

Abstract
The Marburg virus (MARV) envelope consists of a lipid membrane and two major proteins, the matrix protein VP40 and the glycoprotein GP. Both proteins use different intracellular transport pathways: GP utilizes the exocytotic pathway, while VP40 is transported through the retrograde late endosomal pathway. It is currently unknown where the proteins combine to form the viral envelope. In the present study, we identified the intracellular site where the two major envelope proteins of MARV come together as peripheral multivesicular bodies (MVBs). Upon coexpression with VP40, GP is redistributed from the trans-Golgi network into the VP40-containing MVBs. Ultrastructural analysis of MVBs suggested that they provide the platform for the formation of membrane structures that bud as virus-like particles from the cell surface. The virus-like particles contain both VP40 and GP. Single expression of GP also resulted in the release of particles, which are round or pleomorphic. Single expression of VP40 led to the release of filamentous structures that closely resemble viral particles and contain traces of endosomal marker proteins. This finding indicated a central role of VP40 in the formation of the filamentous structure of MARV particles, which is similar to the role of the related Ebola virusVP40. In MARV-infected cells, VP40 and GP are colocalized in peripheral MVBs as well. Moreover, intracellular budding of progeny virions into MVBs was frequently detected. Taken together, these results demonstrate an intracellular intersection between GP and VP40 pathways and suggest a crucial role of the late endosomal compartment for the formation of the viral envelope.
Marburg virus (MARV), a filovirus, is the causative agent of a fatal hemorrhagic fever that causes sporadic outbreaks in central Africa (3, 9, 12, 51). To date, neither a vaccine nor a treatment for MARV infection is available, which is partly due to the limited knowledge of the viral replication cycle. The filamentous, enveloped MARV particles are composed of seven structural proteins and the negative-sense RNA genome (11, 16). The genome is surrounded by a nucleocapsid complex that has four protein constituents, NP, VP35, L, and VP30 (6, 42). Between the nucleocapsid and the lipid envelope, two proteins are detected, the matrix protein VP40 and VP24, whose function is elusive (6, 31). Inserted into the viral lipid envelope is the transmembrane glycoprotein GP (5, 17).
The MARV envelope is composed mainly of a lipid bilayer and the membrane-associated viral proteins VP40 and GP (5, 10, 31). GP is the only surface protein of filoviruses and is assumed to be responsible for binding to cellular receptors and for fusing the viral envelope with the cellular membrane in the course of viral entry into the cells (7). GP is also one of the major targets for the immune response of the infected organism. GP is cotranslationally translocated into the endoplasmic reticulum (ER) and is subjected to heavy N- and O-glycosylation (21). During its transport to the Golgi apparatus, GP is subjected to acylation at two cysteine residues at the border between the membrane anchor and the cytoplasmic tail (19). Serine residues of the ectodomain of GP are phosphorylated in the Golgi apparatus (43). In the trans-Golgi network (TGN), GP is cleaved by the prohormone convertase furin into two subunits, GP1 (170 kDa) and GP2 (46 kDa), that are linked by disulfide bonds (49). When GP was recombinantly expressed in mammalian cells, it was shown to be partially localized at the plasma membrane, indicating that GP in principle does not need the other viral proteins to be correctly transported (5). Further experiments using polarized Madin-Darby canine kidney (MDCK) cells revealed that GP is released exclusively into the culture medium facing the apical membrane, suggesting that the protein contains an autonomous apical transport signal. In MARV-infected polarized MDCK cells, the majority of GP was also transported to the apical membrane; however, the release of infectious progeny virions took place exclusively at the basolateral membrane of the cells. Thus, in the presence of other viral proteins, GP obviously is redirected to an alternative route (43). Another observation indicating a different route of GP transport in the context of the viral infection is intracellular budding of MARV in human macrophages (15). The nature of the cellular membrane compartment, where budding of MARV particles was detected, remains unidentified. However, this observation indicated that the final destination of GP is not exclusively the plasma membrane but may also be an intracellular membrane compartment.
One of the viral factors that is most likely involved in changes to the intracellular route of GP is VP40. When the viral envelope is removed by treatment with a low concentration of detergent, the majority of VP40 as well as GP is found to be associated with the lipid membranes (31). This finding suggested that VP40, together with GP, is involved in the formation of the MARV envelope. VP40 is the major matrix protein of MARV and has recently been shown to use the retrograde late endosomal route for its transport to the plasma membrane. VP40 is initially a soluble protein, which associates with membranes early after synthesis; it then accumulates in multivesicular bodies (MVBs) with a perinuclear location and is subsequently transported to the cell periphery (30). Upon coexpression with GP, VP40 formed virus-like particles (VLPs) that were released into the cell culture supernatant (47). Although it is still unclear whether this process is driven by VP40, GP, or both, it is assumed that the formation of the VLPs is equivalent to the budding process in MARV-infected cells. Morphological studies suggested that the MARV envelope is formed immediately before the budding of virions and is uniquely restricted to the plasma membrane (20). However, the precise site where GP and VP40 are combined and incorporated into the membrane subcompartment that finally serves as the platform for budding of progeny virions remains poorly understood.
To understand in more detail the role of VP40 and GP during viral morphogenesis, we analyzed the mutual impact of the two proteins on their respective intracellular distribution and release from the cells. We were able to show that upon coexpression, VP40 and GP synergistically enhance their release from the cells. This effect was accompanied by a redistribution of GP from the ER or Golgi compartment to peripheral MVBs that contained VP40 and marker proteins of the endosome. These MVBs seem to represent the sites where membrane subcompartments that are enriched simultaneously in VP40 and GP are formed. Ultrastructural analysis suggested that these subcompartments fuse with the plasma membrane to provide the platform from which filamentous VLPs are released into the medium. We also report budding of MARV into MVBs. Our results demonstrate that the late endosomal compartment might serve as a site for the formation of viral envelope.
MATERIALS AND METHODS
Virus.
The Musoke strain of MARV, isolated in 1980 in Kenya (45), was propagated in Vero E6 cells and purified as described previously (19). Vero cells and human macrophages were infected under biosafety level 4 conditions with a multiplicity of infection of approximately 1 PFU per cell and then fixed at 24 or 48 h postinfection.
Cells.
Vero E6 cells, human embryonic kidney 293 cells, and human hepatoma HUH-7 cells were maintained in Dulbecco's modified Eagle medium supplemented with 10% fetal calf serum, l-glutamine, and penicillin-streptomycin solution. The cells were grown in an incubator at 37°C under 5% CO2. Monocytes/macrophages were isolated and cultivated as described previously (15).
Antibodies.
For indirect immunofluorescence (IF) and Western blot (WB) analyses, the following primary antibodies were used: affinity-purified rabbit serum against MARV GP (dilution for IF, 1:100; for WB, 1:2,500), a mouse monoclonal antibody against MARV VP40, which was kindly provided by the Centers for Disease Control, Atlanta, Ga. (dilution for IF, 1:100; for WB, 1:3,000), and affinity-purified rabbit serum against VP40 (dilution for IF, 1:50; for WB, 1:2,500). Monoclonal antibodies against lysosome-associated membrane protein 1 (Lamp-1), integrin alpha 2 (VLA-2α), GM130 (Golgi compartment), early endosome antigen 1 (EEA1), and annexin II were obtained from Transduction Laboratories (BD Biosciences, Heidelberg, Germany) and used in dilution according to the manufacturer's instructions for WB analyses. Rabbit polyclonal serum against calnexin was from Stressgen (dilution for WB, 1:2,000). Secondary antibodies conjugated with fluorescein isothiocyanate (FITC) or rhodamine (Dianova, Hamburg, Germany) or with 7-amino-4-methylcoumarin-3-acetic acid (AMCA; Vector Laboratories) were used for IF in dilutions of 1:100. Secondary antibodies conjugated with horseradish peroxidase (dilution for WB, 1:40,000) or with colloidal gold (dilution for immunoelectron microscopy, 1:30) were from Dianova or from Dako (A/S, Copenhagen, Denmark).
DNA plasmids and molecular cloning.
The open reading frames of VP40 or GP (for reference, see EMBL Nucleotide Sequence Database accession number Z12132) were cloned into the mammalian expression vector pCAGGS, using the restriction enzymes SmaI and NotI. The generated plasmids contain VP40 or GP genes under the control of the chicken beta-actin promoter and were verified via sequencing (29, 30, 38).
Living Colors subcellular localization vectors (pECFP-ER, pECFP-Golgi, and pECFP-Endo) were obtained from BD Biosciences Clontech. pECFP-ER encodes a fusion protein consisting of enhanced cyan fluorescent protein (ECFP), the ER-targeting sequence of calreticulin (18), and the sequence encoding the ER retrieval sequence, KDEL (37, 41). ECFP-ER is a soluble protein that localizes in the lumen of the ER in transfected cells. pECFP-Golgi encodes a fusion protein consisting of ECFP and a sequence encoding the N-terminal 81 amino acids of human β-1,4-galactosyltransferase (50). This region of human β-1,4-galactosyltransferase contains the membrane-anchoring signal peptide that targets the fusion protein to the transmedial region of the Golgi apparatus (24, 34, 52). pECFP-Endo encodes a fusion protein consisting of the human RhoB GTPase containing an N-terminal c-Myc epitope tag and ECFP. The RhoB GTPase localizes the ECFP-c-Myc-RhoB fusion protein to the surface of vesicles of the endocytic pathway (1).
Transfection of cells.
293 cells and HUH-7 cells were transfected with plasmids by using Lipofectamine PLUS reagent (Life Technologies) according to the manufacturer's instructions. Transfected cells were incubated at 37°C for 24 or 48 h. Cotransfection of 293 cells or HUH-7 cells with plasmids pC-VP40 and pC-GP was done with plasmid concentrations that resulted in the proteins being expressed in a ratio that was similar to that of MARV-infected cells. The optimal proportion between pC-VP40 and pC-GP was 4:1 (our unpublished data). For cotransfection of HUH-7 cells with pC-VP40, pC-GP, and Living Colors vectors, equal amounts of plasmids were used (1:1:1). In all experiments, the overall amounts of transfected plasmids were held constant by adding empty vector (pCAGGS).
Electrophoresis and immunoblot analysis.
At 48 h posttransfection, 3 × 106 transfected 293 cells were washed two times with phosphate-buffered saline (PBS), resuspended in 400 μl of PBS, lysed by being mixed with 200 μl of 4× sample buffer (100 mM Tris-HCl [pH 6.8], 4% sodium dodecyl sulfate [SDS], 20% 2-mercaptoethanol, 20% glycerol, 0.2% bromophenol blue), and heated for 5 min to 96°C. Supernatants of the cells were pelleted for 3 h at 35,000 rpm through a 20% sucrose cushion. Pellets were resuspended in 40 μl of PBS, mixed with 20 μl of 4× sample buffer, and heated for 5 min to 96°C. Five microliters of the lysate (corresponding to 2.5 × 104 cells) and 7 μl of the pelleted supernatant (corresponding to 3.5 × 105 cells) were separated by SDS-12% polyacrylamide gel electrophoresis and electrophoretically transferred onto polyvinylidene fluoride membranes. The membranes were blocked at 4°C overnight with 10% milk powder in PBS. The blots were incubated for 1 h with the respective primary antibody diluted in PBS supplemented with 1% milk powder and 0.1% Tween 20, followed by incubation with a secondary antibody complexed with horseradish peroxidase (Dianova). Protein bands were visualized with SuperSignal chemoluminescence substrate as described by the supplier (Pierce).
IF analysis.
The HUH-7 cells were washed with PBS at 24 h posttransfection and fixed with 4% paraformaldehyde for 1 h at 4°C. The cells were then rinsed two times with PBS and incubated with 0.1 M glycine in PBS for 5 min at room temperature. Thereafter, samples were washed once with PBS and permeabilized with PBS containing 0.1% Triton X-100. After a wash with PBS, the cells were incubated in blocking solution (2% bovine serum albumin, 0.2% Tween 20, 5% glycerol in 1× PBS) and stained first with the appropriate primary antibodies and then with secondary antibodies as indicated below (see figure legends). Microscopic analysis was performed with an Axiomat fluorescence microscope (Zeiss).
Electron microscopy.
For immunoelectron microscopy, cells were fixed with 4% paraformaldehyde, and after dehydration, samples were embedded in LR Gold (32). Indirect immunogold labeling was carried out with ultrathin sections. The antibodies used are given below (see figure legends). For negative staining, particulate material from the culture media of 293 cells was collected at 48 h posttransfection onto Formvar-coated nickel grids and incubated with PBS containing 1% bovine serum albumin (BSA) for 10 min. The grids were then probed with rabbit anti-GP polyclonal antibody (1:100) in PBS containing 0.5% BSA and 0.025% Tween 20 (PBS-BSA-Tween) or with monoclonal anti-VP40 antibody (1:100 in PBS-BSA-Tween). Next, the grids were rinsed six times with PBS, followed by incubation with a donkey anti-rabbit immunoglobulin conjugated to 12-nm-diameter gold particles (1:30 dilution; Dianova) or a donkey anti-mouse immunoglobulin conjugated to 6-nm-diameter gold particles (1:30 dilution; Dianova). After being washed, the samples were fixed for 10 min in 0.25% glutaraldehyde, negatively stained with 2% phosphotungstic acid solution, and examined with a Zeiss 109 electron microscope at 80 kV.
RESULTS
Cellular membrane proteins are specifically released upon coexpression of MARV GP and VP40.
It has been published earlier that MARV GP is released into the supernatant of GP-expressing cells (43). A recent study by Swenson et al. confirmed this observation and, in addition, showed that the amount of GP released from the cells was increased by coexpression of the viral matrix protein VP40 (47). To investigate in more detail the release of GP and VP40, as well as the impact of VP40 on the release of GP, we compared the expression of the GP and VP40 proteins singly with the coexpression of both proteins. Expression of the viral proteins was performed in 293 cells by transfection with plasmids containing the respective gene under the control of the beta-actin promoter. At 48 h posttransfection, the culture medium was collected, and the cells were lysed. Particulate material from the culture medium was pelleted through a sucrose cushion. Pellets and cell lysates were then analyzed by Western blotting.
Upon single expression, GP and VP40 were detected both intracellularly and in the particulate material from the supernatant (Fig. 1A and B, lanes 2 and 3). In agreement with previously obtained data (5), both the immature form of GP located in the ER (Fig. 1A, lanes 3 and 4) and the mature cleaved form of GP (GP1 and GP2) were detected inside the cells. In the supernatant, only cleaved GP was observed (Fig. 1B, lanes 3 and 4). This finding suggests that the release of GP specifically involves the molecules that passed the TGN, where cleavage of GP takes place (49). Upon coexpression, VP40 and GP synergistically stimulated their respective release, indicating a mutual influence of the two proteins (Fig. 1B, lanes 2 to 4). These data are in line with previous observations that release of Ebola virus (EBOV) or MARV GP could be enhanced by coexpression with the respective matrix protein VP40 (4, 47).
FIG. 1.
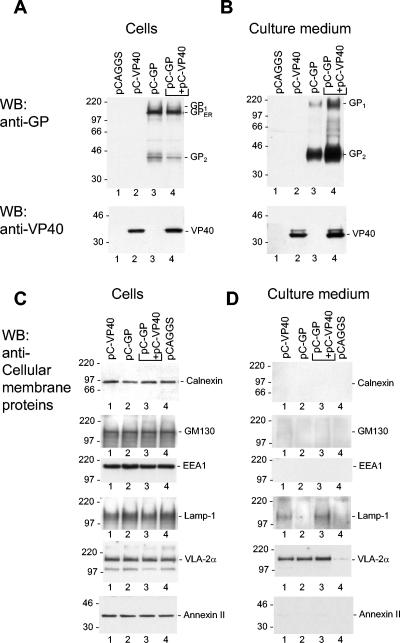
Detection of GP, VP40, and cellular membrane proteins in cell lysate and cellular supernatant by immunoblot analysis. (A to D) 293 cells were transfected with the plasmids encoding the proteins indicated above the lanes. At 48 h posttransfection, the cells and particulate material of cellular supernatant were harvested, diluted as described in Materials and Methods, and subjected to SDS-PAGE (12% polyacrylamide). The positions of the immature form of GP located in the ER (GPER), GP1, GP2, VP40, and cellular membrane proteins are indicated. (A and C) Cell lysate; (B and D) supernatant.
It was of interest to determine whether the release of the viral proteins also resulted in the release of cellular membrane proteins. The intracellular expression levels of the tested marker proteins were not changed upon the expression of the viral proteins (Fig. 1C). In the supernatant, the plasma membrane marker VLA-2α was detected when cells expressed GP, VP40, or both (Fig. 1D, lanes 1 to 3). A weak signal for VLA-2α was also detected in the supernatant of control cells (Fig. 1D, lanes 4). Additionally, the late endosomal marker Lamp-1 was found in supernatants of cells expressing either VP40 or VP40 and GP (Fig. 1D, lanes 1 and 3). The other cellular marker proteins tested, such as the ER-resident protein calnexin, the Golgi marker GM130, early endosomal marker EEA1, and the cytosolic and plasma membrane-associated annexin II, were not present in any detectable amounts in the supernatant. The signal of VLA-2α in the supernatant of control cells (lane 4) most likely represented release of host-derived microvesicles (8). Detection of increased levels of Lamp-1 and VLA-2α in the supernatant suggested that these proteins are incorporated into the VP40- or GP-induced particles.
Morphological characterization of VLPs.
To determine the morphology of particles induced by GP expression, supernatants of GP-expressing cells were pelleted through a 20% sucrose cushion. The pellets were resuspended in PBS and then analyzed by electron microscopy, which revealed round or slightly elongated membrane vesicles with diameters ranging from 50 to 300 nm. The vesicles contained clearly visible surface spikes that were composed of GP, as evidenced by immunoelectron microscopy using a rabbit anti-GP polyclonal antibody (Fig. 2A and B).
FIG. 2.
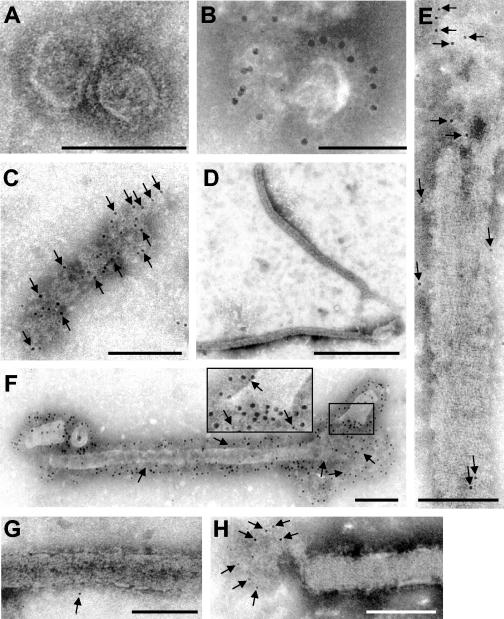
Immunoelectron microscopic analysis of VLPs containing GP, VP40, or both proteins. The supernatants of 293 cells were centrifuged through a 20% sucrose cushion, and pelleted material was negatively stained with 2% phosphotungstic acid solution and analyzed by electron microscopy. (A and B) Round and pleomorphic particles in the supernatant of cells expressing GP. (B) Immunoelectron microscopy of GP-containing particles with a rabbit anti-GP antibody (12-nm-diameter gold beads). (C) Membrane patches and (D and E) filamentous particles in the supernatant of cells expressing VP40. Samples were immunostained with a mouse anti-VP40 antibody (6-nm-diameter gold beads) (arrows). (F) Filamentous particles in the supernatant of cells expressing GP and VP40. Double immunostaining was performed with a rabbit anti-GP antibody (12-nm-diameter gold beads) and a mouse anti-VP40 antibody (6-nm-diameter gold beads) (arrows). (G and H) Filamentous particles in the supernatant of cells expressing GP and VP40. Staining was performed with anti-Lamp-1 antibodies (6-nm-diameter gold beads) (arrows). Bars, 100 nm (A to C, E, G, and H), 1,000 nm (D), and 200 nm (F).
Supernatants of VP40-expressing cells contained membrane patches of pleomorphic shapes and sizes (Fig. 2C) as well as characteristic filamentous particles, 53 to 80 nm in diameter, that closely resembled the morphology of MARV particles (Fig. 2D and E). These particles were further named VLPs. The length of VLPs ranged from 500 to more than 2,000 nm. Immunogold labeling detected VP40 in association with the released membrane patches (Fig. 2C) and with VLPs at places where the lipid bilayer was partially destroyed (Fig. 2E), indicating that VP40 is located beneath the lipid bilayer inside VLPs. A regular striation of 4.2 to 5 nm was observed at the surface of VP40-induced VLPs. Such striation was also found in the envelopes of MARV particles (31, 36). Upon coexpression of GP and VP40, membrane patches (not shown) and filamentous VLPs with diameters of 67 ± 8.5 nm and lengths of up to 2,000 nm were observed. Immunoelectron microscopic analysis showed that VLPs contained GP and VP40 (Fig. 2F). Finally, it was observed that VLPs contained traces of Lamp-1 (Fig. 2G), confirming the results shown in Fig. 1D. Interestingly, the labeling of Lamp-1 by gold particles was more pronounced in the membrane vesicles that are occasionally found to be attached to the VLPs (Fig. 2H). It is presumed that these vesicles represent an incomplete pinching off of the filamentous particles. These results provide evidence that MARV VP40, like EBOV VP40, displays budding activity and has the intrinsic ability to determine the filamentous morphology of the virions (39, 48). These results point further to a specific interaction between GP and VP40 resulting in the incorporation of GP into the VP40-induced VLPs.
GP is redistributed into MVBs containing VP40.
We have shown recently that GP is transported along the exocytotic pathway whereas VP40 is transported along the retrograde late endosomal pathway (5, 30). It is obvious that the routes of GP and VP40 intersect at a particular point, since the two proteins need to be sorted into the same membrane subcompartment from which VLPs containing the two proteins are released. At first glance, the secretory pathway that is used by GP and the retrograde late endosomal pathway used by VP40 do not intersect before they end at the plasma membrane. To determine whether coexpression of GP and VP40 results in changes to their respective intracellular localization, HUH-7 cells expressing GP alone or GP together with VP40 were analyzed by IF microscopy. Several cellular compartments were labeled by expression of fluorescent fusion proteins that are targeted either to the ER (ECFP-ER), the Golgi compartment (ECFP-Golgi), or the endosomal compartment (ECFP-Endo).
As expected, when GP was expressed alone, it was located in the perinuclear region (Fig. 3A), partially colocalized with the ER and partially colocalized with the Golgi apparatus. A weak signal of GP also lined the plasma membrane (5). Coexpression with VP40 resulted in redistribution of GP to VP40-positive clusters located close to the cell periphery (Fig. 3C, upper panels). The distribution of VP40 was not changed upon coexpression with GP (Fig. 3B and C). The VP40- and GP-positive clusters colocalized with a marker of the endosomal compartment but not with markers of the ER or Golgi compartment (Fig. 3C, columns 1 to 3). Colocalization of VP40 and the endosomal marker was almost complete and also comprised GP-negative clusters that had a perinuclear location. We have shown previously that VP40 is intracellularly associated with the late endosomal compartment and can be detected in perinuclear and peripheral MVBs (30, 31). The present findings indicate that GP is redirected to the peripheral but not to the perinuclear VP40-positive MVBs.
FIG. 3.
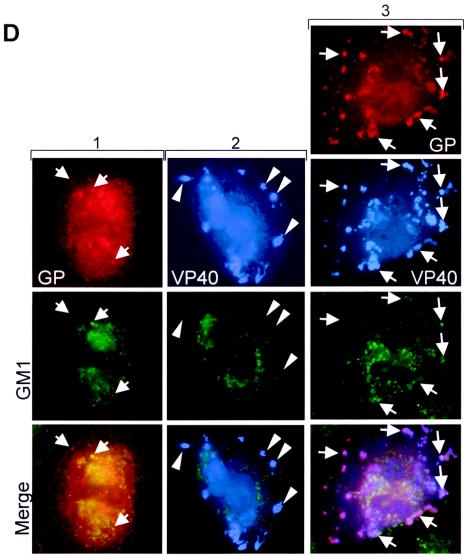
Intracellular distribution of GP in the presence of VP40. (A) HUH-7 cells were transfected with pC-GP and pECFP-ER vector (column 1) or with pC-GP and pECFP-Golgi vector (column 2), fixed at 24 h posttransfection, and immunostained with a rabbit anti-GP and a secondary donkey anti-rabbit IgG conjugated with rhodamine. (B) HUH-7 cells were transfected with pC-VP40 and pECFP-ER vector (column 1) with pC-VP40 and pECFP-Golgi vector (column 2), or with pC-VP40 and pECFP-Endo vector (column 3). After fixation with 4% paraformaldehyde at 24 h posttransfection, cells were immunostained with a mouse anti-VP40 antibody. Bound antibodies were detected using secondary horse anti-mouse antibody conjugated with AMCA. Arrows show peripherally located clusters where Endo marker and VP40 are colocalized. Arrow-heads show peripheral VP40-positive clusters which are not colocalized with the ER and Golgi marker. (C) HUH-7 cells were transfected with pC-GP, pC-VP40, and pECFP-ER vector (column 1), with pC-GP, pC-VP40, and pECFP-Golgi vector (column 2), or with pC-GP, pC-VP40, and pECFP-Endo vector (column 3). After fixation with 4% paraformaldehyde at 24 h posttransfection, cells were immunostained with a rabbit anti-GP antibody and a mouse anti-VP40 antibody. Bound antibodies were detected using secondary donkey anti-rabbit IgG conjugated with rhodamine and horse anti-mouse antibody conjugated with AMCA. Arrows show peripherally located clusters where GP and VP40 are colocalized. (D) HUH-7 cells were transfected with pC-GP and pCAGGS (column 1), with pC-VP40 and pCAGGS (column 2), or with pC-GP, pC-VP40, and pCAGGS (column 3). Fixation and immunostaining of GP and VP40 were done as described for panel C. Immunostaining of ganglioside M1 was performed by using cholera toxin B conjugated with FITC. Arrows show colocalization between GM1 and GP or between GM1 and VP40-GP-positive clusters. Arrowheads show peripheral VP40-positive clusters which are not colocalized with GM1.
It has been earlier published that the formation of filovirus particles is dependent on the association of GP with lipid rafts (4). To determine whether observed GP-VP40-positive clusters are enriched in ganglioside M1 (GM1), a marker of lipid rafts, we analyzed its distribution in cells expressing GP and VP40. GM1, like other gangliosides, is located in the plasma membrane and in intracellular membranes (2, 23, 35) and can be detected by cholera toxin B (26, 27). Partial colocalization between GP and GM1 was detected in the perinuclear region and to a lower degree at the plasma membrane (Fig. 3D, column 1). GM1 was not detected in peripheral VP40 clusters upon single expression of VP40 (Fig. 3D, column 2). However, GM1 accumulated in the peripheral VP40 clusters that contained both GP and VP40 (Fig. 3D, column 3). These findings suggest that lipid sorting accompanies formation of the membrane subcompartment that contains both GP and VP40.
Colocalization of GP and VP40 in peripheral clusters was confirmed by immunoelectron microscopy. VP40- and GP-positive clusters with diameters ranging from 100 to 1,000 nm were detected. These clusters had multivesicular or multilayered structures, which are typical for MVBs. They were located (i) in the cell body (Fig. 4A and B), (ii) very close to the plasma membrane (Fig. 4B and E), and (iii) as filopodium- or lamellipodium-like membrane protrusions at the cell surface (Fig. 4C, F, and G). Most likely, the different localizations of the MVBs represented different steps of their transport to the plasma membrane that result in either filopodium- or lamellipodium-like membrane protrusions and culminate in the release of VLPs or membrane patches. The sizes of the MVBs might limit the sizes of the protrusions at the plasma membrane.
FIG. 4.
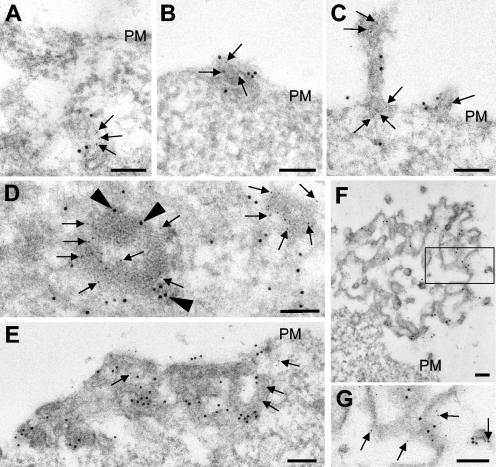
Immunoelectron microscopic analysis of the ultrathin sections of GP- and VP40-expressing 293 cells. (A to G) GP- and VP40-expressing 293 cells were fixed at 24 h posttransfection, dehydrated, and embedded in LR Gold. Double immunostaining of ultrathin sections was performed with a rabbit anti-GP antibody (12-nm-diameter gold beads) (arrowheads) and a mouse anti-VP40 antibody (6-nm-diameter gold beads) (arrows). (G) Enlarged framed fragment of panel F. PM, plasma membrane. Bars, 100 nm.
GP and VP40 are colocalized in MVBs in MARV-infected cells.
To investigate the sites of colocalization for GP and VP40 in MARV-infected cells, Vero cells at 2 days postinfection were subjected to IF analysis. GP was located predominantly in the perinuclear region and was partly colocalized with VP40 in clusters which were distributed both at the plasma membrane and intracellularly (Fig. 5A). This result reinforces the idea that delivery of GP to the cell surface in MARV-infected cells involves an interaction with a VP40-positive compartment before GP and VP40 arrive at the plasma membrane. Immunoelectron microscopy confirmed that sites of GP and VP40 colocalization in MARV-infected cells represent MVBs (Fig. 5B to E). GP and VP40 were detected both in proximity to the viral inclusions (Fig. 5B) and close to the cell surface (Fig. 5C to E). The appearance of GP- and VP40-enriched membrane patches at the site of virus budding was similar to the appearance of those detected in cells coexpressing GP and VP40 (Fig. 4).
FIG. 5.
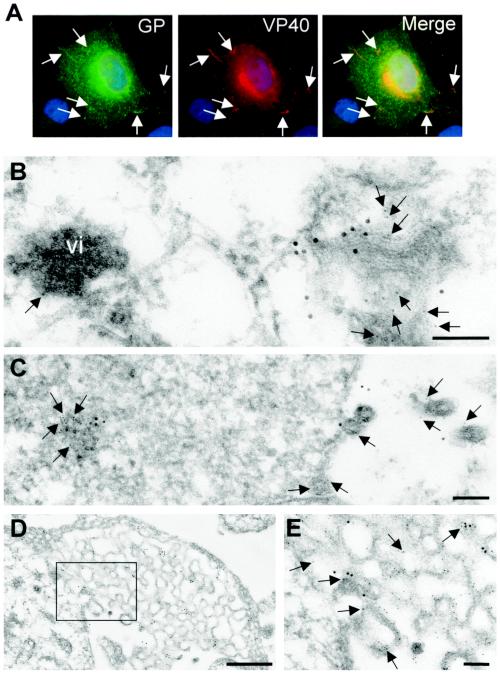
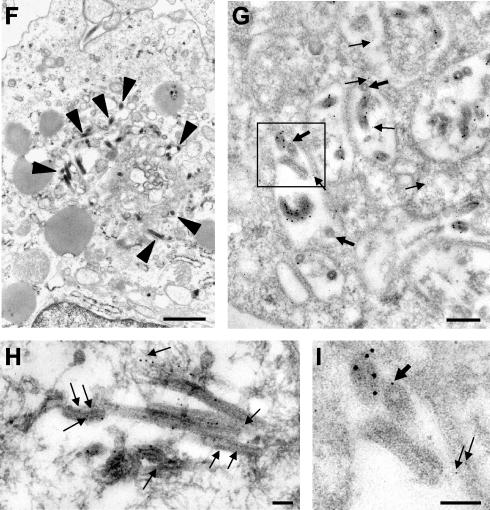
Distribution of GP and VP40 in MARV-infected cells. (A) MARV-infected Vero E6 cells were fixed at 2 days postinfection and immunostained with a rabbit anti-GP antibody and a mouse anti-VP40 antibody. Bound antibodies were detected using secondary donkey anti-rabbit IgG conjugated with FITC and donkey anti-mouse antibody conjugated with rhodamine. The nuclei were counterstained with DAPI (4′,6′-diamidino-2-phenylindole). Arrows show peripherally located clusters where GP and VP40 are colocalized. (B to E and H) MARV-infected Vero cells were fixed at 48 h postinfection, dehydrated, and embedded in LR Gold. Double immunostaining of ultrathin sections was performed with a rabbit anti-GP antibody (12-nm-diameter gold beads) and a mouse anti-VP40 antibody (6-nm-diameter gold beads) (arrows). vi, viral inclusion. (E) Enlarged framed fragment of panel D. (F, G, and I) MARV-infected human macrophages were fixed at 48 h postinfection, dehydrated, and embedded either in Epon (F) or in LR Gold (G and I). (F) MARV budding inside MVBs (arrowheads indicate viral particles). (G and I) Double immunostaining of ultrathin sections was performed with a rabbit anti-VP40 antibody (12-nm-diameter gold beads) and a mouse anti-Lamp-1 antibody (6-nm-diameter gold beads) (arrows). (I) Enlarged framed fragment of panel G. Thick arrows show Lamp-1-bound gold beads in the viral particles. Bars, 100 nm (B, C, E, H, and I), 500 nm (D), 1,000 nm (F), and 200 nm (G).
Interestingly, we found MARV budding not only at the plasma membrane but also intracellularly. Intracellular budding was observed in both Vero cells and human macrophages at 2 days postinfection. Analysis of the sites of intracellular budding of MARV showed that these sites represent MVBs (Fig. 5F to I). Sites of intracellular budding contained multiple profiles of curved membranes with viral particles (Fig. 5F). These curved membranes were Lamp-1 positive (Fig. 5G and I). Some of the released MARV particles also contained Lamp-1 (Fig. 5G and I), indicating that this cellular protein was incorporated into the virion. By use of immunostaining, GP was detected at the surface of the viral particles that bud into the lumen of the MVBs (Fig. 5H). These data indicate that delivery of GP to the endosomal compartment also takes place in MARV-infected cells. Intracellular budding detected within MVBs underlines the importance of the late endosome for MARV morphogenesis.
DISCUSSION
The aim of the present study was to identify the site where the transmembrane protein GP and the peripheral membrane protein VP40 of MARV are combined to form the viral envelope. The formation of VLPs upon coexpression of VP40 and GP suggests that the two proteins are transported to the same membrane compartment that serves as the budding site. However, transport of GP was shown to involve the classical secretory pathway from the ER, via the Golgi apparatus and the TGN, to the plasma membrane (5). On the other hand, transport of VP40 to the plasma membrane involves the late endosomal compartment and is associated with the formation of clusters containing VP40 and late endosomal proteins like Lamp-1. These clusters represent MVBs that appear first in the perinuclear region and then at the cellular periphery (30, 31). Since the two transport routes both end at the plasma membrane, it was tempting to speculate that merging of VP40 and GP takes place at the cell surface. To our surprise, the present study showed that upon coexpression of GP with VP40, a redistribution was detected of the majority of GP from the ER-Golgi compartment to peripheral clusters that were positive for VP40 and an endosomal marker. In ultrastructural analyses, these clusters turned out to be MVBs. Our observations were supported by the fact that in MARV-infected cells, GP and VP40 were also detected as being colocalized in peripheral MVBs.
To date, the mechanism leading to the redistribution of GP into the VP40-containing MVBs is not clear. It is possible that the signal that targets GP to the endosomes is located in either its cytoplasmic, lumenal, or transmembrane domain. On the other hand, a direct interaction between the two proteins might not be necessary if sorting of GP is accomplished by lipid sorting (46). A recent study showed that MARV GP and EBOV GP are colocalized with the lipid raft molecule GM1 and, moreover, that viral particles incorporate GM1 (4). It was then suggested that filoviruses employ lipid microdomains for their release from infected cells. In agreement with this, colocalization between VP40- and GP-positive MVBs and GM1 was also detected in our study. Moreover, localization of GM1 in peripheral MVBs was dependent on the presence of both viral proteins, since neither GP nor VP40 alone could induce a redistribution of GM1. Notably, perinuclear sites with high levels of GM1 were not colocalized with VP40 and GP, indicating that membranes highly enriched in GM1 are not preferred targets of GP and VP40. Conversely, GP- and VP40-positive MVBs seem to recruit GM1. Taken together, our data indicate that lipid sorting takes place simultaneously with the targeting of GP to the peripheral VP40-positive MVBs. Most likely, the accumulation of the viral proteins induces the recruitment of specific lipids that are suitable for the formation of the characteristic filamentous viral particles. The driving force and the targeting signals of this process are currently unknown.
In the present study, we have shown that even upon single expression, GP and VP40 were released into the medium, where GP was found in spherical vesicles and VP40 was found in membrane patches and/or filamentous VLPs. These findings indicate a central role for VP40 in the formation of the filamentous structure of MARV particles. Thus, VP40 displays budding activity similar to that of matrix proteins of other enveloped viruses, like EBOV (37, 46), vesicular stomatitis virus (28, 33), and human parainfluenza virus type 1 (13), and the retroviral Gag protein (14, 22, 25). Coexpression with VP40 led to the incorporation of GP into VP40-induced filamentous VLPs. We have reported previously that traces of endosomal marker proteins, like Lamp-1, are incorporated into MARV particles (31). Trace amounts of Lamp-1 were also detected in the VP40-induced VLPs but not in the GP-induced vesicles. This finding indicates on the one hand that incorporation of Lamp-1 into the viral particles is triggered by VP40 and on the other hand that the late endosomal compartment is strongly involved in assembly of MARV. Indeed, in addition to the fact that VP40 uses the late endosome for its transport (30), we detected budding of MARV particles into MVBs. These particles contained GP, indicating that the surface protein is also channeled into the late endosomal compartment in MARV-infected cells.
We repeatedly observed the formation of large vacuoles filled with multiple viral particles both in human macrophages and in Vero cells infected with MARV, suggesting that along with budding at the plasma membrane, MARV budding into MVBs represents a constitutive pathway in cells of different tissue origin. Moreover, intracellular budding of MARV indicates that not only membrane-associated viral proteins but also nucleocapsid-associated proteins can be targeted to the MVBs. At the late stages of infection, MARV budding into MVBs is observed more frequently, suggesting that putative cellular factors which are responsible for the delivery of viral components to the cell surface are exhausted. Recently, it has been described that human immunodeficiency virus type 1 also buds into MVBs of infected macrophages, and the infectious particles are ultimately released into the medium (40). Sherer et al. reported that in cell types thought to exclusively release retroviruses at the plasma membrane, budding into MVBs also takes place (44). Together with our observations, these results suggest that budding into MVBs may represent a common mechanism of virus assembly. Possibly, the intracellular viral particles serve as a source of infectious units that can be delivered in a signal-dependent manner, e.g., upon cell-to-cell contact as hypothesized by Sherer and colleagues (44).
Taken together, the presented data show for the first time that a viral matrix protein triggers the redistribution of a viral surface transmembrane protein to the endosomal compartment. It is suggested that these VP40- and GP-containing membrane structures represent precursors of the viral envelope whose formation is an intermediate step for the assembly of progeny virions. Either these envelope precursors are transported to the plasma membrane, where budding of progeny virions takes place, or they remain in the endosomal compartment and enable budding of virions into MVBs. Defining the specific subcellular compartment where the viral envelope is assembled may allow the design of antivirals that specifically inhibit the release of infectious virus.
.
Acknowledgments
We thank Angelika Lander for technical assistance, Hans-Dieter Klenk for critical reading of the manuscript and helpful discussions, and Milton Medina for editing the manuscript. We thank Yoshihiro Kawaoka for providing the vector pCAGGS and Stuart Nichol (Centers for Disease Control and Prevention, Atlanta, Ga.) for providing a monoclonal antibody against VP40.
This work was supported by the Deutsche Forschungsgemeinschaft, Sonderforschungsbereich 535 TP A13 and 593 TP B3, and by the Land Hessen through a fellowship to Sandra Bamberg.
REFERENCES
- 1.Adamson, P., H. F. Paterson, and A. Hall. 1992. Intracellular localization of the P21rho proteins. J. Cell Biol. 119:617-627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ardail, D., I. Popa, J. Bodennec, P. Louisot, D. Schmitt, and J. Portoukalian. 2003. The mitochondria-associated endoplasmic-reticulum subcompartment (MAM fraction) of rat liver contains highly active sphingolipid-specific glycosyltransferases. Biochem. J. 371:1013-1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bausch, D. G., M. Borchert, T. Grein, C. Roth, R. Swanepoel, M. L. Libande, A. Talarmin, E. Bertherat, J. J. Muyembe-Tamfum, B. Tugume, R. Colebunders, K. M. Konde, P. Pirad, L. L. Olinda, G. R. Rodier, P. Campbell, O. Tomori, T. G. Ksiazek, and P. E. Rollin. 2003. Risk factors for Marburg hemorrhagic fever, Democratic Republic of the Congo. Emerg. Infect. Dis. 9:1531-1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bavari, S., C. M. Bosio, E. Wiegand, G. Ruthel, A. B. Will, T. W. Geisbert, M. Hevey, C. Schmaljohn, A. Schmaljohn, and M. J. Aman. 2002. Lipid raft microdomains: a gateway for compartmentalized trafficking of Ebola and Marburg viruses. J. Exp. Med. 195:593-602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Becker, S., H.-D. Klenk, and E. Mühlberger. 1996. Intracellular transport and processing of the Marburg virus surface protein in vertebrate and insect cells. Virology 225:145-155. [DOI] [PubMed] [Google Scholar]
- 6.Becker, S., C. Rinne, U. Hofsass, H.-D. Klenk, and E. Mühlberger. 1998. Interactions of Marburg virus nucleocapsid proteins. Virology 249:406-417. [DOI] [PubMed] [Google Scholar]
- 7.Becker, S., M. Spiess, and H.-D. Klenk. 1995. The asialoglycoprotein receptor is a potential liver-specific receptor for Marburg virus. J. Gen. Virol. 76:393-399. [DOI] [PubMed] [Google Scholar]
- 8.Bess, J. W., Jr., R. J. Gorelick, W. J. Bosche, L. E. Henderson, and L. O. Arthur. 1997. Microvesicles are a source of contaminating cellular proteins found in purified HIV-1 preparations. Virology 230:134-144. [DOI] [PubMed] [Google Scholar]
- 9.Borchert, M., J. J. Muyembe-Tamfum, R. Colebunders, M. Libande, M. Sabue, and P. Van Der Stuyft. 2002. Short communication: a cluster of Marburg virus disease involving an infant. Trop. Med. Int. Health 7:902-906. [DOI] [PubMed] [Google Scholar]
- 10.Bukreyev, A., V. E. Volchkov, V. M. Blinov, and S. V. Netesov. 1993. The GP-protein of Marburg virus contains the region similar to the ′immunosuppressive domain' of oncogenic retrovirus P15E proteins. FEBS Lett. 323:183-187. [DOI] [PubMed] [Google Scholar]
- 11.Bukreyev, A. A., V. E. Volchkov, V. M. Blinov, S. A. Dryga, and S. V. Netesov. 1995. The complete nucleotide sequence of the Popp (1967) strain of Marburg virus: a comparison with the Musoke (1980) strain. Arch. Virol. 140:1589-1600. [DOI] [PubMed] [Google Scholar]
- 12.Colebunders, R., H. Sleurs, P. Pirard, M. Borchert, M. Libande, J. P. Mustin, A. Tshomba, L. Kinuani, L. A. Olinda, F. Tshioko, and J. J. Muyembe-Tamfum. 2004. Organisation of health care during an outbreak of Marburg haemorrhagic fever in the Democratic Republic of Congo, 1999. J. Infect. 48:347-353. [DOI] [PubMed] [Google Scholar]
- 13.Coronel, E. C., K. G. Murti, T. Takimoto, and A. Portner. 1999. Human parainfluenza virus type 1 matrix and nucleoprotein genes transiently expressed in mammalian cells induce the release of virus-like particles containing nucleocapsid-like structures. J. Virol. 73:7035-7038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Delchambre, M., D. Gheysen, D. Thines, C. Thiriart, E. Jacobs, E. Verdin, M. Horth, A. Burny, and F. Bex. 1989. The GAG precursor of simian immunodeficiency virus assembles into virus-like particles. EMBO J. 8:2653-2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feldmann, H., H. Bugany, F. Mahner, H.-D. Klenk, D. Drenckhahn, and H. J. Schnittler. 1996. Filovirus-induced endothelial leakage triggered by infected monocytes/macrophages. J. Virol. 70:2208-2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feldmann, H., E. Mühlberger, A. Randolf, C. Will, M. P. Kiley, A. Sanchez, and H.-D. Klenk. 1992. Marburg virus, a filovirus: messenger RNAs, gene order, and regulatory elements of the replication cycle. Virus Res. 24:1-19. [DOI] [PubMed] [Google Scholar]
- 17.Feldmann, H., C. Will, M. Schikore, W. Slenczka, and H.-D. Klenk. 1991. Glycosylation and oligomerization of the spike protein of Marburg virus. Virology 182:353-356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fliegel, L., K. Burns, D. H. MacLennan, R. A. Reithmeier, and M. Michalak. 1989. Molecular cloning of the high affinity calcium-binding protein (calreticulin) of skeletal muscle sarcoplasmic reticulum. J. Biol. Chem. 264:21522-21528. [PubMed] [Google Scholar]
- 19.Funke, C., S. Becker, H. Dartsch, H.-D. Klenk, and E. Mühlberger. 1995. Acylation of the Marburg virus glycoprotein. Virology 208:289-297. [DOI] [PubMed] [Google Scholar]
- 20.Geisbert, T. W., and P. B. Jahrling. 1995. Differentiation of filoviruses by electron microscopy. Virus Res. 39:129-150. [DOI] [PubMed] [Google Scholar]
- 21.Geyer, H., C. Will, H. Feldmann, H.-D. Klenk, and R. Geyer. 1992. Carbohydrate structure of Marburg virus glycoprotein. Glycobiology 2:299-312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gheysen, D., E. Jacobs, F. de Foresta, C. Thiriart, M. Francotte, D. Thines, and M. De Wilde. 1989. Assembly and release of HIV-1 precursor Pr55gag virus-like particles from recombinant baculovirus-infected insect cells. Cell 59:103-112. [DOI] [PubMed] [Google Scholar]
- 23.Gillard, B. K., L. T. Thurmon, and D. M. Marcus. 1993. Variable subcellular localization of glycosphingolipids. Glycobiology 3:57-67. [DOI] [PubMed] [Google Scholar]
- 24.Gleeson, P. A., R. D. Teasdale, and J. Burke. 1994. Targeting of proteins to the Golgi apparatus. Glycoconj. J. 11:381-394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haffar, O., J. Garrigues, B. Travis, P. Moran, J. Zarling, and S. L. Hu. 1990. Human immunodeficiency virus-like, nonreplicating, gag-env particles assemble in a recombinant vaccinia virus expression system. J. Virol. 64:2653-2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harder, T., P. Scheiffele, P. Verkade, and K. Simons. 1998. Lipid domain structure of the plasma membrane revealed by patching of membrane components. J. Cell Biol. 141:929-942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heyningen, S. V. 1974. Cholera toxin: interaction of subunits with ganglioside GM1. Science 183:656-657. [DOI] [PubMed] [Google Scholar]
- 28.Justice, P. A., W. Sun, Y. Li, Z. Ye, P. R. Grigera, and R. R. Wagner. 1995. Membrane vesiculation function and exocytosis of wild-type and mutant matrix proteins of vesicular stomatitis virus. J. Virol. 69:3156-3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kobasa, D., M. E. Rodgers, K. Wells, and Y. Kawaoka. 1997. Neuraminidase hemadsorption activity, conserved in avian influenza A viruses, does not influence viral replication in ducks. J. Virol. 71:6706-6713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kolesnikova, L., S. Bamberg, B. Berghöfer, and S. Becker. 2004. The matrix protein of Marburg virus is transported to the plasma membrane along cellular membranes: exploiting the retrograde late endosomal pathway. J. Virol. 78:2382-2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kolesnikova, L., H. Bugany, H.-D. Klenk, and S. Becker. 2002. VP40, the matrix protein of Marburg virus, is associated with membranes of the late endosomal compartment. J. Virol. 76:1825-1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kolesnikova, L., E. Mühlberger, E. Ryabchikova, and S. Becker. 2000. Ultrastructural organization of recombinant Marburg virus nucleoprotein: comparison with Marburg virus inclusions. J. Virol. 74:3899-3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li, Y., L. Luo, M. Schubert, R. R. Wagner, and C. Y. Kang. 1993. Viral liposomes released from insect cells infected with recombinant baculovirus expressing the matrix protein of vesicular stomatitis virus. J. Virol. 67:4415-4420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Llopis, J., J. M. McCaffery, A. Miyawaki, M. G. Farquhar, and R. Y. Tsien. 1998. Measurement of cytosolic, mitochondrial, and Golgi pH in single living cells with green fluorescent proteins. Proc. Natl. Acad. Sci. USA 95:6803-6808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matyas, G. R., and D. J. Morre. 1987. Subcellular distribution and biosynthesis of rat liver gangliosides. Biochim. Biophys. Acta 921:599-614. [DOI] [PubMed] [Google Scholar]
- 36.Mavrakis, M., L. Kolesnikova, G. Schoehn, S. Becker, and R. W. Ruigrok. 2002. Morphology of Marburg virus NP-RNA. Virology 296:300-307. [DOI] [PubMed] [Google Scholar]
- 37.Munro, S., and H. R. Pelham. 1987. A C-terminal signal prevents secretion of luminal ER proteins. Cell 48:899-907. [DOI] [PubMed] [Google Scholar]
- 38.Niwa, H., K. Yamamura, and J. Miyazaki. 1991. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108:193-199. [DOI] [PubMed] [Google Scholar]
- 39.Noda, T., H. Sagara, E. Suzuki, A. Takada, H. Kida, and Y. Kawaoka. 2002. Ebola virus VP40 drives the formation of virus-like filamentous particles along with GP. J. Virol. 76:4855-4865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pelchen-Matthews, A., B. Kramer, and M. Marsh. 2003. Infectious HIV-1 assembles in late endosomes in primary macrophages. J. Cell Biol. 162:443-455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pelham, H. R. 1996. The dynamic organisation of the secretory pathway. Cell. Struct. Funct. 21:413-419. [DOI] [PubMed] [Google Scholar]
- 42.Sanchez, A., M. P. Kiley, H.-D. Klenk, and H. Feldmann. 1992. Sequence analysis of the Marburg virus nucleoprotein gene: comparison to Ebola virus and other non-segmented negative-strand RNA viruses. J. Gen. Virol. 73:347-357. [DOI] [PubMed] [Google Scholar]
- 43.Sanger, C., E. Mühlberger, E. Ryabchikova, L. Kolesnikova, H.-D. Klenk, and S. Becker. 2001. Sorting of Marburg virus surface protein and virus release take place at opposite surfaces of infected polarized epithelial cells. J. Virol. 75:1274-1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sherer, N. M., M. J. Lehmann, L. F. Jimenez-Soto, A. Ingmundson, S. M. Horner, G. Cicchetti, P. G. Allen, M. Pypaert, J. M. Cunningham, and W. Mothes. 2003. Visualization of retroviral replication in living cells reveals budding into multivesicular bodies. Traffic 4:785-801. [DOI] [PubMed] [Google Scholar]
- 45.Smith, D. H., B. K. Johnson, M. Isaacson, R. Swanapoel, K. M. Johnson, M. Killey, A. Bagshawe, T. Siongok, and W. K. Keruga. 1982. Marburg-virus disease in Kenya. Lancet i:816-820. [DOI] [PubMed] [Google Scholar]
- 46.Sprong, H., P. van der Sluijs, and G. van Meer. 2001. How proteins move lipids and lipids move proteins. Nat. Rev. Mol. Cell. Biol. 2:504-513. [DOI] [PubMed] [Google Scholar]
- 47.Swenson, D. L., K. L. Warfield, K. Kuehl, T. Larsen, M. C. Hevey, A. Schmaljohn, S. Bavari, and M. J. Aman. 2004. Generation of Marburg virus-like particles by co-expression of glycoprotein and matrix protein. FEMS Immunol. Med. Microbiol. 40:27-31. [DOI] [PubMed] [Google Scholar]
- 48.Timmins, J., S. Scianimanico, G. Schoehn, and W. Weissenhorn. 2001. Vesicular release of Ebola virus matrix protein VP40. Virology 283:1-6. [DOI] [PubMed] [Google Scholar]
- 49.Volchkov, V. E., V. A. Volchkova, U. Ströher, S. Becker, O. Dolnik, M. Cieplik, W. Garten, H.-D. Klenk, and H. Feldmann. 2000. Proteolytic processing of Marburg virus glycoprotein. Virology 268:1-6. [DOI] [PubMed] [Google Scholar]
- 50.Watzele, G., and E. G. Berger. 1990. Near identity of HeLa cell galactosyltransferase with the human placental enzyme. Nucleic Acids Res. 18:7174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.World Health Organization. 1999. Marburg fever, Democratic Republic of the Congo. Wkly Epidemiol. Rec. 74:145. [PubMed] [Google Scholar]
- 52.Yamaguchi, N., and M. N. Fukuda. 1995. Golgi retention mechanism of beta-1,4-galactosyltransferase. Membrane-spanning domain-dependent homodimerization and association with alpha- and beta-tubulins. J. Biol. Chem. 270:12170-12176. [DOI] [PubMed] [Google Scholar]