

Abstract
The retroviral nucleocapsid protein (NC) originates by cleavage of the Gag polyprotein. It is highly basic and contains one or two zinc fingers. Mutations in either the basic residues or the zinc fingers can affect several events of the virus life cycle. They frequently prevent the specific packaging of the viral RNA, affect reverse transcription, and impair virion assembly. In this work, we explore the role of NC in murine leukemia virus (MLV) particle assembly and release. A panel of NC mutants, including mutants of the zinc finger and of a basic region, as well as truncations of the NC domain of Gag, were studied. Several of these mutations dramatically reduce the release of virus particles. A mutant completely lacking the NC domain is apparently incapable of assembling into particles, although its Gag protein is still targeted to the plasma membrane. By electron microscopy on thin sections of virus-producing cells, we observed that some NC mutants exhibit various stages of budding defects at the plasma membrane and have aberrant particle morphology; electron micrographs of cells expressing some of these mutants are strikingly similar to those of cells expressing “late-domain” mutants. However, the defects of NC mutants with respect to virus release and infectivity could be complemented by an MLV lacking the p12 domain. Therefore, the functions of NC in virus budding and infectivity are completely distinct from viral late-domain function.
Assembly of retroviruses is dependent on the Gag polyprotein. In all retroviruses, Gag consists of at least three domains: the matrix (MA), capsid (CA), and nucleocapsid (NC) (40). In murine leukemia virus (MLV), a prototype member of the gammaretrovirus family, there is an additional domain between MA and CA, designated p12 (40). During or after the release of a retroviral particle from the host cell, the virion undergoes a maturation process in which the viral protease (PR) cleaves Gag into its respective domains. In the mature particle, MA will be located under the virion membrane that derived from the infected cell membrane, CA will form the outer shell of the mature virion core, and NC will be condensed around the viral RNA genome and form the interior of the virion core. NC is highly conserved among retroviruses. It contains one or two zinc fingers surrounded by basic regions. MLV NC contains one zinc finger, has 60 amino acids, and is located at the C terminus of Gag. Little is known about the function of p12, except that it contains the viral late domain, which participates in the release of the budding particle from the host cell (45, 46).
NC appears to be essential for virus replication and is involved in several steps of the virus life cycle (for reviews, see references 2, 13, 34, and 36). As a domain of Gag, it participates in selection of the genomic RNA for encapsidation into the nascent virus particle and apparently uses RNA as “scaffolding” in particle assembly (6, 7, 11, 28). It also acts as a nucleic acid chaperone, unwinding a cellular tRNA molecule and catalyzing the hybridization of a portion of the tRNA to the viral genome (9, 15). In the mature, infectious virion, NC is complexed with genomic RNA; the nucleic acid chaperone activity of the protein causes a stabilization of the dimeric linkage between the genomic RNA molecules within the particle (16, 27). When the particle infects a new host cell, this chaperone activity plays a crucial role in reverse transcription and possibly in integration as well (8). Thus, NC interacts with nucleic acids in remarkably diverse ways during the retroviral replication cycle.
The RNA content of MLV particles that lacked viral RNA because of mutations in the NC domain was previously characterized (29). It was found that the predominant RNA species in these particles is rRNA, which is present in ribosomes within the virions. In the present study, we have explored the role of NC in MLV particle formation. We found that mutations in NC lead to defects in both the assembly and release steps of virion production and, in some cases, cause the formation of aberrant particles. However, our results show that, in contrast to results with human immunodeficiency virus type 1 (HIV-1) (38), the NC domain is not required for the targeting of Gag at the plasma membrane.
MATERIALS AND METHODS
Cell culture.
293T cells used in this study were maintained in Dulbecco's modified Eagle's medium (Gibco, Inc.) supplemented with 10% fetal calf serum, 2 mM glutamine, and penicillin (100 U/ml)-streptomycin (100 μg/ml) (Gibco).
Plasmid constructs.
Proviral clones of wild-type Moloney MLV and the PR− active-site mutant D32L used in this study are in a plasmid vector, termed pGCcos3neo, that is derived from pSV2Neo (39) and have been described previously (20). The C39S, C17-, C37-, and Δ16-23 NC mutant clones have been described previously (19, 22, 29, 35). The C60- mutant clone contains a termination codon at the end of the CA coding region and thus encodes a truncated Gag protein completely lacking its NC domain. All of these mutants were in the context of the full-length pRR88 plasmid (20). Since the translational termination codons introduced into gag in these truncations are not accompanied by a readthrough signal (17), none of the truncations expresses PR or any other pol gene product (19).
Mammalian cell transfection and virus production.
Culture fluids containing virus particles were harvested at 24, 48, and 72 h after transient transfection of 293T cells by the calcium phosphate method (23) or by TransIT (Mirrus) and were clarified by filtration (pore size, 0.45 μm; Nalge). Ten to 21 μg of plasmid DNA was used to transfect 106 cells per 10-cm-diameter culture dish.
Virus preparation.
Virions were purified from filtered culture supernatants by pelleting through a cushion of 20% sucrose-TNE (100 mM NaCl, 10 mM Tris hydrochloride [pH 7.4], 1 mM EDTA), at 25,000 rpm for 50 min at 4°C in a Beckman SW28 rotor. The virus pellets were resuspended for 1 h at 4°C in TNE and stored at −20°C for protein quantification. A “mock” preparation was obtained from 293T cells transfected with the empty pCGcos3neo vector and was treated exactly like the viral preparations.
Immunoblotting.
Relative amounts of virus were measured by immunoblotting with a rabbit antiserum against MLV p30CA by using the ECL reagent (Amersham Life Science) as described elsewhere (28). Quantification of the ECL signal on the membrane was performed either by scanning the X-ray film or directly, by measuring the chemiluminescence with a phosphorimager (Bio-Rad), using the Quantity One quantification program (Bio-Rad). Wild-type, NC(Δ16-23), and C39S mutant virions were compared by using p30CA, while the PR−, C17-, C37-, and C60- mutants were compared with respect to levels of the uncleaved Gag protein.
For measurement of intracellular Gag levels, cells were lysed without removal from the monolayer, by using 500 μl of 2× sodium dodecyl sulfate loading dye (Novex) followed by 30 s of sonication using a VCX 500 autotuned instrument (Vibracell). Viral proteins were then detected by immunoblotting as described above.
Infectivity assay.
Infectivity was assayed with pLZRS-EGFP, an MLV-based vector that directs the synthesis of green fluorescent protein (GFP) (12) (a gift from Vineet KewalRamani, National Cancer Institute), by using a fluorescence-activated cell sorter (FACS) as described previously (30). Each 10-cm-diameter culture dish was transfected with a mixture of 5 μg of pLZRS-EGFP DNA and 8 μg of each of the two MLV-derived plasmids that were being tested for complementation. In control dishes transfected with a single MLV-derived plasmid, 8 μg of pGCcos3neo DNA was included in the DNA mixture. We estimate that ∼1.5 × 103 infectious units (IU)/ml is the minimum titer detectable in our assay.
Buoyant-density determination.
Viral pellets were resuspended in TNE and layered on 15 to 60% (wt/wt) sucrose gradients. Gradients were centrifuged for 18 to 36 h at 40,000 rpm in an SW41 rotor (Beckman). Fractions were collected and analyzed by immunoblotting with an anti-p30CA antiserum.
Transmission EM.
Detailed procedures for transmission electron microscopy (EM) have been described previously (41). These procedures were slightly modified, as follows. 293T cells were trypsinized, washed once with phosphate-buffered saline (PBS), and pelleted by centrifugation at 185 × g for 5 min at 4°C in a J-75 Beckman rotor. Cells were fixed in 2% glutaraldehyde in cacodylate buffer (0.1 M; pH 7.4). The cell pellet was rinsed in cacodylate buffer, postfixed in 1% osmium tetroxide in the same buffer, and dehydrated in graded ethanol and propylene oxide. The pellet was infiltrated overnight in a 1:1 mixture of 100% propylene oxide and epoxy resin (Electron Microscope Science). The pellet was then embedded in pure resin and cured at 55°C. The cured block was thin-sectioned and stained with uranyl acetate and lead citrate.
For immunoelectron microscopy, the cells were prepared as described above but were fixed with 4% paraformaldehyde in PBS. The cell pellet was then rinsed in cold PBS, dehydrated in cold ethanol, infiltrated with a 1:1 mixture of 100% ethanol-LR White resin, and then incubated with pure LR White resin (Polysciences, Inc.) overnight at 4°C. The embedded pellet was then cured at 55°C. The cured block was thin-sectioned and mounted on naked 200-mesh nickel grids. For immunogold labeling, the grids were blocked for 30 min in PBS containing 10% (wt/vol) goat serum, 0.1% (wt/vol) bovine serum albumin, and 0.05% Tween 20 and were then incubated overnight at 4°C with the rabbit anti-CA serum at a 1:100 dilution. The grids were washed for 2 h in the same buffer and incubated for 1 h with an immunogold (particle diameter, 15 nm)-conjugated donkey anti-rabbit secondary antibody (Amersham Pharmacia Biotech). After a last wash, the grids were stained in uranyl acetate and lead citrate.
Digital images were obtained with an electron microscope (Hitachi H7000) equipped with a digital camera system (Gatan).
Immunofluorescence and image analysis.
Transfected 293T cells were grown on 35-mm-diameter coverglass-bottom dishes (MatTek Corporation, Ashland, Mass.) and processed at room temperature for immunofluorescence staining 48 h after transfection. Cells were washed with PBS and fixed with 3% paraformaldehyde in PBS for 15 min, followed by 0.2% Triton X-100 permeabilization for 5 min. Fixed cells were incubated for 1 h with a 1:20,000 dilution of a rabbit anti-p30CA antibody in 1% bovine serum albumin-PBS and were then washed and stained for 1 h with an Alexa Fluor 488-conjugated donkey anti-rabbit antibody (0.5 μg/ml) (Molecular Probes, Inc.). The cells were washed twice with PBS and mounted on microscope slides with Prolong antifade reagent (Molecular Probes). Images were acquired with an LSM 510 confocal microscope attached to an Axiovert 200 inverted microscope (Carl Zeiss Inc., Thornwood, N.Y.) and a 40× 1.3 NA plan-NEOFLUAR oil objective lens. The pixel sizes were 0.15 by 0.15 by 2.5 μm3. The optical thickness of 2.5 μm was chosen as a compromise between the sharpness of the image and the need to encompass the whole cytoplasm of the cell. Alexa Fluor 488 was excited with an argon laser line at 488 nm, and emission was collected with an LP505 long-pass filter. The image and statistical analyses were performed with Matlab (MathWorks Inc., Natick, Mass.) and DIPimage (image processing toolbox for Matlab, Delft University of Technology, Delft, The Netherlands), using in-house imaging software, where segmentation of each cell was done semiautomatically. The mean intensity of each cell was computed. All intensity measurements were made using the same laser power and gain; thus, the linear relationship between Gag concentration and fluorescence intensity was the same for each sample. Since optical sections were thick enough to encompass the full cytoplasm and the Gag protein was present mainly in the cytoplasm, the mean intensity will be proportional to the cellular Gag concentration. Intensity measurements were corrected for background by subtracting the average cellular intensity of 15 mock-transfected cells stained as described above.
RESULTS
Mutations in the MLV NC domain impair virus release.
This study used MLV mutants with mutations within the NC domain of Gag, or with C-terminal truncations of NC (diagrammed in Fig. 1). In order to evaluate the efficiency of virus release by mutant MLVs, we used immunoblotting to compare the level of p30CA (or, where appropriate, Gag) present in pelletable form in culture fluids with the amount of Gag in cell lysates. We observed efficient release of wild-type virions and PR− Gag virus-like particles into the medium (V) (Fig. 2, lanes 1 and 7), so that the levels of viral proteins in the medium were equal to or greater than those in the cell lysate (C) (Fig. 2, lanes 2 and 8). The same was observed for the C17- mutant (Fig. 2, lanes 9 and 10). However, for all the other mutants, Gag was much more abundant in the cell (Fig. 2, lanes 4, 6, 12, and 14) than in pelletable form in the medium (Fig. 2, lanes 3, 5, 11, and 13). We calculated “efficiency of release” as the amount of material released into the medium at 24 and 48 h after transfection relative to the intracellular Gag level. We noted that the V/C ratios are similar at 24 and 48 h, suggesting that our observations were made while viral protein production and particle release were at steady state (data not shown). Average values from three independent experiments are presented in Table 1. We can observe that particle release by the C39S and C17- mutants is 2- to 3-fold less efficient than that by the wild type, while release by the NC(Δ16-23) and C37- mutants is 30- to 70-fold less efficient than that from wild-type or PR− particles. Thus, all of the NC mutants show some retention of viral protein in the cell relative to the wild type. Mutants lacking significant numbers of basic amino acids show far more severe defects than those altering the zinc finger (C39S) or truncating the NC domain distal to the zinc finger (C17-).
FIG. 1.
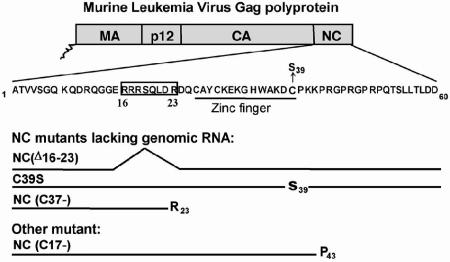
MLV NC mutants used in this study. The NC domain is located at the C terminus of the MLV Gag polyprotein. The amino acid sequence of MLV NC, which contains only 60 residues, is shown in 1-letter code. The location of the zinc finger is indicated. The NC mutants used in this study are the C39S mutant, carrying a point mutation in the zinc finger (22); the NC(Δ16-23) mutant, with a deletion of residues R16 through R23 (35); and the C37- mutant, which is truncated after R23. These mutants fail to encapsidate genomic RNA efficiently. Another mutant, C17- (19), is missing the 17 C-terminal amino acids of the NC domain of Gag but still packages some viral RNA.
FIG. 2.
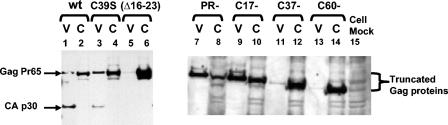
Virus release by MLV NC mutants. MLV Pr65Gag and p30CA released from transfected cells into culture fluid in pelletable form (V) and Pr65Gag in cell lysates (C) are analyzed by immunoblotting with an anti-p30CA antiserum. Lanes 1 and 2, wild type; lanes 3 and 4, C39S; lanes 5 and 6, NC(Δ16-23); lanes 7 and 8, PR−; lanes 9 and 10, C17-; lanes 11 and 12, C37-; lanes 13 and 14, C60-; lane 15, lysate of mock-transfected cells.
TABLE 1.
Virus release by mutants
| MLV | Virus release (%)a |
|---|---|
| wtb | 100 |
| C39S | 45 ± 10 |
| ΔNC(16-23) | 2.5 ± 1 |
| PR− | 127 ± 23 |
| C17- | 58 ± 25 |
| C37- | 2.3 ± 0.7 |
| C60- | 0.35 ± 0.25 |
The amount of virus released in pelletable form over a 24-h period was compared with the amount of viral protein remaining in the cells. Virus and viral protein levels were measured by immunoblotting with anti-p30CA. The released/cell-associated ratio was set at 100% for wild-type MLV.
wt, wild type.
Effects of NC mutations on MLV Gag cellular localization.
To extend our observations on intracellular Gag retention, we examined transfected 293T cells by immunofluorescence staining using an antibody against MLV CA (Fig. 3A). While the antibody gave a negligible signal on mock-transfected cells (Fig. 3A, mock), virus-expressing cells exhibited fluorescence mainly at the periphery of the cell.
FIG. 3.
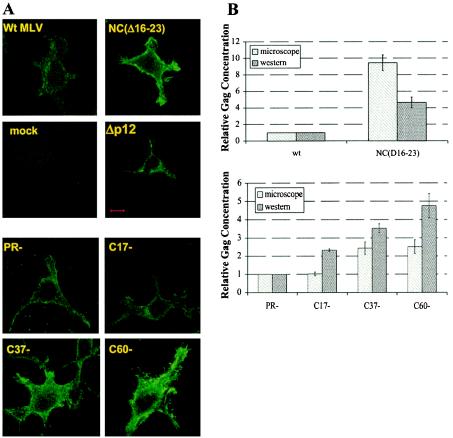
Localization and quantitation of mutant Gag proteins by immunofluorescence and immunoblotting. Cells were first transfected with the indicated viral clones and then fixed and stained for detection of CA as described in Materials and Methods. (A) Immunofluorescence images. Bar, 10 μm. (B) Relative concentrations of anti-CA-reactive material in the cells, measured either by integrating immunofluorescence intensity over the cells as described in Materials and Methods (microscope) or by immunoblotting (Western). wt, wild type.
Cells expressing wild-type Gag showed punctate foci of fluorescence both at the plasma membrane and in the cytoplasm, with additional perinuclear staining (Fig. 3A). The C39S mutant showed the same distribution of staining as the wild type (data not shown). In cells expressing the NC(Δ16-23) mutant, Gag was present all along the plasma membrane, with intense clusters at the extremities of the cell pseudopodia. Cells expressing this mutant were stained much more brightly than those expressing the wild type, confirming the retention of the mutant protein within the cell (Fig. 3A). A similar pattern was seen with C37-, while C17- resembled the wild type (and/or the PR− mutant) in its distribution and intensity (Fig. 3A).
We integrated the total fluorescence in individual cells, as described in Materials and Methods. As shown in Fig. 3B, the results of these measurements were in good qualitative agreement with those obtained by immunoblotting, with the severely truncated Gag proteins and NC(Δ16-23) protein retained within the cell to a far greater extent than wild-type proteins. The mutant Gag proteins were concentrated at the plasma membrane, indicating that particle release is blocked at a step after trafficking of Gag to the plasma membrane.
It was of considerable interest to compare the NC mutants with a mutant in which particle release is blocked because of the absence of the “late domain, ” a motif in Gag known to participate in particle budding (42). Figure 3 also shows immunofluorescence results obtained with Δp12 MLV (45, 46). While it is evident that the level of Δp12 Gag protein within the cell is dramatically elevated, its distribution in the cytoplasm is somewhat different from that of the NC mutants: it appears to be concentrated within cytoplasmic vacuoles, with very little fluorescence at the cell periphery (Fig. 3A). This distribution suggests that, while Δp12 Gag is known to assemble into tubular particles at the cell surface (45), a much larger fraction of this mutant protein is shunted into internal vacuoles.
Effects of NC mutations on virion size and morphogenesis.
Since the mutant Gag proteins which fail to direct efficient virion release accumulate at the plasma membrane, it was of considerable interest to determine whether they had assembled into virions and how closely these virions resembled wild-type MLV particles. Therefore, thin sections of cells expressing wild-type or mutant Gag proteins were examined by EM. We found (Fig. 4) (see also reference 29) that cells expressing each of the mutants described above had an accumulation of budding particles at the plasma membrane. We also measured the sizes of the particles observed and, for immature particles, the sizes of the cores. The results of these measurements are summarized in Table 2. We observed that C39S virions are similar to wild-type virions in size and morphology (Fig. 4A): the average size of the particle is close to that of the wild type (Table 2). In contrast, NC(Δ16-23) particles are substantially larger than wild-type virions, with an average diameter ∼20% greater than that of wild-type particles (Table 2). The NC(Δ16-23) particles have an abnormal, nonuniform spherical shape; many of the mature NC(Δ16-23) particles contain multiple, irregularly shaped cores (Fig. 4A). Many particles, including highly aberrant particle-like structures, were also visible at the plasma membranes of cells expressing NC(Δ16-23) MLV (Fig. 4B). The results show that the basic region upstream of the zinc finger is critical for correct particle assembly, while a point mutation in the zinc finger has little effect on particle morphogenesis.
FIG. 4.
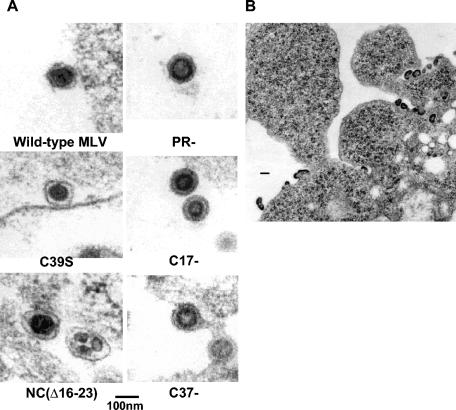
Morphology of NC mutant particles. (A) Thin sections of cells transfected with the indicated viral clones. (B) Thin section of cells transfected with the NC(Δ16-23) mutant. Bar in panel B, 200 nm.
TABLE 2.
Characteristics of NC mutant virions
| MLV virions | Packaging of genomic RNAa | Particle density (g/ml) | Particle diam (nm)b | Capsid core diam (nm) |
|---|---|---|---|---|
| Wild-type | ++ | 1.14-1.16 | 105 ± 4 | ND |
| C39S | −c | 1.14-1.16 | 107 ± 9 | ND |
| ΔNC(16-23) | −d | 1.13-1.15 | 126 ± 11 | Irregular cores |
| PR− | ++e | 1.14-1.16 | 112 ± 6 | 61 ± 5 |
| C17- | +e | 1.14-1.16 | 101 ± 6 | 54 ± 4 |
| C37- | ND | 1.11-1.13 (light) | 99 ± 5 | 50 ± 4 |
| C60- | ND | No visible particle |
The presence (++, 100%; +, ∼50%) or the absence (−, ≤1%) of viral genomic RNA is indicated. ND, not determined.
All the mutant particle sizes except that of C39S are significantly different from wild-type (P < 0.001) particle sizes.
See reference 22.
See reference 35.
Viral RNA content was determined by Northern blotting (data not shown) or by RNA end labeling (29).
Particles produced by the truncation mutants, i.e., C17- and C37-, are somewhat smaller than the PR− control particles assembled from full-length Gag (Table 2). In addition, C17- and C37- cores are significantly smaller in diameter than PR− cores, indicating that the truncated Gag proteins in these mutants assemble into particles with a reduced radius of curvature. However, immature C17- and PR− particles appear very similar in morphology, as seen by EM (Fig. 4A), indicating that the last 17 amino acids of MLV Gag are not essential for proper particle formation. In contrast, C37- appears to exhibit a late budding defect, with particles tethered together at the plasma membrane (Fig. 4A).
We also measured the buoyant density of the particles released by the mutants. All mutants produced particles with the same density as wild-type particles, with the exception of C37- (Table 2). Thus, the C-terminal 37 residues of Gag (which include the zinc finger in NC) are required for production of particles with normal density.
Deletion of the entire NC domain of MLV Gag prevents virus assembly.
We also generated a mutant in which the entire NC domain of MLV Gag, spanning the C-terminal 60 amino acids of Gag, is deleted. Release of pelletable particles from cells expressing this truncation mutant was completely undetectable (Table 1). Furthermore, we were totally unable to find C60- particles at the plasma membrane by electron microscopy. Thus, the absence of NC drastically reduces or eliminates particle assembly.
In order to locate the C60- Gag protein, immunofluorescence and immunogold staining were performed. Immunofluorescence of C60- showed that the truncated Gag protein was uniformly distributed along the plasma membrane (Fig. 3A). The plasma membrane localization of the C60- Gag was also confirmed by immunoelectron microscopy (Fig. 5). However, neither virions nor other multimeric structures were visible in the electron micrographs; indeed, while the protein was detectable immunologically (Fig. 3A and 5), there was no accumulation of densely staining material at the membrane. The data suggest that the NC domain is absolutely required for the initiation of particle assembly.
FIG. 5.
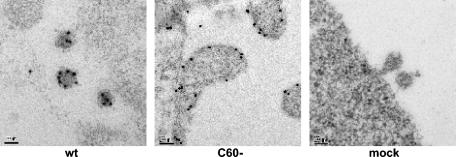
Immunoelectron microscopy of the C60- Gag mutant. Cells transfected with the indicated viral clones were fixed in 4% paraformaldehyde and stained with anti-p30CA as described in Materials and Methods. Wild-type MLV (wt), C60-, and negative-control (mock) cells are shown. Bars, 100 nm.
Complementation between NC mutants and a late-domain mutant.
As shown above, the NC(Δ16-23) and C37- mutants exhibit budding defects. Their phenotype is remarkably similar to that of MLV late-domain (L-domain) mutants, which form viral particles whose release from the budding site on the plasma membrane is impaired (18, 45). It was of great interest to test the possibility that the same function is blocked in these NC mutants as in L-domain mutants with changes in the p12 domain (45). We therefore tested the ability of the NC mutants to complement an MLV mutant in which the p12 domain has been completely deleted. We measured both virus release and infectivity following cotransfection of the NC mutant genomes with the Δp12 mutant genome (45). Results of these studies are shown in Fig. 6 and Table 3. We found that the level of virus production induced by the Δp12 mutant was only ∼10% that of the wild type, while the NC(Δ16-23) mutant directed only ∼3% as much virion release as the wild type. However, the cotransfected cells produced approximately the same amount of virus as the wild-type controls, measured either as total pelletable p30CA-reactive material (Fig. 6; also data not shown) or as fully matured p30CA (Table 3). Further, whereas no infectious virus is produced by either of these mutants individually, the cotransfected culture released virus with ∼1/10 of the infectivity of the wild-type control.
FIG. 6.
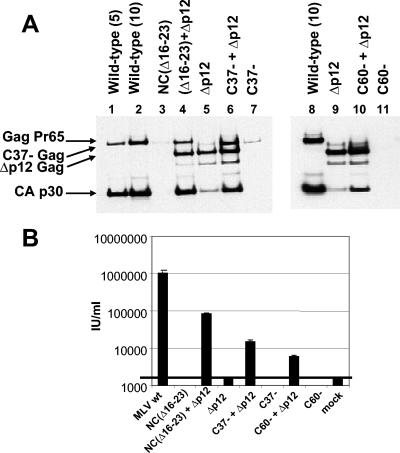
Complementation between NC mutants and the Δp12 mutant. (A) Cells were transfected with the indicated viral clones. Pelletable material in culture fluids was analyzed by immunoblotting with an anti-p30CA antiserum. An equal volume of culture fluid was loaded in each lane. Numbers in parentheses above lanes 1, 2, and 8 are micrograms of wild-type MLV DNA transfected. (B) Infectivity titers of culture fluids from cells transfected with the indicated viral clones.
TABLE 3.
Virus production by cells expressing NC mutants together with Δp12 MLVa
| Particle type | % Pelletable mature virions | IU/ml |
|---|---|---|
| Wild type | 100 | 106 ± 5 × 104 |
| Δp12 + NC(Δ16-23) | 94 ± 37 | 8.5 × 104 ± 4 × 103 |
| NC(Δ16-23) | 3 ± 2 | <1.5 × 103 |
| C37- (or C60-) | <0.5 | <1.5 × 103 |
| Δp12 + C37- | 56 ± 6 | 1.5 × 104 ± 2 × 103 |
| Δp12 + C60- | 34 ± 8 | 6 × 103 ± 5 × 102 |
| Δp12 alone | 11 ± 8 | <1.5 × 103 |
The amount of mature particle release was evaluated by measuring p30CA by immunoblotting. Results are averages from three independent experiments; the amount released by wild-type MLV was set at 100%. Infectivity was measured by GFP transduction as described elsewhere (30).
Similar, although somewhat less dramatic, restorations of virus production and of infectivity were observed when the C37- or C60- mutant was cotransfected with the Δp12 mutant. The differences between the different NC mutants with respect to the level of synergy with the Δp12 mutant suggest that the interaction with the latter represents complementation rather than recombination. These results demonstrate that the functional defects of the NC mutants are completely distinct from those, including L-domain function, that are absent in the p12 deletion mutant.
DISCUSSION
The results presented here show that the NC domain of MLV plays a critical role in the ability of the Gag protein to assemble into budding virus particles. Thus, a truncation mutant lacking the C-terminal 17 residues and a point mutant disrupting the zinc finger exhibit mild defects in virus production. Mutants with a deletion of a basic region on the N-terminal side of the zinc finger, or a truncation in which Gag is terminated just before the zinc finger, show severely impaired virus release; large numbers of virus particles are observed tethered to the cell surface in these cases, reminiscent of the phenotype of a late-domain mutant (18, 45). Finally, a truncation mutant in which the Gag protein contains no NC sequences does not assemble into organized structures visible in the electron microscope.
In all of these cases, Gag protein is observed concentrated at the plasma membrane or in punctate sites within the cytoplasm, as previously reported by others (24). The results suggest that the NC domain is not involved in the targeting of Gag to the plasma membrane but is essential for its assembly into large multimeric structures. The presence of fully formed virions tethered to the plasma membrane in the C37- and NC(Δ16-23) mutants is highly reminiscent of the L-domain phenotype (18, 32). The impairment in budding of L-domain mutants is due to their inability to interact efficiently with cellular proteins in the multivesicular body pathway; these proteins evidently play a crucial role in the release of retrovirus particles from the cell (18, 32). The ability of the NC mutants to complement a p12 deletion (Fig. 6; Table 3), as well as the difference in targeting between the NC mutants and the p12 deletion mutant (Fig. 3A), suggests that the defect of NC mutants is entirely distinct from that of L-domain mutants. This complementation also implies that the interactions between the MA and/or CA domains of multiple Gag molecules are the only homotypic interactions required for the assembly and release of infectious MLV particles.
It is interesting to compare the present results with those of prior studies on other retroviruses. There have been many reports documenting defects in HIV-1 production caused by mutations in the NC domain (e.g., references 14, 21, 25, and 47). However, the correct interpretation of these results is, in some cases, still somewhat unclear. For example, in some studies the reduction in virus production did not seem to be compensated for by an increase in the level of Gag protein within the cell. Therefore, the reduction might reflect degradation of the mutant Gag protein within the cell or the disruption of particles following their release (42, 43) and would then indicate that an intact NC domain is important for Gag or virion stability. With MLV, on the other hand, the Gag protein that is not released in virions is retained within the cell, principally at the cell surface (Fig. 2, 3A, and 4B), demonstrating unequivocally that the mutations in NC interfere with efficient virus assembly or release.
It has been reported previously that immature virus-like particles of NC mutants are considerably more fragile than those formed from wild-type Gag (29). With one of our mutants, the NC(Δ16-23) mutant, we found that the cell-associated particles tend to be abnormal with respect to size and morphology. This observation is quite reminiscent of those of Cimarelli and Luban (10) for HIV-1 and those of Yovandich et al. (44) for SIV(Mne). Taken together, the results show that in both primate lentiviruses and gammaretroviruses, the NC domain contributes to the determination of the radius of curvature of the nascent virion and to the formation of the core of the mature virion. One notable characteristic of these mutant particles is the frequent presence of multiple cores. Briggs et al. (4) have suggested that genomic RNA molecules act as nucleation sites in the formation of mature cores and that wild-type particles with multiple cores probably contain multiple dimers of genomic RNA. In the case of MLV, it is interesting that the most abundant RNA species in NC mutant particles is rRNA, which is present in ribosomes within the virion (29).
How does alteration of the NC domain lead to a defect in retroviral particle production? Many lines of evidence show that the NC domain of retroviral Gag proteins is crucial in the interactions of Gag with nucleic acids (2). Thus, one interpretation of the present results would be that the mutants tested here are impaired in their interactions with RNA, and that this defect is responsible for their inability to assemble in or escape from the cell as efficiently as wild-type MLV. This interpretation would be consistent with prior work showing that RNA is essential for the structural integrity of immature MLV particles (28). However, the data reported here do not allow us to exclude the alternative possibility that the mutations affect other interactions, such as those between Gag protein molecules or between Gag and other cellular components, in virus assembly or release. Indeed, mutational studies with several retroviruses have given rise to the designation of regions within NC as “interaction” domains (“I” domains) (40). I domains have been held responsible for the formation of particles of the correct buoyant density (1), for the efficient trafficking of Gag-GFP fusion proteins to the cell surface (5, 37, 38), and for the reversal of assembly defects of severely truncated Gag proteins (3). It is still not clear whether I domains influence particle structure directly, by participating in protein-protein interactions, or by facilitating the interactions with RNA required for optimal Gag-Gag interactions in the assembling virion.
The most striking difference between the present results with MLV and prior results with HIV-1 is the phenotype of an NC− PR− mutant. In HIV-1, the near-total elimination of the NC domain still does not prevent virion assembly and release, provided that it is coupled with a defect in the viral PR (31). This surprising result suggests that the defects in virus production observed in other studies (14, 21, 25, 47) may actually be due, at least in part, to the degradation of the mutant protein by PR. In contrast, NC− PR− MLV is evidently completely unable to assemble into virions (Fig. 2 and 5; Tables 1 and 2). We do not know whether this difference between HIV-1 and MLV is due to differences in their Gag proteins or reflects differences in targeting of the two proteins. It has been suggested (11, 28) that RNA is a structural element in retroviral particles; thus, if all interactions of Gag protein with nucleic acids are mediated by the NC domain, we would expect Gag proteins lacking NC to be unable to assemble. This model may well apply to MLV. However, there are several lines of evidence supporting the idea that in HIV-1, the MA domain, as well as the NC domain, can bind to RNA (26, 33). Thus, one possible resolution of the discrepancy between results with NC− PR− mutants of MLV and those with corresponding HIV-1 mutants is that the MA domain of HIV-1 can bind RNA and, in the absence of NC, facilitate virion formation, while that of MLV cannot. While the particles formed by HIV-1 NC− PR− Gag are devoid of genomic RNA (31), their total nucleic acid content is not yet known; it will be of considerable interest to determine this.
In conclusion, we have shown that the NC domain in MLV Gag plays a crucial role not only in selective encapsidation of viral RNA (2) but also in particle assembly and release. Alteration of this domain, particularly of its basic residues, leads to assembly of somewhat abnormal particles and to a drastic impairment of the release of these particles from the plasma membrane. The function of the NC domain in particle release is distinct from that of the late domain of the virus. Total elimination of the NC domain appears to completely prevent particle assembly, although, in contrast to HIV-1, it does not interfere with the targeting of the protein to the plasma membrane.
Acknowledgments
We thank Michelle Gignac and Kelly Stanard for help in electron microscopy imaging, Zandrea Ambrose for helping with the FACS analysis, Vineet KewalRamani for the pLZRS-EGFP plasmid and the use of the FACS apparatus, and Sandra Ernst for plasmid construction.
This project has been funded in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract no. NO1-CO12400.
REFERENCES
- 1.Bennett, R. P., T. D. Nelle, and J. W. Wills. 1993. Functional chimeras of the Rous sarcoma virus and human immunodeficiency virus Gag proteins. J. Virol. 67:6487-6498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berkowitz, R., J. Fisher, and S. P. Goff. 1996. RNA packaging. Curr. Top. Microbiol. Immunol. 214:177-218. [DOI] [PubMed] [Google Scholar]
- 3.Bowzard, J. B., R. P. Bennett, N. K. Krishna, S. M. Ernst, A. Rein, and J. W. Wills. 1998. Importance of basic residues in the nucleocapsid sequence for retrovirus Gag assembly and complementation rescue. J. Virol. 72:9034-9044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Briggs, J. A., T. Wilk, R. Welker, H. G. Krausslich, and S. D. Fuller. 2003. Structural organization of authentic, mature HIV-1 virions and cores. EMBO J. 22:1707-1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Callahan, E. M., and J. W. Wills. 2003. Link between genome packaging and rate of budding for Rous sarcoma virus. J. Virol. 77:9388-9398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Campbell, S., and A. Rein. 1999. In vitro assembly properties of human immunodeficiency virus type 1 Gag protein lacking the p6 domain. J. Virol. 73:2270-2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Campbell, S., and V. M. Vogt. 1995. Self-assembly in vitro of purified CA-NC proteins from Rous sarcoma virus and human immunodeficiency virus type 1. J. Virol. 69:6487-6497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carteau, S., R. J. Gorelick, and F. D. Bushman. 1999. Coupled integration of human immunodeficiency virus type 1 cDNA ends by purified integrase in vitro: stimulation by the viral nucleocapsid protein. J. Virol. 73:6670-6679. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
9.Cen, S., Y. Huang, A. Khorchid, J. L. Darlix, M. A. Wainberg, and L. Kleiman. 1999. The role of Pr55gag in the annealing of
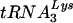 to human immunodeficiency virus type 1 genomic RNA. J. Virol. 73:4485-4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
to human immunodeficiency virus type 1 genomic RNA. J. Virol. 73:4485-4488. [DOI] [PMC free article] [PubMed] [Google Scholar] - 10.Cimarelli, A., and J. Luban. 2000. Human immunodeficiency virus type 1 virion density is not determined by nucleocapsid basic residues. J. Virol. 74:6734-6740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cimarelli, A., S. Sandin, S. Hoglund, and J. Luban. 2000. Basic residues in human immunodeficiency virus type 1 nucleocapsid promote virion assembly via interaction with RNA. J. Virol. 74:3046-3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dardalhon, V., N. Noraz, K. Pollok, C. Rebouissou, M. Boyer, A. Q. Bakker, H. Spits, and N. Taylor. 1999. Green fluorescent protein as a selectable marker of fibronectin-facilitated retroviral gene transfer in primary human T lymphocytes. Hum. Gene Ther. 10:5-14. [DOI] [PubMed] [Google Scholar]
- 13.Darlix, J. L., M. Lapadat-Tapolsky, H. de Rocquigny, and B. P. Roques. 1995. First glimpses at structure-function relationships of the nucleocapsid protein of retroviruses. J. Mol. Biol. 254:523-537. [DOI] [PubMed] [Google Scholar]
- 14.Dawson, L., and X. F. Yu. 1998. The role of nucleocapsid of HIV-1 in virus assembly. Virology 251:141-157. [DOI] [PubMed] [Google Scholar]
- 15.Feng, Y. X., S. Campbell, D. Harvin, B. Ehresmann, C. Ehresmann, and A. Rein. 1999. The human immunodeficiency virus type 1 Gag polyprotein has nucleic acid chaperone activity: possible role in dimerization of genomic RNA and placement of tRNA on the primer binding site. J. Virol. 73:4251-4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feng, Y. X., T. D. Copeland, L. E. Henderson, R. J. Gorelick, W. J. Bosche, J. G. Levin, and A. Rein. 1996. HIV-1 nucleocapsid protein induces “maturation” of dimeric retroviral RNA in vitro. Proc. Natl. Acad. Sci. USA 93:7577-7581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feng, Y. X., H. Yuan, A. Rein, and J. G. Levin. 1992. Bipartite signal for read-through suppression in murine leukemia virus mRNA: an eight-nucleotide purine-rich sequence immediately downstream of the gag termination codon followed by an RNA pseudoknot. J. Virol. 66:5127-5132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Freed, E. O. 2002. Viral late domains. J. Virol. 76:4679-4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fu, W., B. A. Ortiz-Conde, R. J. Gorelick, S. H. Hughes, and A. Rein. 1997. Placement of tRNA primer on the primer-binding site requires pol gene expression in avian but not murine retroviruses. J. Virol. 71:6940-6946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fu, W., and A. Rein. 1993. Maturation of dimeric viral RNA of Moloney murine leukemia virus. J. Virol. 67:5443-5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gheysen, D., E. Jacobs, F. de Foresta, C. Thiriart, M. Francotte, D. Thines, and M. De Wilde. 1989. Assembly and release of HIV-1 precursor Pr55gag virus-like particles from recombinant baculovirus-infected insect cells. Cell 59:103-112. [DOI] [PubMed] [Google Scholar]
- 22.Gorelick, R. J., L. E. Henderson, J. P. Hanser, and A. Rein. 1988. Point mutants of Moloney murine leukemia virus that fail to package viral RNA: evidence for specific RNA recognition by a “zinc finger-like” protein sequence. Proc. Natl. Acad. Sci. USA 85:8420-8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Graham, F. L., and A. J. van der Eb. 1973. A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology 52:456-467. [DOI] [PubMed] [Google Scholar]
- 24.Hansen, M., L. Jelinek, R. S. Jones, J. Stegeman-Olsen, and E. Barklis. 1993. Assembly and composition of intracellular particles formed by Moloney murine leukemia virus. J. Virol. 67:5163-5174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jowett, J., D. Hockley, M. Nermut, and I. Jones. 1992. Distinct signals in human immunodeficiency virus type 1 Pr55 necessary for RNA binding and particle formation. J. Gen. Virol. 73:3079-3086. (Erratum, 74:943, 1993.) [DOI] [PubMed] [Google Scholar]
- 26.Lochrie, M. A., S. Waugh, D. G. Pratt, Jr., J. Clever, T. G. Parslow, and B. Polisky. 1997. In vitro selection of RNAs that bind to the human immunodeficiency virus type-1 Gag polyprotein. Nucleic Acids Res. 25:2902-2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muriaux, D., H. De Rocquigny, B. P. Roques, and J. Paoletti. 1996. NCp7 activates HIV-1Lai RNA dimerization by converting a transient loop-loop complex into a stable dimer. J. Biol. Chem. 271:33686-33692. [DOI] [PubMed] [Google Scholar]
- 28.Muriaux, D., J. Mirro, D. Harvin, and A. Rein. 2001. RNA is a structural element in retrovirus particles. Proc. Natl. Acad. Sci. USA 98:5246-5251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Muriaux, D., J. Mirro, K. Nagashima, D. Harvin, and A. Rein. 2002. Murine leukemia virus nucleocapsid mutant particles lacking viral RNA encapsidate ribosomes. J. Virol. 76:11405-11413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oshima, M., D. Muriaux, J. Mirro, K. Nagashima, K. Dryden, M. Yeager, and A. Rein. 2004. Effects of blocking individual maturation cleavages in murine leukemia virus Gag. J. Virol. 78:1411-1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ott, D. E., L. V. Coren, E. N. Chertova, T. D. Gagliardi, K. Nagashima, R. C. Sowder II, D. T. Poon, and R. J. Gorelick. 2003. Elimination of protease activity restores efficient virion production to a human immunodeficiency virus type 1 nucleocapsid deletion mutant. J. Virol. 77:5547-5556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pornillos, O., J. E. Garrus, and W. I. Sundquist. 2002. Mechanisms of enveloped RNA virus budding. Trends Cell Biol. 12:569-579. [DOI] [PubMed] [Google Scholar]
- 33.Purohit, P., S. Dupont, M. Stevenson, and M. R. Green. 2001. Sequence-specific interaction between HIV-1 matrix protein and viral genomic RNA revealed by in vitro genetic selection. RNA 7:576-584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rein, A. 1994. Retroviral RNA packaging: a review. Arch. Virol. Suppl. 9:513-522. [DOI] [PubMed] [Google Scholar]
- 35.Rein, A., D. P. Harvin, J. Mirro, S. M. Ernst, and R. J. Gorelick. 1994. Evidence that a central domain of nucleocapsid protein is required for RNA packaging in murine leukemia virus. J. Virol. 68:6124-6129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rein, A., L. E. Henderson, and J. G. Levin. 1998. Nucleic acid chaperone activity of retroviral nucleocapsid proteins: significance for viral replication. Trends Biochem. Sci. 23:297-301. [DOI] [PubMed] [Google Scholar]
- 37.Sandefur, S., R. M. Smith, V. Varthakavi, and P. Spearman. 2000. Mapping and characterization of the N-terminal I domain of human immunodeficiency virus type 1 Pr55Gag. J. Virol. 74:7238-7249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sandefur, S., V. Varthakavi, and P. Spearman. 1998. The I domain is required for efficient plasma membrane binding of human immunodeficiency virus type 1 Pr55Gag. J. Virol. 72:2723-2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Southern, P. J., and P. Berg. 1982. Transformation of mammalian cells to antibiotic resistance with a bacterial gene under control of the SV40 early region promoter. J. Mol. Appl. Genet. 1:327-341. [PubMed] [Google Scholar]
- 40.Swanstrom, R., and J. W. Wills. 1997. Synthesis, assembly, and processing of viral proteins, p. 263-334. In J. M. Coffin, S. H. Hughes, and H. E. Varmus (ed.), Retroviruses. Cold Spring Harbor Laboratory Press, Plainview, N.Y. [PubMed]
- 41.Tobin, G. J., K. Nagashima, and M. A. Gonda. 1996. Immunologic and ultrastructural characterization of HIV pseudovirions containing Gag and Env precursor proteins engineered in insect cells. Methods 10:208-218. [DOI] [PubMed] [Google Scholar]
- 42.Wang, S. W., and A. Aldovini. 2002. RNA incorporation is critical for retroviral particle integrity after cell membrane assembly of Gag complexes. J. Virol. 76:11853-11865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang, S.-W., K. Noonan, and A. Aldovini. 2004. Nucleocapsid-RNA interactions are essential to structural stability but not to assembly of retroviruses. J. Virol. 78:716-723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yovandich, J. L., E. N. Chertova, B. P. Kane, T. D. Gagliardi, J. W. Bess, Jr., R. C. Sowder II, L. E. Henderson, and R. J. Gorelick. 2001. Alteration of zinc-binding residues of simian immunodeficiency virus p8NC results in subtle differences in Gag processing and virion maturation associated with degradative loss of mutant NC. J. Virol. 75:115-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yuan, B., S. Campbell, E. Bacharach, A. Rein, and S. P. Goff. 2000. Infectivity of Moloney murine leukemia virus defective in late assembly events is restored by late assembly domains of other retroviruses. J. Virol. 74:7250-7260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yuan, B., X. Li, and S. P. Goff. 1999. Mutations altering the Moloney murine leukemia virus p12 Gag protein affect virion production and early events of the virus life cycle. EMBO J. 18:4700-4710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang, Y., and E. Barklis. 1997. Effects of nucleocapsid mutations on human immunodeficiency virus assembly and RNA encapsidation. J. Virol. 71:6765-6776. [DOI] [PMC free article] [PubMed] [Google Scholar]