mutation of particular residues. Swapping of nucleotides at positions −63 and −59 between human and gibbon was realized in the context of human (UREmutGi-LTR) and gibbon (UREmutHu-LTR) wild-type URE-LTRs. Mutations that specifically eliminated putative transcription factor binding sites for AP-2, AP-2-Sp-1, AP-2-Sp-1-GCMa, and GCMa were introduced into the human wild-type URE-LTR construct, as shown on the left by crosses inside boxes. Promoter activities were measured in BeWo (black bars) and LC5 (white bars) cells. The mean and standard deviation from at least three independent experiments are shown.
To further delineate regions responsible for this activity, sequential 5′ deletions (Fig. 5b) and point mutations (Fig. 5c) were constructed. Deletion of a 5′ URE subregion (HW-129/310) and the MaLR (HW-67/310) only slightly affected the high level of transcription observed with the 436-bp fragment (HW-129/310). The relative activity remained 2.9- to 5.1-fold higher than that of the HW-35/310 reference sequence, compared to 5.6-fold for the complete URE. More precisely, in BeWo cells, deletion of the 5′ URE region (positions −436 to −129) caused a 47% reduction in URE transcriptional activity, and additional deletion of the MaLR (positions −129 to −67) restored almost complete URE activity. These results indicated that the sequences at positions −436 to −129 and positions −129 to −67 act as positive and negative regulatory regions, respectively. No variation was observed in the LC5 control cell experiment. Hence, the region from positions −67 to −35 region appears to define the minimal URE domain showing TSE activity. Moreover, sequencing of this minimal URE domain showed strict conservation among 100 alleles from 50 human individuals (data not shown).
A comparison of gibbon and other hominoid regions from positions −67 to −35 shows that the gibbon URE displays a transition from G to A at position −63 and a transition from C to T at position −59 that disrupt a consensus binding site putatively recognized by members of the AP-2 family. In order to determine whether these two bases may account for the opposite behaviors of the human and gibbon UREs in BeWo cells, both nucleotides were swapped between human and gibbon luciferase expression plasmids in their overall URE-LTR species contexts. The introduction of the AT gibbon-specific nucleotides within the human backbone caused a substantial 3.1-fold decrease in human URE-LTR activity that was close to that seen with the LTR alone (Fig. 5c). Conversely, the introduction of the GC human-specific nucleotides within the gibbon backbone caused a substantial 9.3-fold increase in gibbon URE-LTR activity that reached the same level as that seen with the native human URE-LTR (Fig. 5c). Taken together, these observations suggest that the CG-rich sequence overlapping the AP-2 consensus binding site is the primary contributor to human placenta-specific expression of the ERVWE1 env gene. Nevertheless, although greatly decreased, the activity of the human URE-LTR containing the gibbon signature remained slightly higher than the activity of the LTR alone. This result was paradoxical compared to the result obtained in the gibbon context, where the URE-LTR activity was lower than the LTR activity.
Thus, a directed mutational strategy was designed to specifically alter one or several human URE putative binding sites which were located within the region from positions −67 to −35 and which were divergent between gibbons and humans (AP-2 and Sp-1) or conserved between the species (GCMa). Single AP-2 and GCMa, double AP-2-Sp-1, and triple AP-2-Sp-1-GCMa putative consensus binding sites were inactivated (Fig. 5c). In agreement with the results of the gibbon-human sequence swapping experiment, mutations that solely disrupted the AP-2 putative consensus binding site caused a 3.1-fold reduction in transcriptional activity in placental cells. The AP-2-Sp-1 double mutant led to a 5.2-fold decrease, and the AP-2-Sp-1-GCMa triple mutant led to a 7.3-fold decrease, to the level seen with the LTR. These results indicated that all three sites supplied the complete URE activity. Thus, the region from positions −67 to −35, containing all three cooperative sites, was designated the TSE. Although the AP-2 binding site accounted for the majority of the enhancer function of the URE, a complex synergistic process seemed to be involved, as illustrated by the significant 1.9-fold decrease produced by mutations that disrupted only the GCMa binding site.
Identification of the cAMP-responsive domains of URE-LTR.
In vitro stimulation of BeWo or primary trophoblastic cell fusion and differentiation by cAMP is associated with a concomitant increase in HERV-W env mRNA expression (14, 24, 34). In addition, it was previously shown that cAMP-dependent protein kinase plays a role in human trophoblastic cell fusion and differentiation (22). Thus, we evaluated the effects of both forskolin, an inducer of cAMP, and H89, an inhibitor of the protein kinase A (PKA) pathway, on syncytin URE-LTR promoter activity by using the luciferase reporter gene assay and BeWo cells (Fig. 6a). A 1.8-fold increase in integral syncytin promoter activity was caused by the addition of forskolin. Pretreatment with H89, before the addition of forskolin, abolished the increased activity due to the forskolin. Analysis of HERV-W env expression in primary trophoblasts treated with 8-bromo-cAMP and H89 confirmed that native HERV-W mRNA expression was also induced by cAMP and hampered by H89 (J. L. Frendo, personal communication). These results suggested that the PKA pathway was directly involved in syncytin promoter regulation.
FIG. 6.
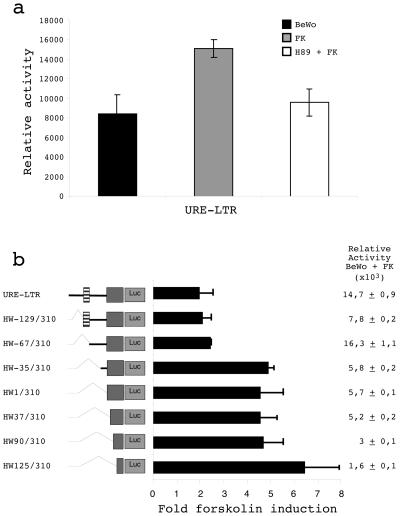
Identification of cAMP-responsive regions on the syncytin promoter. (a) Effect of cAMP on the syncytin promoter. The activities of the URE-LTR (HW-436/310) in BeWo cells (black bars), BeWo cells treated with 50 μM forskolin (FK) for 9 h after transfection (gray bars), and BeWo cells pretreated with 20 μM H89 for 30 min before the addition of 50 μM forskolin (white bars) are shown. Normalized luciferase activities from at least three experiments (mean and standard deviation) are shown. (b) Effect of cAMP on 5′ URE sequential deletion mutants. The constructs schematically represented on the left were used to transfect BeWo cells incubated with or without 50 μM forskolin for 9 h. The fold increase in the presence of forskolin represented the ratio of the relative luciferase activity with forskolin to the relative luciferase activity without forskolin for each construct. Luciferase relative activities after forskolin treatment are indicated for each construct on the right. Data represent the mean and standard deviation of three independent transfection experiments.
The URE-LTR regions involved in cAMP/PKA upregulation were localized by deciphering the responses of URE-LTR deletion mutants to forskolin stimulation (Fig. 6b). First, consistent with the above results, constructs containing the TSE (URE-LTR, HW-129/310, and HW-67/310) exhibited the highest activities after treatment with forskolin. Positive and negative roles for the 5′ distal URE and the MaLR region were confirmed. The TSE-restricted construct (HW-67/310) showed 2.8-fold higher activity than the reference construct (HW-35/310). Second, a clear dichotomy was observed between TSE-containing constructs and LTR-restricted constructs with regard to the effect of forskolin. A 1.9- to 2.1-fold induction by forskolin and a 4.4- to 6.4-fold induction by forskolin were observed for TSE-LTR constructs and LTR-only constructs, respectively. A similar 4.1-fold induction by forskolin was observed for a 3′ deletion mutant in which the R domain of the LTR was eliminated. These results suggest that the minimal domain for cAMP/PKA upregulation is located between nt 125 and nt 247. Interestingly, AP-2, AP-2-Sp-1, and AP2-Sp-1-GCMa mutations that were shown to gradually decrease URE-LTR efficiency concomitantly caused an increase in forskolin induction (2.0-, 3.6-, and 3.9-fold, respectively) of TSE-containing constructs, similar to the level of induction seen with the LTR (data not shown). This result apparently correlated with the equivalent levels of induction observed with the nonfunctional gibbon URE-LTR (3.2-fold) containing an altered AP-2-Sp-1 nucleic acid motif and the gibbon LTR alone (3.8-fold). Surprisingly, the two mutations that were shown to restore gibbon URE activity not only decreased the effect of forskolin but also completely impeded this effect (0.8-fold) (data not shown). These results suggested a complex situation with, on the one hand, an (H)ERV-W LTR domain basically stimulated by the cAMP/PKA pathway and, on the other hand, a TSE region interfering with the regulation of the coopted ERVWE1 LTR.
DISCUSSION
In this study, we showed that the promoter controlling the placental expression of the ERVWE1 env gene is a bipartite element composed of (i) a retroviral component consisting of the ERVWE1 5′ LTR and (ii) a cellular URE conferring placental specificity. The LTR provides the basal placental promoter activity and cAMP-responsive elements. The URE provides a TSE inducing a high level of tissue-specific expression. We observed complex regulatory mechanisms involving the retroviral element (ERVWE1 versus HERV-W LTRs), the cellular context (placental versus nonplacental cells), the chromosomal environment, and consequently the benefits and disadvantages resulting from cellular-retroviral coopted functions. The overall scheme is summarized in Fig. 7.
FIG. 7.
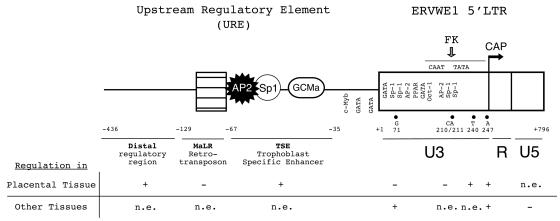
Schematic representation of regulatory elements controlling ERVWE1 env gene expression. The URE is composed of a distal regulatory region (black line), truncated MaLR (hatched box), and a TSE region containing putative transcription factor AP-2, Sp-1, and GCMa binding sites. The ERVWE1 5′ LTR (white box) is composed of the U3, R, and U5 regions. The U3 region contains particular regulatory nucleotides (71, 210-211, 240, and 247) and the CAAT and TATA boxes. The CAP transcription initiation site (arrow) is located at the 5′ end of the R region. The forskolin (FK)-responsive domain of the U3 region corresponds to the basal promoter region (stippled line). The positive (+) or negative (−) involvement or the absence of an effect (n.e.) of regulatory domains and nucleotides on syncytin promoter activity in placental and other tissues is annotated below the schematic representation. The GCMa site in the TSE region and the Oct-1 site in the LTR were proven to interact with their relative transcription factors (9, 52).
Like infectious retroviruses, HERVs possess all signals required for the initiation and regulation of transcription within their LTRs. The U3 region of the ERVWE1 5′ LTR, like other HERV-W LTR U3 regions (5, 27, 44), possesses promoter activity (32). The identified HERV-W core promoter (positions +125 to +247) contains a CAAT box (positions +167 to +175) and a TATA box (positions +219 to +224) located −23 bp and −72 bp upstream from the CAP site corresponding to the R region. It should be noted that this definition of the transcription initiation site (5) contradicts the proposal that it is localized upstream from the TATA box (9), although different assay systems and the presence of several HERV-W loci expressed in the placenta may explain this discrepancy. Analysis of the 5′ ends of various HERV-W processed pseudogenes (36) supported the R region as being the primary initiation site, independent of the parental-proviral chromosomal contexts. The 5′ end of the U3 region (positions +1 to +124) contains binding sites contributing to overall promoter efficiency and cell type specificity. Notably, GATA (26), Sp-1 (43), AP-2 (2, 37, 41), Oct-1 (9), and PPAR-γ/RXR (46) factors have been shown to be important in trophoblast-specific gene transcription. Nevertheless, this dichotomy between 5′ and 3′ U3 domains is not as clear-cut as the single-site mutant analysis reveals (Fig. 7). Thus, the different activities of these LTRs may be due to a combination of regulatory sites. Nevertheless, the G71 and T240 nucleotides belong to placental negative and positive regulatory sites, respectively. The U5 region also seems to be involved in syncytin regulation. A silencer effect restricted to nonplacental cells was observed. A similar U5 silencer effect has been reported for another HERV-W LTR in HeLa cells (27) and for HERV-K (13) and human T-cell leukemia virus type 1 (35) as well.
Analysis of the complete ERVWE1 5′ LTR (U3-R-U5) in 11 cell types revealed a surprising profile. The LTR transcriptional activities in three types of nonplacental cell lines, including testis and breast cancer cell lines, were higher than that in the BeWo trophoblastic model. The highest activity was still observed in TCL-1 and Jeg-3 placental cells. These results suggest that the relative amounts of ubiquitous and specific factors produce precise control of the LTR. Even higher LTR activity was observed in differentiated BeWo cells upon forskolin treatment, a finding which may correspond to a drastic shift of this equilibrium. Chimpanzee, gorilla, orangutan, and gibbon ERVWE1 5′ LTRs were also induced by the cAMP activator forskolin (data not shown). Inhibition of cAMP stimulation by the PKA inhibitor H89 demonstrated the pivotal role of the cAMP/PKA pathway in LTR regulation. The minimal region required for cAMP stimulation was located within the core LTR promoter (positions +125 to +247). The absence of a cAMP-responsive element (51) suggests the involvement of other cAMP-modulated transcription factors. Interestingly, the broadly distributed AP-2 and Sp-1 factors (16, 42) are regulated by cAMP/PKA, correlating with AP-2 and Sp-1 putative binding sites in the core LTR (Fig. 7).
In addition to their promoter activities, HERV LTRs exhibit enhancer capacities which confer tissue tropism to cellular genes. An HERV-E LTR mediates salivary gland expression of the amylase gene in hominoids (47). Placental expression of the human leptin (3) and insulin-like growth factor 4 (4) genes relies on MER11 and HERV-K enhancer elements, respectively. The 7-kb cellular region located upstream from the ERVWE1 provirus contains two HERV-H LTRs and one HERV-P LTR. These three LTRs did not significantly affect ERVWE1 proviral expression, at least in BeWo and LC5 cells. This finding contradicted the proposed requirement of the region from positions −1343 to −808 (covering 87% of the HERV-H 3′ LTR and 34% of the juxtaposed HERV-P LTR) for maximal expression (9). Additional analyses of isolated HERV-H 5′ and 3′ LTRs (out of their chromosomal context) did not reveal any promoter or enhancer-silencer activity in BeWo cells (data not shown). Nevertheless, evaluation of these sequences in other cell types deserves further investigation due to the codetection of HERV-W (38)- and HERV-H (10)-related sequences in multiple sclerosis patients and the related hypothesis of local coactivation of ERVWE1 and upstream HERV-H elements (39). Conversely, a URE encompassing nt −436 to +1 displayed enhancer activity (i) upstream from a heterologous promoter and (ii) in a reverse orientation far downstream from the 5′ LTR promoter. Furthermore, a comparison of the URE activities in 11 cell types classified the URE as a TSE. The URE is composed of three subdomains, distal (positions −436 to −129) and proximal (positions −67 to +1) positive regulatory regions which flank a truncated nonautonomous MaLR (positions −129 to −67) (Fig. 7). The silencer activity of the MaLR was observed only in BeWo cells. The proximal region was the minimal region essential for the high transcriptional activity in placental cells and was therefore qualified as a TSE more accurately delimited by positions −67 and −35. The TSE is indeed included in the region from positions −119 to +28 that is involved in a high level of expression in primary trophoblastic cells (9).
The genetic and functional conservation of hominoid URE sequences supported a crucial role in syncytin regulation. As the gibbon URE was not functionally active in human BeWo cells, a comparison of all hominoid URE sequences allowed us to identify AP-2 and Sp-1 putative binding sites as critical constituents of TSE activity. Their involvement was confirmed by site-directed mutagenesis experiments. In addition, a conserved GCMa binding site was proven to contribute to TSE activity. Indeed, this site was previously shown to interact with the GCMa transcription factor in BeWo and Jeg-3 cells (52). A second site located in the pol pseudogene of the HERV-H provirus and able to bind to GCMa was proposed to act synergistically with the TSE GCMa binding site in adenovirus GCMa-transduced BeWo cells (52). Nevertheless, a large deletion including this GCMa binding site indicated that it was apparently not essential for enhancer activity in BeWo cells. The ubiquitous related AP-2 (2, 37, 41) and Sp-1 (43) transcription factors and the GCMa (1, 50) trophoblast-specific factor were shown to be important in trophoblast-specific gene transcription. Tissue-specific expression of human chorionic gonadotropin hormone produced during cytotrophoblastic cell fusion and differentiation was shown to be dependent on an enhancer controlled by the AP-2, CREB (cAMP binding protein), GATA, and Dlx3 transcription factors (28, 29). The regulation of 17β-hydroxysteroid dehydrogenase type 1 was shown to involve the GATA, AP-2, and Sp-1 enhancer elements (40). Syncytium-specific CYP-19 or aromatase expression was shown to be controlled by GCMa, AP-2, and Sp-1 (50). Unidentified transcription factors binding to all three sites of the syncytin TSE acted synergistically. Thus, AP-2, AP-2-Sp-1, and AP-2-Sp-1-GCMa TSE mutants lost 81, 93.5, and 99.6% of the native enhancer activity, respectively. Mutation of the GCMa binding site nevertheless accounted for 54% of the TSE activity. AP-2 and Sp-1 binding sites consisting of GC-rich domains (18, 49) are found either as close neighbors or as overlapping domains within a variety of promoters (7, 12, 19, 40). AP-2- and Sp-1-related factors can function additively or antagonistically, depending on the promoter context and the relative ratios of both proteins (7, 40). Note that it was recently proposed that neither a dominant-negative form of AP-2α nor the AP-2α protein itself influences the syncytin mRNA level (8). Further studies will be required to determine the exact nature of the transcription factors that bind the GC-rich AP-2 consensus binding site.
Both benefits and disadvantages appear to be derived from the cooperation of the URE and the LTR. The ca. 2.5-fold increase in URE-LTR activity upon forskolin treatment was compatible with the 4-fold increase in syncytin mRNA expression observed upon differentiation of BeWo cells (34) and primary trophoblastic cells (14). Nevertheless, the very high URE-LTR activity observed in differentiated BeWo cells did not correlate with the gain expected from forskolin treatment of the LTR (fivefold increase) together with URE juxtaposition (sixfold increase). This finding may have resulted from an interference phenomenon due to the presence of AP-2 and Sp-1 putative binding sites on the same TSE and LTR components. TSE elements might compete with cAMP-responsive elements present in the LTR by sequestering cAMP-inducible transcription factor binding to AP-2- and Sp-1-related sites. This hypothesis is supported by results obtained with AP-2 and AP-2-Sp-1 TSE mutants, which exhibited gradually reduced activity along with enhanced forskolin susceptibility. Conversely, mutation of the cAMP-independent GCMa-related binding site did not alter the forskolin effect. Gibbon TSE represented a natural mutant of the AP-2 and Sp-1 binding sites. In agreement with the above hypothesis, the URE-LTR and the LTR alone behaved similarly upon forskolin treatment. Intriguingly, mutation of gibbon AP-2 and Sp-1 binding sites, which restored TSE functionality, not only reduced but also completely inhibited URE-LTR forskolin susceptibility. The contrast between the inefficiency of the gibbon URE and the potency of the gibbon LTR among hominoids may reflect such a dual situation. However, we cannot exclude the possibility that these specific observations are attributable to the specific context of human BeWo cells. Consistently, the human LTR exhibited the highest activity in trophoblastic TCL-1 cell lines (among 11 cell types), whereas the URE was proven to be almost inefficient. The variable activities of the URE-LTR and the LTR observed among the three trophoblastic cell lines, BeWo, Jeg-3, and TCL-1, probably reflect different differentiation stages or distinct lineages. The data suggest that accurate mechanisms coregulate the host URE sequence and the ERVWE1 LTR element to maintain adequate and optimal syncytin expression during trophoblast differentiation and possibly within the syncytiotrophoblast layer.
Cooperation of cellular and retroviral elements is a relatively frequent event during primate evolution. Retroviral insertion commonly confers alternate tropism to the cellular promoter. We describe here a reverse situation where a provirus inserted downstream from a host genomic sequence either exhibited a TSE or coevolved to provide such a tropism. The context of the Hylobatidae versus the Hominidae may thus reflect a coevolution process, although the possibility of drift of the branch of the Hylobatidae cannot be excluded. Whatever the explanations, fine mechanisms which regulate the TSE LTR deserve scrutiny in order to address potential syncytin-associated pathologies. Thus, down-regulation of ERVWE1 env mRNA in placental dysfunction, such as preeclampsia or HELLP (hemolysis, elevated enzymes, low platelets) syndrome (23), may be related to an altered context of TSE or transcription factors. Lessons from this analysis could also contribute to the understanding of HERV-W and ERVWE1 expression in nonplacental contexts, such as schizophrenia and multiple sclerosis.
Acknowledgments
We are grateful to K. Ruel, J. Fitamant, and J. Beliaeff for technical assistance and to R. Buckland for helping us to improve the manuscript. We thank J. Strauss III, C. Leib-Mösch, M. H. Sullivan, and D. Mager for providing cell lines.
S.P. is supported by a doctoral fellowship from La Fondation Mérieux and le Ministère de la Recherche.
REFERENCES
- 1.Anson-Cartwright, L., K. Dawson, D. Holmyard, S. J. Fisher, R. A. Lazzarini, and J. C. Cross. 2000. The glial cells missing-1 protein is essential for branching morphogenesis in the chorioallantoic placenta. Nat. Genet. 25:311-314. [DOI] [PubMed] [Google Scholar]
- 2.Ben Zimra, M., M. Koler, and J. Orly. 2002. Transcription of cholesterol side-chain cleavage cytochrome P450 in the placenta: activating protein-2 assumes the role of steroidogenic factor-1 by binding to an overlapping promoter element. Mol. Endocrinol. 16:1864-1880. [DOI] [PubMed] [Google Scholar]
- 3.Bi, S., O. Gavrilova, D. W. Gong, M. M. Mason, and M. Reitman. 1997. Identification of a placental enhancer for the human leptin gene. J. Biol. Chem. 272:30583-30588. [DOI] [PubMed] [Google Scholar]
- 4.Bieche, I., A. Laurent, I. Laurendeau, L. Duret, Y. Giovangrandi, J. L. Frendo, M. Olivi, J. L. Fausser, D. Evain-Brion, and M. Vidaud. 2003. Placenta-specific INSL4 expression is mediated by a human endogenous retrovirus element. Biol. Reprod. 68:1422-1429. [DOI] [PubMed] [Google Scholar]
- 5.Blond, J. L., F. Beseme, L. Duret, O. Bouton, F. Bedin, H. Perron, B. Mandrand, and F. Mallet. 1999. Molecular characterization and placental expression of HERV-W, a new human endogenous retrovirus family. J. Virol. 73:1175-1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blond, J. L., D. Lavillette, V. Cheynet, O. Bouton, G. Oriol, S. Chapel-Fernandes, B. Mandrand, F. Mallet, and F. L. Cosset. 2000. An envelope glycoprotein of the human endogenous retrovirus HERV-W is expressed in the human placenta and fuses cells expressing the type D mammalian retrovirus receptor. J. Virol. 74:3321-3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen, T. T., R. L. Wu, F. Castro-Munozledo, and T. T. Sun. 1997. Regulation of K3 keratin gene transcription by Sp1 and AP-2 in differentiating rabbit corneal epithelial cells. Mol. Cell. Biol. 17:3056-3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheng, Y. H., B. J. Aronow, S. Hossain, B. Trapnell, S. Kong, and S. Handwerger. 2004. Critical role for transcription factor AP-2′ in human trophoblast differentiation. Physiol. Genomics 18:99-107. [DOI] [PubMed]
- 9.Cheng, Y. H., B. D. Richardson, M. A. Hubert, and S. Handwerger. 2004. Isolation and characterization of the human syncytin gene promoter. Biol. Reprod. 70:694-701. [DOI] [PubMed] [Google Scholar]
- 10.Christensen, T., P. Dissing Sorensen, H. Riemann, H. J. Hansen, and A. Moller-Larsen. 1998. Expression of sequence variants of endogenous retrovirus RGH in particle form in multiple sclerosis. Lancet 352:1033. [DOI] [PubMed] [Google Scholar]
- 11.De Parseval, N., V. Lazar, J. F. Casella, L. Benit, and T. Heidmann. 2003. Survey of human genes of retroviral origin: identification and transcriptome of the genes with coding capacity for complete envelope proteins. J. Virol. 77:10414-10422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Descheemaeker, K. A., S. Wyns, L. Nelles, J. Auwerx, T. Ny, and D. Collen. 1992. Interaction of AP-1-, AP-2-, and Sp1-like proteins with two distinct sites in the upstream regulatory region of the plasminogen activator inhibitor-1 gene mediates the phorbol 12-myristate 13-acetate response. J. Biol. Chem. 267:15086-15091. [PubMed] [Google Scholar]
- 13.Domansky, A. N., E. P. Kopantzev, E. V. Snezhkov, Y. B. Lebedev, C. Leib-Mosch, and E. D. Sverdlov. 2000. Solitary HERV-K LTRs possess bi-directional promoter activity and contain a negative regulatory element in the U5 region. FEBS Lett. 472:191-195. [DOI] [PubMed] [Google Scholar]
- 14.Frendo, J. L., D. Olivier, V. Cheynet, J. L. Blond, O. Bouton, M. Vidaud, M. Rabreau, D. Evain-Brion, and F. Mallet. 2003. Direct involvement of HERV-W Env glycoprotein in human trophoblast cell fusion and differentiation. Mol. Cell. Biol. 23:3566-3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fujinami, R. S., and J. E. Libbey. 1999. Endogenous retroviruses: are they the cause of multiple sclerosis? Trends Microbiol. 7:263-264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garcia, M. A., M. Campillos, A. Marina, F. Valdivieso, and J. Vazquez. 1999. Transcription factor AP-2 activity is modulated by protein kinase A-mediated phosphorylation. FEBS Lett. 444:27-31. [DOI] [PubMed] [Google Scholar]
- 17.Gaudin, P., H. Perron, G. Favre, B. Mandrand, R. Juvin, F. Marcel, F. Beseme, F. Bedin, F. Mallet, B. Mougin, J. Fauconnier, J. M. Seigneurin, and X. Phelip. 1997. Detection of retrovirus RNA in plasma from rheumatoid arthritis. Arthritis Rheum. 40:S245. [Google Scholar]
- 18.Hagen, G., S. Muller, M. Beato, and G. Suske. 1992. Cloning by recognition site screening of two novel GT box binding proteins: a family of Sp1 related genes. Nucleic Acids Res. 20:5519-5525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson, W., and J. L. Jameson. 1999. AP-2 (activating protein 2) and Sp1 (selective promoter factor 1) regulatory elements play distinct roles in the control of basal activity and cyclic adenosine 3′,5′-monophosphate responsiveness of the human chorionic gonadotropin-beta promoter. Mol. Endocrinol. 13:1963-1975. [DOI] [PubMed] [Google Scholar]
- 20.Johnston, J. B., C. Silva, J. Holden, K. G. Warren, A. W. Clark, and C. Power. 2001. Monocyte activation and differentiation augment human endogenous retrovirus expression: implications for inflammatory brain diseases. Ann. Neurol. 50:434-442. [DOI] [PubMed] [Google Scholar]
- 21.Karlsson, H., S. Bachmann, J. Schroder, J. McArthur, E. F. Torrey, and R. H. Yolken. 2001. Retroviral RNA identified in the cerebrospinal fluids and brains of individuals with schizophrenia. Proc. Natl. Acad. Sci. USA 98:4634-4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Keryer, G., E. Alsat, K. Taskén, and D. Evain-Brion. 1998. Cyclic AMP-dependent protein kinases and human trophoblast cell differentiation in vitro. J. Cell Sci. 111:995-1004. [DOI] [PubMed] [Google Scholar]
- 23.Knerr, I., E. Beinder, and W. Rascher. 2002. Syncytin, a novel human endogenous retroviral gene in human placenta: evidence for its dysregulation in preeclampsia and HELLP syndrome. Am. J. Obstet. Gynecol. 186:210-213. [DOI] [PubMed] [Google Scholar]
- 24.Kudo, Y., C. A. Boyd, I. L. Sargent, and C. W. Redman. 2003. Hypoxia alters expression and function of syncytin and its receptor during trophoblast cell fusion of human placental BeWo cells: implications for impaired trophoblast syncytialisation in pre-eclampsia. Biochim. Biophys. Acta 1638:63-71. [DOI] [PubMed] [Google Scholar]
- 25.Landry, J. R., A. Rouhi, P. Medstrand, and D. L. Mager. 2002. The Opitz syndrome gene Mid1 is transcribed from a human endogenous retroviral promoter. Mol. Biol. Evol. 19:1934-1942. [DOI] [PubMed] [Google Scholar]
- 26.LaVoie, H. A. 2003. The role of GATA in mammalian reproduction. Exp. Biol. Med. (Maywood) 228:1282-1290. [DOI] [PubMed] [Google Scholar]
- 27.Lee, W. J., H. J. Kwun, and K. L. Jang. 2003. Analysis of transcriptional regulatory sequences in the human endogenous retrovirus W long terminal repeat. J. Gen. Virol. 84:2229-2235. [DOI] [PubMed] [Google Scholar]
- 28.LiCalsi, C., S. Christophe, D. J. Steger, M. Buescher, W. Fischer, and P. L. Mellon. 2000. AP-2 family members regulate basal and cAMP-induced expression of human chorionic gonadotropin. Nucleic Acids Res. 28:1036-1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Loregger, T., J. Pollheimer, and M. Knofler. 2003. Regulatory transcription factors controlling function and differentiation of human trophoblast—a review. Placenta 24(Suppl. A):S104-S110. [DOI] [PubMed] [Google Scholar]
- 30.Löwer, R., R. R. Tönjes, C. Korbmacher, R. Kurth, and J. Löwer. 1995. Identification of a Rev-related protein by analysis of spliced transcripts of the human endogenous retrovirus HTDV/HERV-K. J. Virol. 69:141-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Magin, C., R. Löwer, and J. Löwer. 1999. cORF and RcRE, the Rev/Rex and RRE/RxRE homologues of the human endogenous retrovirus family HTDV/HERV-K. J. Virol. 73:9496-9507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mallet, F., O. Bouton, S. Prudhomme, V. Cheynet, G. Oriol, B. Bonnaud, G. Lucotte, L. Duret, and B. Mandrand. 2004. The endogenous retroviral locus ERVWE1 is a bona fide gene involved in hominoid placental physiology. Proc. Natl. Acad. Sci. USA 101:1731-1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Medstrand, P., J. R. Landry, and D. L. Mager. 2001. Long terminal repeats are used as alternative promoters for the endothelin B receptor and apolipoprotein C-I genes in humans. J. Biol. Chem. 276:1896-1903. [DOI] [PubMed] [Google Scholar]
- 34.Mi, S., X. Lee, X. Li, G. M. Veldman, H. Finnerty, L. Racie, E. LaVallie, X. Y. Tang, P. Edouard, S. Howes, J. C. Keith, Jr., and J. M. McCoy. 2000. Syncytin is a captive retroviral envelope protein involved in human placental morphogenesis. Nature 403:785-789. [DOI] [PubMed] [Google Scholar]
- 35.Okumura, K., G. Sakaguchi, S. Takagi, K. Naito, T. Mimori, and H. Igarashi. 1996. Sp1 family proteins recognize the U5 repressive element of the long terminal repeat of human T cell leukemia virus type I through binding to the CACCC core motif. J. Biol. Chem. 271:12944-12950. [DOI] [PubMed] [Google Scholar]
- 36.Pavlicek, A., J. Paces, D. Elleder, and J. Hejnar. 2002. Processed pseudogenes of human endogenous retroviruses generated by LINEs: their integration, stability, and distribution. Genome Res. 12:391-399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peng, L., and A. H. Payne. 2002. AP-2 gamma and the homeodomain protein distal-less 3 are required for placental-specific expression of the murine 3 beta-hydroxysteroid dehydrogenase VI gene, Hsd3b6. J. Biol. Chem. 277:7945-7954. [DOI] [PubMed] [Google Scholar]
- 38.Perron, H., J. A. Garson, F. Bedin, F. Beseme, G. Paranhos-Baccala, F. Komurian-Pradel, F. Mallet, P. W. Tuke, C. Voisset, J. L. Blond, B. Lalande, J. M. Seigneurin, B. Mandrand, et al. 1997. Molecular identification of a novel retrovirus repeatedly isolated from patients with multiple sclerosis. Proc. Natl. Acad. Sci. USA 94:7583-7588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Perron, H., J. P. Perin, F. Rieger, and P. M. Alliel. 2000. Particle-associated retroviral RNA and tandem RGH/HERV-W copies on human chromosome 7q: possible components of a ′chain-reaction' triggered by infectious agents in multiple sclerosis? J. Neurovirol. 6(Suppl. 2):S67-S75. [PubMed] [Google Scholar]
- 40.Piao, Y. S., H. Peltoketo, P. Vihko, and R. Vihko. 1997. The proximal promoter region of the gene encoding human 17beta-hydroxysteroid dehydrogenase type 1 contains GATA, AP-2, and Sp1 response elements: analysis of promoter function in choriocarcinoma cells. Endocrinology 138:3417-3425. [DOI] [PubMed] [Google Scholar]
- 41.Richardson, B. D., R. A. Langland, C. J. Bachurski, R. G. Richards, C. A. Kessler, Y. H. Cheng, and S. Handwerger. 2000. Activator protein-2 regulates human placental lactogen gene expression. Mol. Cell. Endocrinol. 160:183-192. [DOI] [PubMed] [Google Scholar]
- 42.Rohlff, C., S. Ahmad, F. Borellini, J. Lei, and R. I. Glazer. 1997. Modulation of transcription factor Sp1 by cAMP-dependent protein kinase. J. Biol. Chem. 272:21137-21141. [DOI] [PubMed] [Google Scholar]
- 43.Schanke, J. T., M. Durning, K. J. Johnson, L. K. Bennett, and T. G. Golos. 1998. SP1/SP3-binding sites and adjacent elements contribute to basal and cyclic adenosine 3′,5′-monophosphate-stimulated transcriptional activation of the rhesus growth hormone-variant gene in trophoblasts. Mol. Endocrinol. 12:405-417. [DOI] [PubMed] [Google Scholar]
- 44.Schön, U., W. Seifarth, C. Baust, C. Hohenadl, V. Erfle, and C. Leib-Mosch. 2001. Cell type-specific expression and promoter activity of human endogenous retroviral long terminal repeats. Virology 279:280-291. [DOI] [PubMed] [Google Scholar]
- 45.Schulte, A. M., S. Lai, A. Kurtz, F. Czubayko, A. T. Riegel, and A. Wellstein. 1996. Human trophoblast and choriocarcinoma expression of the growth factor pleiotrophin attributable to germ-line insertion of an endogenous retrovirus. Proc. Natl. Acad. Sci. USA 93:14759-14764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tarrade, A., K. Schoonjans, J. Guibourdenche, J. M. Bidart, M. Vidaud, J. Auwerx, C. Rochette-Egly, and D. Evain-Brion. 2001. PPAR gamma/RXR alpha heterodimers are involved in human CG beta synthesis and human trophoblast differentiation. Endocrinology 142:4504-4514. [DOI] [PubMed] [Google Scholar]
- 47.Ting, C. N., M. P. Rosenberg, C. M. Snow, L. C. Samuelson, and M. H. Meisler. 1992. Endogenous retroviral sequences are required for tissue-specific expression of a human salivary amylase gene. Genes Dev. 6:1457-1465. [DOI] [PubMed] [Google Scholar]
- 48.Voisset, C., O. Bouton, F. Bedin, L. Duret, B. Mandrand, F. Mallet, and G. Paranhos-Baccala. 2000. Chromosomal distribution and coding capacity of the human endogenous retrovirus HERV-W family. AIDS Res. Hum. Retrovir. 16:731-740. [DOI] [PubMed] [Google Scholar]
- 49.Wada, N., and J. Y. Chou. 1993. Characterization of upstream activation elements essential for the expression of germ cell alkaline phosphatase in human choriocarcinoma cells. J. Biol. Chem. 268:14003-14010. [PubMed] [Google Scholar]
- 50.Yamada, K., H. Ogawa, S. Honda, N. Harada, and T. Okazaki. 1999. A GCM motif protein is involved in placenta-specific expression of human aromatase gene. J. Biol. Chem. 274:32279-32286. [DOI] [PubMed] [Google Scholar]
- 51.Yamamoto, K. K., G. A. Gonzalez, W. H. Biggs III, and M. R. Montminy. 1988. Phosphorylation-induced binding and transcriptional efficacy of nuclear factor CREB. Nature 334:494-498. [DOI] [PubMed] [Google Scholar]
- 52.Yu, C., K. Shen, M. Lin, P. Chen, C. Lin, G. D. Chang, and H. Chen. 2002. GCMa regulates the syncytin-mediated trophoblastic fusion. J. Biol. Chem. 277:50062-50068. [DOI] [PubMed] [Google Scholar]