

Abstract
Influenza virus neuraminidase (NA) plays an essential role in release and spread of progeny virions, following the intracellular viral replication cycle. To test whether NA could also facilitate virus entry into cell, we infected cultures of human airway epithelium with human and avian influenza viruses in the presence of the NA inhibitor oseltamivir carboxylate. Twenty- to 500-fold less cells became infected in drug-treated versus nontreated cultures (P < 0.0001) 7 h after virus application, indicating that the drug suppressed the initiation of infection. These data demonstrate that viral NA plays a role early in infection, and they provide further rationale for the prophylactic use of NA inhibitors.
It is believed that the major function of viral neuraminidase (NA) is at the final stage of infection when NA cleaves sialic acid from cell surface and progeny virions facilitating virus release from infected cells (1, 2). Less is known about NA functions during virus entry into the cell. It has long been assumed that NA promotes virus access to target cells in airways by mucus degradation (3). However, this concept has never been formally proven due to the lack of an adequate experimental system. Moreover, some evidence arguing against the role of NA at the early stages of infection has been reported (reviewed in reference 2).
To address this issue, we studied the effects of the NA inhibitor oseltamivir carboxylate (OC) (9) on influenza virus entry into cultures of human airway epithelium. Primary human tracheobronchial epithelial cells (HTBE; Clonetics) and primary nasal epithelial cells (PromoCell GmbH) were grown on membrane supports (12-mm Transwell-Clear; Corning, Inc.) at the air-liquid interface in serum-free growth factor and hormone-supplemented medium (6, 8). Fully differentiated 4- to 8-week-old cultures were used for all experiments. These cultures were pseudostratified and polarized; contained basal, ciliated, and mucus-secreting cells; and closely resembled human airway epithelium in vivo (Fig. 1). OC (1 μM, if not indicated otherwise) was added to virus suspensions and to basolateral compartments of the cultures shortly before infecting two replicate cultures from the apical side. Two control cultures were infected in the absence of inhibitor. One hour postinfection, we removed the virus inoculum and incubated cultures at the air-liquid interface for additional 6 h to allow intracellular virus replication. The cultures were then fixed, and infected cells were identified by staining with polyclonal antisera to whole viruses followed by corresponding peroxidase-labeled secondary antibodies (Dianova) and aminoethylcarbazole substrate (Sigma). Positive staining indicated successful virus entry in the cell. The cultures were analyzed en face at a magnification of ×300 (Olympus IMT-2). A total number of cells expressing viral antigen was counted in the epithelial segment that included all consecutive microscopic views (0.28 by 0.42 mm) along the diameter of the culture (segment surface area, 3 mm2; number of cells per segment, about 30,000). Four segments per culture were counted by rotating the culture clockwise by 45o. The data for eight segments of two replicate cultures were averaged.
FIG. 1.
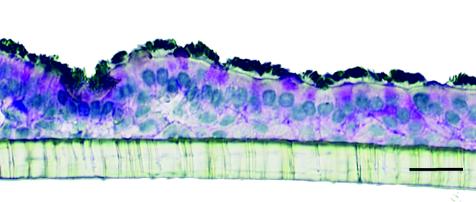
Cross-section of HTBE culture on a membrane support. Shown are immunostaining for cilia (black), periodic acid-Schiff′s reaction for goblet cell mucin (magenta), and counterstaining with hematoxylin. Bar = 20 μm.
In two experiments using HTBE cultures, the human virus A/Memphis/14/96 (H1N1) infected 22- and 65-fold fewer cells in the presence of NA inhibitor as compared to controls (Fig. 2; Table 1) (P < 0.0001). Viruses A/Duck/Alberta/119/98 (H1N1) from a wild aquatic bird and A/Turkey/Italy/2379/99 (H7N1) from domestic poultry were also markedly sensitive to OC in these cultures. In nasal epithelial cultures, OC reduced human and duck virus infection 120- and 520-fold, respectively. These results indicated that inhibition of viral NA suppresses the initiation of virus infection.
FIG. 2.
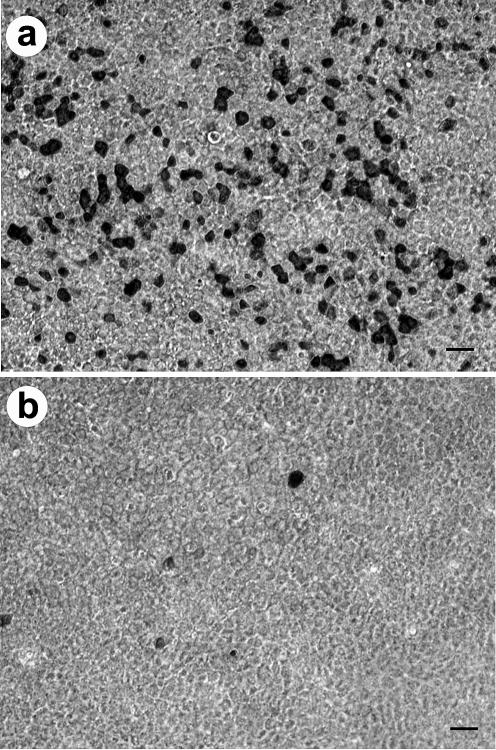
En face view of HTBE cultures infected with human influenza virus A/Memphis/14/96 in the presence of OC (b) and in its absence (a). Shown is immunostaining for virus antigen (black). Bar = 20 μm.
TABLE 1.
Inhibition of influenza virus infection in HTBE and human nasal epithelial cell cultures by OC
| Virus | Culture | MOIa | Mean (SD) no. of infected cells/epithelial segment
|
Control/treated ratio | Pb | |
|---|---|---|---|---|---|---|
| Control | Drug treated | |||||
| A/Memphis/14/96 (HINI) | HTBE | 0.07 | 560 (60) | 26 (6) | 22 | <0.0001 |
| HTBE | 0.25 | 3,100 (600) | 48 (20) | 65 | <0.0001 | |
| HNE | 0.25 | 230 (80) | 2.0 (1.3) | 120 | <0.0001 | |
| A/Duck/Alberta/119/98 (HINI) | HTBE | 0.08 | 2,800 (200) | 44 (15) | 64 | <0.0001 |
| HTBE | 0.04 | 1,500 (300) | 36 (13) | 42 | <0.0001 | |
| HNE | 0.04 | 260 (90) | 0.50 (0.53) | 520 | <0.0001 | |
| A/Turkey/Italy/2379/99 (H7NI) | HTBE | 0.25 | 1,500 (200) | 30 (12) | 50 | <0.0001 |
| A/Chicken/Germany/R28/03 (H7N7) | HTBE | 0.002 | 36 (16) | 0.25 (0.46)c | 140 | <0.0001 |
| 0.88 (0.64)d | 40 | <0.0001 | ||||
| A/Sydney/5/97-like (H3N2) | HTBE | 0.3 | 222 (22) | 4.8 (1.8) | 47 | <0.0001 |
| 154 (39)e | 1.5 | 0.007 | ||||
| 210 (35)f | 1.1 | 0.42 | ||||
| A/Sydney/5/97-like (H3N2) (drug-resistant NA mutant) | HTBE | 0.35 | 360 (93) | 270 (52) | 1.3 | 0.017 |
MOI, multiplicity of infection calculated on the basis of virus infectivity determined in MDCK cells.
P value for comparison between drug-treated and control cultures (unpaired t test).
Infection in the presence of 1 μM OC.
Infection in the presence of 0.1 μM OC.
OC was added to the culture 1 h after virus inoculation.
OC was added to the culture 4 h after virus inoculation.
A/Chicken/Germany/R28/03 (H7N7) belongs to the highly pathogenic avian influenza virus lineage that caused outbreak of fowl plague in commercial poultry farms in The Netherlands, Belgium, and Germany in 2003. These viruses were transmitted to at least 89 humans, most of whom presented with conjunctivitis, but one fatal case of pneumonia occurred also (5). The H7N7 chicken virus infection in HTBE cultures was decreased 140- and 40-fold in the presence of 1 and 0.1 μM OC, respectively. These concentrations represented typical plasma maximum and minimum concentrations of drug achieved in humans following administration of 75 mg of oseltamivir phosphate, the recommended dose for prophylaxis (9).
To confirm the hypothesis that infection inhibition in our experiments was directly related to the inhibition of viral NA enzymatic activity, we used human A/Sydney/5/97-like virus (H3N2) and its oseltamivir-resistant mutant with an R292K substitution in the NA, which renders the NA 9,000-fold less sensitive to inhibition by OC (4). Consistent with the hypothesis, the parent human virus was strongly inhibited by OC, whereas only a marginal level of inhibition of the resistant mutant was observed when both viruses were tested in the same experiment in HTBE cultures (Table 1). Until now, no adequate cell culture assay has been available for monitoring influenza virus resistance to NA inhibitors because of the mismatch between sialic acid receptors in humans and in conventional laboratory cell lines (9, 11). Our results suggest that differentiated cultures of human airway epithelium may be a suitable cell culture system for detection of influenza virus resistance to NA inhibitors.
Finally, using the drug-sensitive A/Sydney/5/97-like virus, we tested infection inhibition by OC added at different times after virus inoculation. Whereas 47-fold fewer cells were infected when the drug was added shortly before the infection, a 1-h delay in addition of OC resulted in only 1.5-fold inhibition with respect to the nontreated control (Table 1). If the drug was added 4 h after virus inoculation, no statistically significant inhibition was observed. These data suggested that OC affected the earliest stages of infection preceding virus replication.
In summary, we have provided here for the first time direct experimental evidence for the essential role of NA at the stage of virus invasion of the ciliated epithelium of human airways. The NA function at this stage is most likely removal of decoy receptors on mucins, cilia, and cellular glycocalix, strong binding to each of which would impede virus access to functional receptors on surface membrane of target cells. However, other NA functions—for example, promotion of hemagglutinin-mediated fusion (7)—cannot be excluded, and further studies are needed to specify the exact mechanisms by which NA promotes virus entry into airway epithelial cells.
With no vaccine available yet against highly pathogenic H7N7 and H5N1 avian influenza viruses, antiviral drugs remain the only option to combat avian flu (10). Compared with other anti-influenza agents, the NA inhibitors are well tolerated and effective against all strains of influenza A and B viruses. There has been little evidence of the emergence of viral resistance, and the infectivity of mutant viruses is usually compromised (9, 11). The ability of NA inhibitors to suppress infection before virus entry into cells underlines their high potential for preventive measures. In particular, our data provide the scientific basis to support the prophylactic use of these compounds for persons at high risk of infection with avian influenza viruses.
Acknowledgments
We thank Robert Webster (St. Jude Children′s Research Hospital, Memphis, Tenn.), Ilaria Capua (Istituto Zooprofilattico Sperimentale delle Venezie, Legnaro, Padua, Italy), and Thomas Mettenleiter (Federal Research Centre for Virus Diseases of Animals, Greifswald-Insel Riems, Germany) for providing influenza viruses.
The study was supported by Roche Products, UK.
REFERENCES
- 1.Air, G. M., and W. G. Laver. 1989. The neuraminidase of influenza virus. Proteins 6:341-356. [DOI] [PubMed] [Google Scholar]
- 2.Bucher, D., and P. Palese. 1975. The biologically active proteins of influenza virus: neuraminidase, p. 83-123. In E. D. Kilbourne (ed.), The influenza viruses and influenza. Academic Press, New York, N.Y.
- 3.Burnet, F. M. 1960. Principles of animal virology. Academic Press, New York, N.Y.
- 4.Carr, J., J. Ives, L. Kelly, R. Lambkin, J. Oxford, D. Mendel, L. Tai, and N. Roberts. 2002. Influenza virus carrying neuraminidase with reduced sensitivity to oseltamivir carboxylate has altered properties in vitro and is compromised for infectivity and replicative ability in vivo. Antivir. Res. 54:79-88. [DOI] [PubMed] [Google Scholar]
- 5.Fouchier, R. A., P. M. Schneeberger, F. W. Rozendaal, J. M. Broekman, S. A. Kemink, V. Munster, T. Kuiken, G. F. Rimmelzwaan, M. Schutten, G. J. Van Doornum, G. Koch, A. Bosman, M. Koopmans, and A. D. Osterhaus. 2004. Avian influenza A virus (H7N7) associated with human conjunctivitis and a fatal case of acute respiratory distress syndrome. Proc. Natl. Acad. Sci. USA 101:1356-1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gray, T. E., K. Guzman, C. W. Davis, L. H. Abdullah, and P. Nettesheim. 1996. Mucociliary differentiation of serially passaged normal human tracheobronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 14:104-112. [DOI] [PubMed] [Google Scholar]
- 7.Huang, R. T., R. Rott, K. Wahn, H.-D. Klenk, and T. Kohama. 1980. The function of the neuraminidase in membrane fusion induced by myxoviruses. Virology 107:313-319. [DOI] [PubMed] [Google Scholar]
- 8.Matrosovich, M. N., T. Y. Matrosovich, T. Gray, N. A. Roberts, and H.-D. Klenk. 2004. Human and avian influenza viruses target different cell types in cultures of human airway epithelium. Proc. Natl. Acad. Sci. USA 101:4620-4624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roberts, N. A. 2001. Anti-influenza drugs and neuraminidase inhibitors. Prog. Drug Res. 56:195-237. [DOI] [PubMed] [Google Scholar]
- 10.Webby, R. J., and R. G. Webster. 2003. Are we ready for pandemic influenza? Science 302:1519-1522. [DOI] [PubMed] [Google Scholar]
- 11.Zambon, M., and F. G. Hayden. 2001. Position statement: global neuraminidase inhibitor susceptibility network. Antivir. Res. 49:147-156. [DOI] [PubMed] [Google Scholar]