

Abstract
In human immunodeficiency virus type 1 (HIV-1)-infected cells, a cell cycle arrest in G2 increases viral expression and may represent a strategy for the virus to optimize its expression. In latently infected cells, balance between viral silencing and reactivation relies on the nucleosomal organization of the integrated long terminal repeat (LTR). It is shown here that nucleosome nuc-1, which is located downstream of the TATA box, is specifically modified when latently infected cells are arrested in G2 by chemical inducers. Notably, histones H3 and H4 are hyperacetylated, and this modification is associated with an increased LTR-driven transcription. nuc-1 hyperacetylation is also associated with the recruitment of histone acetyltransferase CBP and transcription factors NF-κB and c-Jun. NF-κB and/or c-Jun binding to the LTR in G2-arrested cells appears to be required for CBP recruitment as well as for nuc-1 remodeling and viral reactivation.
Epigenetic control of transcription is a key mechanism for the regulation of gene expression in eukaryotes. This control results at least in part from structural modifications of the basic unit of chromatin, the nucleosome, which is composed of DNA tightly wrapped around an octamer of histones. This structure may limit the access of several transcription factors to their binding sites as well as the efficacy of the formation of transcription initiation complexes within gene promoters. The permanent modulation of the chromatin conformation allows dynamic control of transcriptional activity. This modulation involves posttranslational modifications of the tail of core histones, such as methylation, phosphorylation, and acetylation (7) and the action of ATP-dependant chromatin remodeling proteins, such as the SWI/SNF complex (32). Histone acetylation is regulated by histone acetyl transferases and histone deacetylases that are recruited close to nucleosomes by transcription factors (4, 8).
Postintegration latency is an important mechanism of human immunodeficiency virus type 1 (HIV-1) silencing. It is observed in a large proportion of cells infected in vivo that are located in viral sanctuaries. These cells are preserved from current antiviral therapies and impede virus eradication (24). A better knowledge of the molecular mechanisms controlling latency and reactivation is therefore crucial to raise strategies to purge HIV-1 from the organism. In its integrated form, HIV-1 DNA is packaged into chromatin, and this may contribute to the control of viral expression. In that respect, latent T-cell clones that survive acute infection frequently contain viral DNA integrated within or near haploid repeat elements in heterochromatin (19).
In various cell lines that are latently infected with HIV-1, a specific array of three nucleosomes, nuc-0, nuc-1, and nuc-2, are positioned within the 5′ long terminal repeat (LTR) (Fig. 1) (35). Modification of nuc-1, which is located downstream of the TATA box, is thought to be essential for HIV-1 transcription because it is rapidly disrupted upon viral reactivation induced by Tat, phorbol esters, or tumor necrosis factor alpha (9, 35). Importantly nuc-1 is also modified during viral activation induced by histone deacetylase inhibitors (34), suggesting a direct link between histone acetylation, nuc-1 conformation, and HIV-1 expression. In that respect, the recruitment of histone deacetylase 1 (HDAC-1) to nuc-1 by YY1 and LSF contributes to the repression of the LTR (6). This interaction is counterregulated by histone deacetylase inhibitors as well as by Tat (16), which has been shown to recruit histone acetyltransferases such as (i) p300 and its homologous cyclic AMP-responsive binding protein (CREB) binding protein (CBP); (ii) p300/CBP-associated factor (pCAF); and (iii) the general control nonderepressible 5 (GCN5) protein to the LTR (3, 18).
FIG. 1.
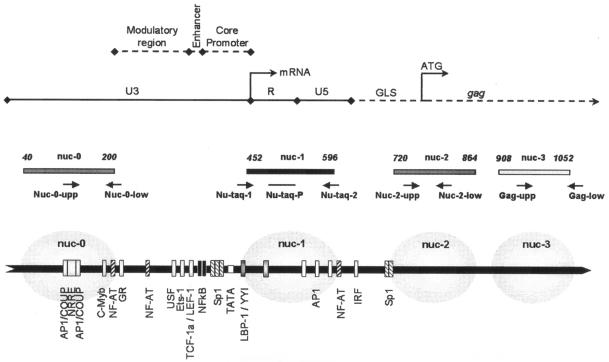
Organization of HIV-1 LTR. Schematic representation of the 5′ LTR and the start of the gag region. The known binding sites for transcription factors, the TATA box, and the nucleosome position are depicted. Adapted from reference 27. The positions of the primers and probe used for real-time PCR are indicated by black arrows.
Alteration of the cell cycle may represent a strategy for HIV-1 to optimize its expression. Notably, the viral protein Vpr induces a cell cycle arrest in G2 (17, 29) that leads to the reactivation of HIV-1 (13, 36). This effect is highly conserved in various HIV-1 isolates and in other lentiviruses (25) and has been observed in both infected and transfected cell lines as well as in infected primary CD4+ T cells (15). HIV-1 activation was proved to be directly linked to the G2 arrest because (i) it can be induced by a dominant negative mutant of the p34cdc2 kinase (13) or by chemical inducers of G2 arrest (14) and (ii) it is not observed with Vpr mutants that are unable to induce G2 arrest (36). Finally, separation of cycling cells by counterflow centrifugal elutriation demonstrated maximal LTR-driven transcription in cells located in the G2 phase of the cell cycle (15). Altogether, these results lead to the assumption that the LTR chromatin adopts a specific conformation during the G2 phase that is favorable to HIV-1 transcription.
In the present paper, we asked whether molecular events leading to HIV-1 induction upon G2 arrest were similar to those observed in infected cells activated by Tat or mitogens. Considering the key role of nuc-1 in the control of HIV-1 transcription, we developed a chromatin immunoprecipitation assay to analyze nuc-1 modifications in G2-arrested cells. It is shown here that the acetylation level of histones H3 and H4 significantly increased at nuc-1 in chronically infected cells arrested in G2, contemporaneous to the transcriptional induction of HIV-1. We also demonstrated an increased recruitment of CBP, NF-κB, and c-Jun to the LTR during the G2 arrest. Finally, it is shown that nuc-1 acetylation and the subsequent proviral activation that occur in G2-arrested cells relied on NF-κB and/or c-Jun binding to the LTR. These results provide evidence for nuc-1 remodeling during the G2 phase and a better understanding of viral reactivation linked to the cell cycle.
MATERIALS AND METHODS
Cell culture and HIV-1 induction.
The ACH-2, U1, and OM-10.1 cell lines were obtained from the AIDS Research and Reference Reagent Program (National Institute of Allergy and Infectious Disease) and maintained in RPMI medium (Life Technologies) supplemented with 10% calf serum, 2 mM glutamine, 100 μg of ampicillin per ml, 200 μg of kanamycin per ml, 20 μg of vancomycin per ml, and 10 mM HEPES buffer in the presence of 5% CO2. In ACH-2 and U1 cells, postintegration latency results from mutations in the TAR sequence and the tat gene, respectively (10, 11), that disrupt the Tat-TAR axis.
Induction of cell cycle arrest in G2 was obtained by treating the cells with genistein (ACH-2, 50 μM; U1 and OM-10.1, 25 μM) or psi-tectorigenin (20 μg/ml) for 24 h. Alleviation of the G2 arrest was based on the concomitant use of pentoxifylline (ACH-2, 2 mM; U1 and OM-10.1, 1 mM). Inhibition of histone deacetylases was obtained by treating the cells with trichostatin A (0.5 μM) for 24 h. Inhibition of NF-κB and c-Jun was obtained by treating the cells with aurintricarboxylic acid (200 μM). Pentoxifylline and aurintricarboxylic acid were added at the same time as genistein for cotreatments of the cells. All chemicals were purchased from Sigma.
Cell cycle analysis.
To determine DNA content, 105 cells were washed in phosphate-buffered saline and fixed with 70% ethanol for 1 h at 4°C. After fixation, the cells were incubated with propidium iodide and RNase (DNA prep kit, Beckman Coulter) for 30 min at room temperature. Propidium iodide fluorescence analysis was performed with an Epics XL flow cytometer (Beckman Coulter).
p24 antigen quantification.
The production of HIV-1 p24 antigen in cell culture supernatants was determined by an enzyme-linked immunosorbent assay (Ingen) according to the manufacturer's protocol.
Real-time RT-PCR for HIV-1 transcripts.
A real-time reverse transcription-PCR (RT-PCR) assay was used to quantify viral RNA transcribed in cycling or in G2-arrested ACH-2 cells. Total RNA was extracted from 106 cells (RNAble, Eurobio) and quantified by UV photospectrometry. RNA (0.2 μg) was reverse transcribed for 1 h at 37°C in the presence of 100 U of Moloney murine leukemia virus reverse transcriptase (Invitrogen), and serial dilutions of cDNA were amplified by real-time PCR on an ABI Prism 7700 sequence detector system (Applied Biosystems) with SYBR green dye (SYBR green PCR master mix, Applied Biosystems). The primers used were MS-1 (5′-CTT AGG CAT CTC CTA TGG CAG GAA-3′) and MS-2 (5′-TTC CTT CGG GCC TGT CGG GTC CC-3′) for the multiply spliced HIV-1 RNA sequence and US-1 (5′-TCT CTA GCA GTG GCG CCC GAA CA-3′) and US-2 (5′-TCT CCT TCT AGC CTC CGC TAG TC-3′) for unspliced HIV-1 RNA.
Chromatin immunoprecipitation assay.
Experiments were performed with the chromatin immunoprecipitation assay kit (Upstate), according to the manufacturer's procedure. Briefly, 5 × 106 cells were treated with 1% formaldehyde for 10 min at 37°C. Subsequent procedures were performed on ice in the presence of protease inhibitors. Cross-linked cells were harvested, washed with phosphate-buffered saline, and lysed in sodium dodecyl sulfate lysis buffer (1% sodium dodecyl sulfate, 10 mM EDTA, 50 mM Tris-HCl, pH 8.1) for 10 min at 4°C. Chromatin was sonicated with five 10-s pulses, 30% amplitude (Sonifier, Branson Ultrasonic Corp). After centrifugation (10 min, 4°C, 14,000 × g), the supernatant was diluted 10-fold with chromatin immunoprecipitation dilution buffer (0.01% sodium dodecyl sulfate, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris-HCl, pH 8.1, 167 mM NaCl).
Diluted extracts were precleared in the presence of salmon sperm DNA-protein A-agarose beads (chromatin immunoprecipitation assay kit, Upstate). One tenth of the diluted extract was kept for a direct quantitative PCR (input). The remaining extracts were incubated for 16 h at 4°C in the presence of 1 μg of specific antibodies per ml followed by 1 h of incubation with salmon sperm DNA-protein A-agarose beads. Antibodies were purchased from Upstate Biotechnology (anti-acetylated histone H4 and anti-acetylated histone H3; anti-c-Jun anti-HDAC-1; anti-NFAT-1; anti-SP1; anti-pCAF and anti-p300) or from Santa Cruz (anti-YY1 clone C20; anti-NF-κB p50; anti-CBP and anti-p300). Following extensive washing (details available upon request), bound DNA fragments were eluted with a 30-min incubation in elution buffer (1% sodium dodecyl sulfate, 0.1 M NaHCO3). The DNA was recovered for 4 h at 65°C in elution buffer containing 200 mM NaCl and then incubated in the presence of proteinase K (20 μg/ml) for 1 h at 45°C. DNA was extracted in the presence of phenol-chloroform and chloroform-isoamyl alcohol and ethanol precipitated before being subjected to real-time PCR.
Real-time PCR.
Purified DNA was quantified after chromatin immunoprecipitation by real-time PCR (ABI Prism 7700 sequence detector system, Applied Biosystems) with the Taqman technology (Taqman Universal Master Mix, Applied Biosystems). The oligonucleotide primers for amplification of proviral DNA and the cellular gene for glyceraldehhyde-3-phosphate dehydrogenase (GAPDH) are listed in Table 1. The nuc-1 sequence was amplified in the presence of the internal 6-carboxy-fluorescein (FAM)/6-carboxy-tetramethyl-rhodamine (TAMRA) target probe Nu-taq-P with Taqman PCR Master Mix (Applied Biosystems). Others PCRs were performed in the presence of SYBR green dye (SYBR green PCR Master Mix, Applied Biosystems). Copy numbers were calculated by reference to a log-linear standard curve constructed from the number of cycles necessary to detect product accumulation after amplification of plasmids harboring the target sequences.
TABLE 1.
Primers and probe used for real-time PCRa
| Primer or probe | Position | Sequence |
|---|---|---|
| Nuc-0-upp | 145 | 5′-TAG TAC CAG TTG AAC CAG AGC AAG-3′ |
| Nuc-0-low | 212 | 5′-CAT CCC ATG CTG GCT CAT A-3′ |
| Nu-taq-1 | 432 | 5′-CAG CTG CT TTT GCC TGT ACT G-3′ |
| Nu-taq-2 | 598 | 5′-TCC ACA CTG ACT AAA AGG GTC TGA-3′ |
| Nu-taq-P | 490 | 5′-TCT CTG GCT AAC TAG GGA ACC CAC TGC TTA A-3′ |
| Nuc-2-upp | 739 | 5′-GAC TGG TGA GTA CGC CAA A-3′ |
| Nuc-2-low | 796 | 5′-TAA TAC CGA CGC TCT CGC-3′ |
| Gag-upp | 966 | 5′-AAT ACT GGG ACA GCT ACA A-3′ |
| Gag-low | 1118 | 5′-TGT GCC TTT TTC TTA CTT T-3′ |
| H-GAPDH-1 | 5′-GGA CCT GAC CTG CCG TCT AGA A-3′ | |
| H-GAPDH-2 | 5′-GGT GTC GCT GTT GAA GTC AGA G-3′ |
Four HIV-1 regions (three in the LTR and one in the gag region) were selected for amplification as well as the human gene GAPDH. The position of HIV-1 primers refers to the LAV/BRU provirus sequence. Position +1 correspond to the 5′ LTR start.
The reference plasmids were p229-LTR-luc (kindly provided by C. Van Lint), pCR2.1-nuc-0, pCR2.1-nuc-2, and pCR2.1-gag (generated with the respective PCR products and the Original TA cloning kit from Invitrogen), and pGAPDH (kindly provided by A. Harel-Bellan), used as standards for nuc-1, nuc-0, nuc-2, nuc-gag, and human GAPDH amplification, respectively. The quantified amount of immunoprecipitated material was normalized to the input DNA. Results were expressed as the enrichment of immunoprecipitated material from the treated cells relative to the untreated control cells.
Western blotting for acetylated H3 histone.
Total proteins were extracted from ACH-2 cells in denaturing buffer (50 mM Tris-HCl, pH 6.8, 2%sodium dodecyl sulfate, 2% β-mercaptoethanol) and separated on a sodium dodecyl sulfate-15% polyacrylamide gel before electric transfer onto a nitrocellulose membrane. Membranes were incubated for 1 h in blocking buffer (phosphate-buffered saline, 0.2% Tween, 5% milk), washed, and incubated for 3 h with a 1:5,000 dilution of an antibody against acetylated histone 3 (Upstate biotechnologies). Donkey anti-rabbit immunoglobulin-horseradish peroxidase conjugate (Amersham) was used as the secondary antibody at a 1:2,000 dilution. Immune complexes were visualized the ECL detection reagents (Amersham).
Statistical evaluation.
Values are expressed as the mean of at least three independent experiments (details are indicated in the figure legends). Confidence interval analysis was used, with a P value of <0.05 considered statistically significant.
RESULTS
G2 arrest induces HIV-1 activation in ACH-2 cells.
ACH-2 cells were used as a model of HIV-1 postintegration latency in T cells. ACH-2 cells contain a single integrated copy of viral DNA (LAI strain) and exhibit a low basal level of HIV-1 expression, with a predominance of multiply spliced viral RNAs (5).
In order to investigate the molecular process that lead to HIV-1 reactivation in G2-arrested cells, ACH-2 cells were treated with two known chemical inducers of G2 arrest, genistein and psi-tectorigenin (14). As expected, both drugs mediated a cell cycle arrest in G2 (Fig. 2A). More importantly, this arrest was concomitant with a strong viral reactivation, as reflected by increased levels of p24 antigen in the cell culture supernatants (Fig. 2B). HIV-1 activation was demonstrated to occur at the transcriptional level, as indicated by the parallel increase of unspliced and multiply spliced viral RNAs in treated cells (Fig. 2C). To confirm that proviral activation was a direct consequence of the G2 arrest, genistein- and psi-tectorigenin-treated cells were exposed to pentoxifylline, a drug that is known to relieve G2 arrest (21). As shown in Fig. 2A, pentoxifylline efficiently reversed the G2 arrest induced by genistein or psi-tectorigenin. Concomitantly, pentoxifylline reduced HIV-1 expression close to basal levels, as indicated by the reduced production of p24 antigen in the cell culture supernatant (Fig. 2B), indicating that HIV-1 reactivation was indeed a consequence of the G2 arrest.
FIG. 2.
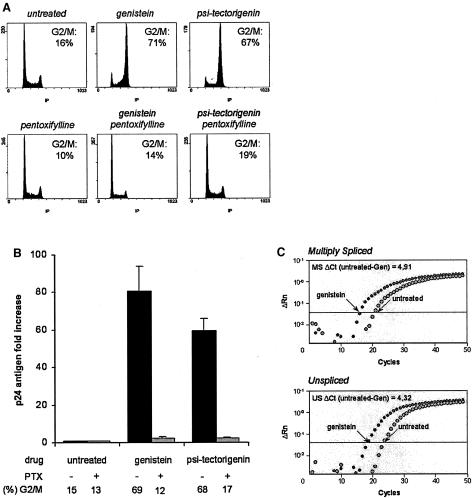
G2 arrest induces HIV-1 transcriptional activation in ACH-2 cells. ACH-2 cells were treated with genistein or psi-tectorigenin in the absence or presence of the G2 arrest inhibitor pentoxifylline (PTX). (A) Flow cytometry analysis of the nuclear DNA content, performed after propidium iodide staining 24 h after treatment. The proportion of cells in the G2 and M phases is indicated. (B) HIV-1 p24 antigen production in cell culture supernatants, quantified at day 3 by enzyme-linked immunosorbent assay. The cell culture conditions and the respective proportion of G2-arrested cells are indicated. Results, normalized to the number of cells, are expressed as the increase relative to the untreated control cells. Values are the mean of at least three independent experiments. (C) Real-time RT-PCR amplification plots for HIV-1 multiply spliced (MS) and unspliced (US) RNAs performed 1 day after treatment. The PCR cycle threshold difference between control and treated cells (ΔCt) is indicated.
Specific hyperacetylation of histones H3 and H4 occurs at nuc-1 in G2-arrested cells.
Previous studies have shown that remodeling of nucleosome nuc-1 was necessary for viral reactivation induced by various agents such as Tat, tumor necrosis factor alpha, or phorbol esters. Consequently, we wondered whether activation of viral expression upon G2 arrest might also be associated with histone acetylation at nuc-1. To address this question, we designed a chromatin immunoprecipitation assay directed against acetylated histone H3 or H4, followed by quantitative real-time PCR assays. One set of primers was designed to amplify nuc-1, whereas two others were chosen to amplify nuc-0 and nuc-2. Control amplifications were also performed in the gag region and in the constitutively expressed cellular gene for GAPDH (11).
This assay was first validated in cells treated with trichostatin A, a specific inhibitor of histone deacetylases. Western blot experiments confirmed that trichostatin A induced a major increase in the total amount of acetylated histone H3 (Fig. 3A). Chromatin immunoprecipitation assays demonstrated that acetylation of histones H3 and H4 increased significantly at nuc-1 in this condition (Fig. 3B).
FIG. 3.
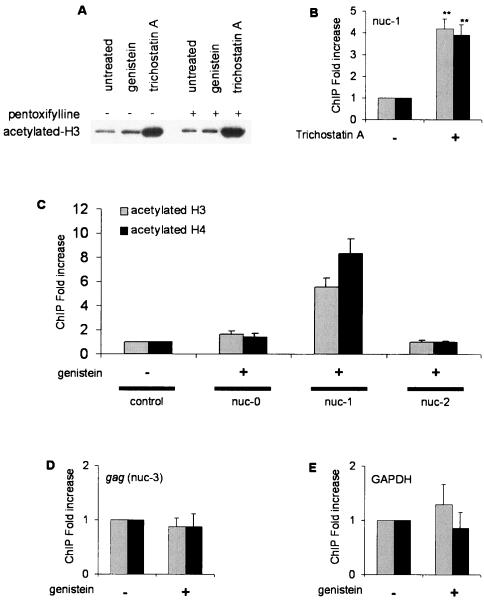
Genistein induces specific acetylation of histones H3 and H4 at nuc-1. (A) Total proteins were extracted from ACH-2 cells treated with trichostatin A or genistein in the presence or absence of pentoxifylline. Proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis on a 15% acrylamide gel and transferred to a nitrocellulose membrane. Western blot analysis was performed with an antibody directed against acetylated histone H3. (B to E) Chromatin immunoprecipitation assays were performed with antibodies directed against acetylated histone H3 (grey bars) or H4 (black bars). ACH-2 cells were treated with trichostatin A (B) or genistein (C, D, and E). DNA obtained from the input or from the immunoprecipitated DNA was applied to real-time PCR, performed in the three nucleosomes located in the LTR (C), a nucleosome positioned in the coding gag region (D), and the cellular gene for GAPDH (E). The amount of immunoprecipitated material was normalized to the input DNA. The results, expressed as the relative enrichment of immunoprecipitated material compared to the untreated control cells, are the means of three to five independent experiments. Confidence interval analysis was used, with a P value of <0.05 considered statistically significant.
In cells arrested in G2 by genistein, a five- to eightfold increase in histone H3 and H4 acetylation was observed at nuc-1 (Fig. 3C), although genistein induced only a mild increase in the total amount of acetylated histone H3 (Fig. 3A). Within the LTR, the acetylation appeared to be restricted to nuc-1 because no significant increase was observed for nuc-0 or nuc-2 (Fig. 3C). This effect proved to be specific, because genistein did not affect histone acetylation on the viral and cellular control genes (Fig. 3D and 3E).
To prove that histone acetylation at nuc-1 was specifically linked to the G2 arrest, we analyzed the level of histone acetylation in genistein-treated cells in the presence or absence of pentoxifylline. As shown in Fig. 4, pentoxifylline abolished the G2 arrest induced by genistein and concomitantly reversed histone acetylation at nuc-1 as well as HIV-1 reactivation. Altogether, these results demonstrate that HIV-1 induction linked to G2 arrest is associated with specific acetylation of nucleosome nuc-1.
FIG. 4.
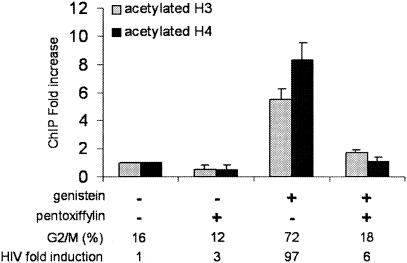
Histone acetylation is alleviated when G2 arrest is abrogated by pentoxifylline. Chromatin immunoprecipitation assays with antibodies directed against acetylated histone H3 or H4 were performed on ACH2 cells treated with genistein in the presence or absence of pentoxifylline. DNA obtained from the input or from the immunoprecipitated DNA was applied to real-time PCR on the nuc-1 region. Results, normalized for the input DNA and expressed as the relative enrichment of immunoprecipitated material compared to the untreated control cells, are the means of three independent experiments. The proportion of cells arrested at the G2 phase of the cell cycle and the increase in HIV-1 expression, evaluated by the release of p24 antigen, in cell supernatants are indicated below the figure.
nuc-1 acetylation and viral reactivation upon G2 arrest requires recruitment of CBP, NF-κB, and/or c-Jun to the LTR.
To further characterize the molecular events leading to the acetylation of nuc-1 during G2 arrest, the chromatin immunoprecipitation assay was modified to precipitate DNA associated with several histone acetyltransferases, including CBP, p300, and pCAF, and with the histone deacetylase HDAC-1. Although the experiments were repeated at least three times, no significant binding of HDAC-1, p300, or pCAF could be detected at nuc-1 in either cycling or G2-arrested cells (data not shown). Conversely, recruitment of CBP at nuc-1 was observed in cycling cells and significantly increased in G2-arrested cells (Fig. 5). Importantly, pentoxifylline both abolished G2 arrest and reversed the binding of CBP to nuc-1.
FIG. 5.
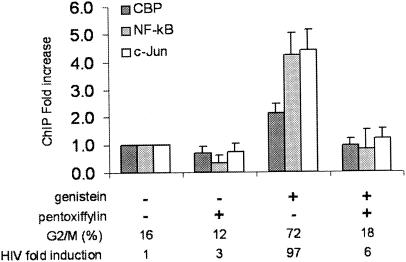
CBP, NF-κB, and c-Jun are recruited to nuc-1 in G2-arrested cells. Chromatin immunoprecipitation assays were performed with anti-CBP (dark grey bars), anti-p50 NF-κB (pale grey bars), or anti-c-Jun (white bars) on ACH2 cells treated with genistein. DNA obtained from the input or from the immunoprecipitated material was applied to real-time PCR in the nuc-1 region. Results, normalized for the input DNA and expressed as the relative enrichment of immunoprecipitated material compared to the untreated control cells, are the means of three independent experiments. The proportion of cells arrested at the G2 phase of the cell cycle and the increase in HIV-1 expression, evaluated by the release of p24 antigen, in cell supernatants are indicated below the figure.
CBP may be recruited to nuc-1 by various transcription factors.
As shown in Fig. 1, several binding sites for cellular factors have been mapped in the vicinity of or inside nuc-1. Therefore, we asked whether some of these factors might be involved in the acetylation of nuc-1 or recruited to nuc-1 after chromatin remodeling. Preliminary experiments indicated that Sp-1 associates weakly but reproductively to the LTR region. Nevertheless, binding of Sp1 to nuc-1 was unaffected by G2 arrest (data not shown). However, no binding was observed for AP3/NFAT or YY1 in cycling or G2-arrested cells (data not shown).
Three AP-1 binding sites are located within nuc-1 (Fig. 1). As AP-1 is composed of Jun-Fos or Jun-Jun dimers, an antibody directed against c-Jun was used for the chromatin immunoprecipitation assays. As illustrated in Fig. 5, c-Jun binding was detected in cycling cells. Most importantly, a four- to fivefold increase in c-Jun binding was observed in cells that were arrested in G2 by genistein.
NF-κB is known to play a critical role in HIV-1 activation (28). Using an antibody directed against the p50 subunit of NF-κB complex, a 4.5-fold increase in the binding of p50 was observed in genistein-arrested cells compared to cycling cells (Fig. 5). As was previously observed for CBP, alleviation of the G2 arrest by pentoxifylline also reversed the increased binding of c-Jun and NF-κB to the LTR (Fig. 5). Therefore, these results demonstrate that CBP, NF-κB, and c-Jun are recruited close to nuc-1 in a specific manner when ACH-2 cells are arrested at the G2 phase of the cell cycle.
To assess the actual involvement of NF-κB and/or c-Jun in CBP recruitment, histone acetylation, and viral induction, ACH-2 cells were exposed to genistein in the presence or absence of aurintricaboxylic acid. Aurintricarboxylic acid inhibits NF-κB and AP-1 activation by blocking upstream kinases such as IκB kinase, extracellular signal-regulated kinase, and p38 mitogen-activated protein kinase (31). As expected, aurintricarboxylic acid inhibited NF-κB and c-Jun binding to nuc-1 (Fig. 6A). More strikingly, aurintricarboxylic acid also reversed CBP binding (Fig. 6A) and histone acetylation at nuc-1 (Fig. 6B), as well as G2-related HIV-1 induction (Fig. 6C). Importantly, aurintricarboxylic acid did not alleviate genistein-induced G2 arrest (Fig. 6D). Altogether, these experiments demonstrate the key role of NF-κB and c-Jun in the molecular events that lead to nuc-1 remodeling and viral reactivation upon G2 arrest.
FIG. 6.
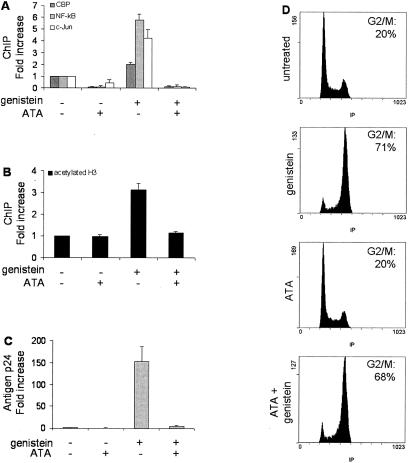
Inhibition of NF-κB and c-Jun activation by aurintricarboxylic acid alleviates CBP recruitment, and histone acetylation at nuc-1, as well as viral reactivation. ACH2 cells were treated with genistein in the presence or absence of aurintricarboxylic acid (ATA). (A and B) Chromatin immunoprecipitation assays were performed with anti-CBP, anti-p50 NF-kB, and anti-c-Jun (A) or anti-acetylated histone H3 (B). DNA obtained from the input or from the immunoprecipitated material was applied to real-time PCR specific for the nuc-1 region. Results, normalized for the input DNA and expressed as the enrichment of immunoprecipitated material relative to the untreated control cells, are the means of four independent experiments. (C) HIV-1 p24 antigen production was quantified at day 3 by enzyme-linked immunosorbent assay in cell culture supernatants. (D) Flow cytometry analysis of the nuclear DNA content, performed after propidium iodide staining 24 h after the treatment. The proportion of cells in the G2 and M phases is indicated.
nuc-1 acetylation and recruitment of CBP, NF-κB, and c-Jun occurs in G2-arrested monocytic cells.
OM-10.1 is a chronically infected promyelocytic clone (LAI strain) harboring a single copy of proviral DNA, whereas U1 is a more differentiated promonocytic clone which contains two copies of provirus. Both cell lines display low basal viral expression and are frequently used as a model of postintegration latency.
As previously demonstrated (14), a G2 arrest mediated by genistein induces a strong proviral induction in these cells, as reflected by the increased release of p24 antigen (Fig. 7A and B). Similar to ACH-2 cells, a clear increase in the acetylation level of histones H3 and H4 was demonstrated at nuc-1, which was reversed when the G2 arrest was alleviated by pentoxifylline (Fig. 7C and D).
FIG.7.
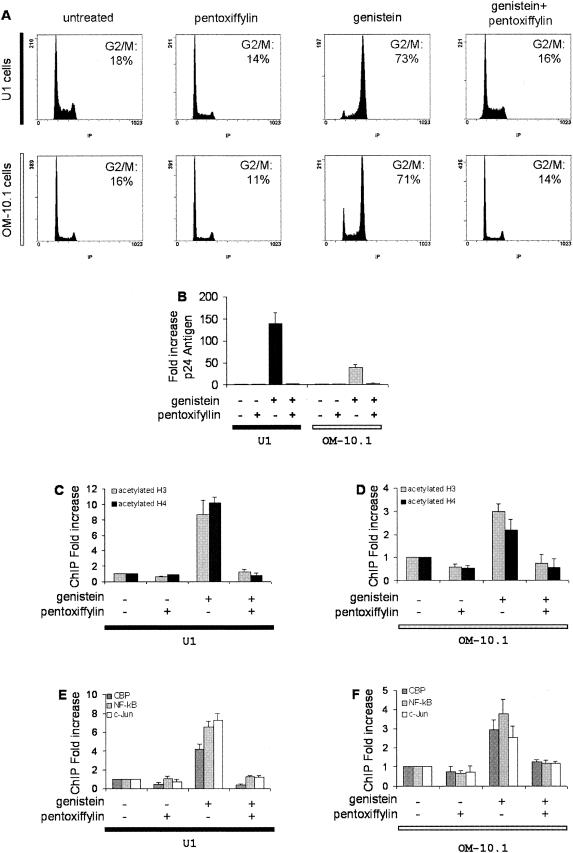
Acetylation of nuc-1 and recruitment of cellular factors to the LTR in chronically infected monocytic cells arrested in G2. U1 and OM-10.1 cells were treated with genistein and/or pentoxifylline (PTX) as indicated. (A) Flow cytometry analysis of the nuclear DNA content, performed after propidium iodide staining 24 h after treatment. The cell culture conditions and the proportion of cells in the G2 and M phases are indicated. (B) HIV-1 p24 antigen production in cell culture supernatants, quantified at day 3 by enzyme-linked immunosorbent assay. The cell culture conditions are indicated below the figure. Results, normalized for the input DNA, are expressed as the relative enrichment of immunoprecipitated material compared to the untreated control cells. Values are the means of at least three independent experiments. Chromatin immunoprecipitation assays with antibodies directed against acetylated histone H3 or H4 (C and D) or against CBP, p50 NF-κB or c-Jun (E and F). DNA obtained from the input or from the immunoprecipitated material was applied to real-time PCR in the nuc-1 region. Results, normalized for the input DNA and expressed as the relative enrichment of immunoprecipitated material compared to the untreated control cells, are the means of three independent experiments.
Similarly, a significant increase in the binding of CBP, NF-κB p50, and c-Jun to nuc-1 was observed in U1 and OM-10.1 cells arrested in G2 and this effect was reversed by pentoxifylline (Fig. 7E and F). These results demonstrate that similar molecular events are involved in HIV-1 reactivation upon G2 arrest in both T cells and cells of myelocytic lineage.
DISCUSSION
HIV-1 expression is closely linked to the cell cycle, and most virus production takes place in activated CD4 T cells. Notably, cells arrested in the G2 phase by the viral protein Vpr or by chemicals exhibit an enhanced level of HIV-1 transcription. Importantly, a significant proportion of circulating T cells arrested in G2 have been reported in HIV-1-infected individuals, suggesting that the virus is also able to alter the cell cycle progression in vivo (30).
In this paper, we investigated specific chromatin modifications that occur following G2 arrest in chronically infected cells. Using a quantitative chromatin immunoprecipitation assay, we demonstrated increased acetylation of histones H3 and H4 at nucleosome nuc-1 in ACH-2 cells treated with genistein or psi-tectorigenin. nuc-1 acetylation was associated with strong transcriptional activation of HIV-1, which confirms the role of nuc-1 as a major hurdle for initiation of viral transcription. In our model, nuc-1 is the only nucleosome to be significantly modified within the LTR. This is in agreement with previous works that described specific disruption of nuc-1 following HIV-1 activation in latently infected cells (34, 35), although Lusic and collaborators reported rapid histone acetylation at nuc-0 and nuc-2 following stimulation of U1 cells by phorbol esters (22). There are two key distinctions with our studies that would account for the apparent discrepancy. First, Lusic et al. used phorbol ester inducers as opposed to cell cycle arrest induction. Second, they analyzed early steps of activation (from 1 to 5 h postinduction), whereas our studies analyzed G2-arrested cells 24 h after treatment.
Our results obtained with ACH-2 cells were extended to two other latently infected cells of the myelocytic lineage, showing that these observations were not restricted to lymphocytic cells.
Despite the pleiotropic effects of the chemicals that we used, our data demonstrate that the molecular processes that lead to G2 arrest are, at least in part, responsible for nuc-1 remodeling and HIV-1 expression. Indeed, reversion of both histone acetylation and viral induction was observed when G2 arrest was alleviated by pentoxifylline.
Histone H3 and H4 acetylation is correlated with increased recruitment of CBP in the vicinity of nuc-1. This result is in agreement with a recent work in which CBP was shown to be recruited to nuc-1 in chronically infected U1 cells activated by phorbol esters (22). Altogether, these data underscore the likely role of CBP in the remodeling of nuc-1.
Coactivators are usually recruited by transcription factors. It is shown here that NF-κB and c-Jun binding is significantly increased in the vicinity of nuc-1 in G2-arrested cells. Inhibition of NF-κB and c-Jun activation by aurintricarboxylic acid concomitantly suppressed CBP binding to the LTR, indicating that NF-κB and/or c-Jun is required for CBP recruitment. Aurintricarboxylic acid also prevented histone acetylation at nuc-1 and viral reactivation, showing that nuc-1 remodeling and subsequent HIV-1 induction rely on the NF-κB and/or AP-1 signaling pathways. However, these pathways act downstream of the molecular events that lead to G2 arrest, since aurintricarboxylic acid did not affect the cell cycle arrest mediated by genistein.
The link between NF-κB and histone acetylation is well documented. Indeed, NF-κB synergistically enhances LTR activation mediated by histone deacetylase inhibitors (26) and its transcriptional activity relies on its ability to recruit coactivators such as CBP/p300 to the LTR (27, 20). In U1 cells treated with phorbol ester, the recruitment of CBP, pCAF, and GCN5 to nuc-1 was also associated with increased binding of the p65 subunit of NF-κB to the promoter (22). Although these results are consistent with the hypothesis that NF-κB recruits CBP upon G2 arrest, one may also suggest that CBP acetylates the p50 subunit of NF-κB itself and therefore increases its affinity for the HIV-1 LTR as well as its transcriptional activity (12).
The relationship between c-Jun binding to nuc-1 and HIV-1 activation is still unclear but may be important, considering the role of the downstream regulatory elements in LTR activity and viral replication (33). Since three AP-1 sites are located within nuc-1, one may assume that NF-κB activity, histone acetylation, and nuc-1 remodeling are a prerequisite for c-Jun binding to the LTR during G2 arrest. Conversely, c-Jun could participate directly in histone acetylation, since it has also been demonstrated to interact with some histone acetyltransferases, including CBP (2).
A multistep process involving the sequential binding of nuclear factors and coactivators has been proved to be essential for activation of the murine mammary tumor virus LTR (1). Determining if a similar model involving NF-κB, c-Jun, and CBP can be proposed for the control of HIV-1 expression upon cell cycle progression is a stimulating issue that is currently under investigation.
Acknowledgments
LTR-luc and HGAPDH were kindly provided by C. Van Lint and A. Harel-Bellan, respectively. We thank M. Raymondjean and A. Bhushan for carefully reading the manuscript.
Research in the laboratory was supported by the Universite Pierre et Marie Curie, Sidaction, the Association pour la Recherche contre le Cancer, and the Programme de Recherche Fondamentale en Microbiologie et Maladies Infectieuses et Parasitaires. S. Thierry is a recipient of a fellowship from the Ministère de l'Education Nationale, de la Recherche et des Technologies.
REFERENCES
- 1.Archer, T. K., P. Lefebvre, R. G. Wolford, and G. L. Hager. 1992. Transcription factor loading on the MMTV promoter: a bimodal mechanism for promoter activation. Science 255:1573-1576. [DOI] [PubMed] [Google Scholar]
- 2.Bannister, A. J., T. Oehler, D. Wilhelm, P. Angel, and T. Kouzarides. 1995. Stimulation of c-Jun activity by CBP: c-Jun residues Ser63/73 are required for CBP induced stimulation in vivo and CBP binding in vitro. Oncogene 11:2509-2514. [PubMed] [Google Scholar]
- 3.Benkirane, M., R. F. Chun, H. Xiao, V. V. Ogryzko, B. H. Howard, Y. Nakatani, and K. T. Jeang. 1998. Activation of integrated provirus requires histone acetyltransferase. p300 and P/CAF are coactivators for HIV-1 Tat. J. Biol. Chem. 273:24898-24905. [DOI] [PubMed] [Google Scholar]
- 4.Berger, S. L. 2002. Histone modifications in transcriptional regulation. Curr. Opin. Genet. Dev. 12:142-148. [DOI] [PubMed] [Google Scholar]
- 5.Butera, S. T., B. D. Roberts, L. Lam, T. Hodge, and T. M. Folks. 1994. Human immunodeficiency virus type 1 RNA expression by four chronically infected cell lines indicates multiple mechanisms of latency. J. Virol. 68:2726-2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coull, J. J., G. He, C. Melander, V. C. Rucker, P. B. Dervan, and D. M. Margolis. 2002. Targeted derepression of the human immunodeficiency virus type 1 long terminal repeat by pyrrole-imidazole polyamides. J. Virol. 76:12349-12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davie, J. R., and V. A. Spencer. 1999. Control of histone modifications. J. Cell Biochem. Suppl. 32-33:141-148. [DOI] [PubMed] [Google Scholar]
- 8.Eberharter, A., and P. B. Becker. 2002. Histone acetylation: a switch between repressive and permissive chromatin. Second in review series on chromatin dynamics. EMBO Rep. 3:224-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.El Kharroubi, A., G. Piras, R. Zensen, and M. A. Martin. 1998. Transcriptional activation of the integrated chromatin-associated human immunodeficiency virus type 1 promoter. Mol. Cell. Biol. 18:2535-2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Emiliani, S., W. Fischle, M. Ott, C. Van Lint, C. A. Amella, and E. Verdin. 1998. Mutations in the tat gene are responsible for human immunodeficiency virus type 1 postintegration latency in the U1 cell line. J. Virol. 72:1666-1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Emiliani, S., C. Van Lint, W. Fischle, P. Paras, Jr., M. Ott, J. Brady, and E. Verdin. 1996. A point mutation in the HIV-1 Tat responsive element is associated with postintegration latency. Proc. Natl. Acad. Sci. USA 93:6377-6381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Furia, B., L. Deng, K. Wu, S. Baylor, K. Kehn, H. Li, R. Donnelly, T. Coleman, and F. Kashanchi. 2002. Enhancement of nuclear factor-kappa B acetylation by coactivator p300 and HIV-1 Tat proteins. J. Biol. Chem. 277:4973-4980. [DOI] [PubMed] [Google Scholar]
- 13.Goh, W. C., M. E. Rogel, C. M. Kinsey, S. F. Michael, P. N. Fultz, M. A. Nowak, B. H. Hahn, and M. Emerman. 1998. HIV-1 Vpr increases viral expression by manipulation of the cell cycle: a mechanism for selection of Vpr in vivo. Nat. Med. 4:65-71. [DOI] [PubMed] [Google Scholar]
- 14.Gozlan, J., J. L. Lathey, and S. A. Spector. 1998. Human immunodeficiency virus type 1 induction mediated by genistein is linked to cell cycle arrest in G2. J. Virol. 72:8174-8180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gummuluru, S., and M. Emerman. 1999. Cell cycle- and Vpr-mediated regulation of human immunodeficiency virus type 1 expression in primary and transformed T-cell lines. J. Virol. 73:5422-5430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.He, G., and D. M. Margolis. 2002. Counterregulation of chromatin deacetylation and histone deacetylase occupancy at the integrated promoter of human immunodeficiency virus type 1 (HIV-1) by the HIV-1 repressor YY1 and HIV-1 activator Tat. Mol. Cell. Biol. 22:2965-2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.He, J., S. Choe, R. Walker, P. Di Marzio, D. O. Morgan, and N. R. Landau. 1995. Hum. immunodeficiency virus type 1 viral protein R (Vpr) arrests cells in the G2 phase of the cell cycle by inhibiting p34cdc2 activity. J. Virol. 69:6705-6711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hottiger, M. O., L. K. Felzien, and G. J. Nabel. 1998. Modulation of cytokine-induced HIV gene expression by competitive binding of transcription factors to the coactivator p300. EMBO J. 17:3124-3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jordan, A., D. Bisgrove, and E. Verdin. 2003. HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J. 22:1868-1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jowett, J. B., V. Planelles, B. Poon, N. P. Shah, M. L. Chen, and I. S. Chen. 1995. The human immunodeficiency virus type 1 vpr gene arrests infected T cells in the G2 and M phases of the cell cycle. J. Virol. 69:6304-6313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li, Y. X., K. Weber-Johnson, L. Q. Sun, N. Paschoud, R. O. Mirimanoff, and P. A. Coucke. 1998. Effect of pentoxifylline on radiation-induced G2-phase delay and radiosensitivity of human colon and cervical cancer cells. Radiat. Res. 149:338-342. [PubMed] [Google Scholar]
- 22.Lusic, M., A. Marcello, A. Cereseto, and M. Giacca. 2003. Regulation of HIV-1 gene expression by histone acetylation and factor recruitment at the LTR promoter. EMBO J. 22:6550-6561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marzio, G., M. Tyagi, M. I. Gutierrez, and M. Giacca. 1998. HIV-1 tat transactivator recruits p300 and CREB-binding protein histone acetyltransferases to the viral promoter. Proc Natl Acad Sci USA 95:13519-13524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pierson, T., J. McArthur, and R. F. Siliciano. 2000. Reservoirs for HIV-1: mechanisms for viral persistence in the presence of antiviral immune responses and antiretroviral therapy. Annu. Rev. Immunol. 18:665-708. [DOI] [PubMed] [Google Scholar]
- 25.Planelles, V., J. B. Jowett, Q. X. Li, Y. Xie, B. Hahn, and I. S. Chen. 1996. Vpr-induced cell cycle arrest is conserved among primate lentiviruses. J. Virol. 70:2516-2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Quivy, V., E. Adam, Y. Collette, D. Demonte, A. Chariot, C. Vanhulle, B. Berkhout, R. Castellano, Y. de Launoit, A. Burny, J. Piette, V. Bours, and C. Van Lint. 2002. Synergistic activation of human immunodeficiency virus type 1 promoter activity by NF-κB and inhibitors of deacetylases: potential perspectives for the development of therapeutic strategies. J. Virol. 76:11091-11103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Quivy, V., and C. Van Lint. 2002. Diversity of acetylation targets and roles in transcriptional regulation: the human immunodeficiency virus type 1 promoter as a model system. Biochem. Pharmacol. 64:925-934. [DOI] [PubMed] [Google Scholar]
- 28.Rabson, A. B., and H. C. Lin. 2000. NF-kappa B and HIV: linking viral and immune activation. Adv. Pharmacol. 48:161-207. [DOI] [PubMed] [Google Scholar]
- 29.Re, F., D. Braaten, E. K. Franke, and J. Luban. 1995. Human immunodeficiency virus type 1 Vpr arrests the cell cycle in G2 by inhibiting the activation of p34cdc2-cyclin B. J. Virol. 69:6859-6864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sherman, M. P. 2002. HIV infection results in G2 cell cycle arrest in vivo. Presented at the 9th Conference of Retroviruses and Opportunistic Infections, Washington, D.C.
- 31.Tsi, C. J., Y. Chao, C. W. Chen, and W. W. Lin. 2002. Aurintricarboxylic acid protects against cell death caused by lipopolysaccharide in macrophages by decreasing inducible nitric-oxide synthase induction via IkappaB kinase, extracellular signal-regulated kinase, and p38 mitogen-activated protein kinase inhibition. Mol. Pharmacol. 62:90-101. [DOI] [PubMed] [Google Scholar]
- 32.Tsukiyama, T. 2002. The in vivo functions of ATP-dependent chromatin-remodelling factors. Nat. Rev. Mol. Cell. Biol. 3:422-429. [DOI] [PubMed] [Google Scholar]
- 33.Van Lint, C., C. A. Amella, S. Emiliani, M. John, T. Jie, and E. Verdin. 1997. Transcription factor binding sites downstream of the human immunodeficiency virus type 1 transcription start site are important for virus infectivity. J. Virol. 71:6113-6127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Van Lint, C., S. Emiliani, M. Ott, and E. Verdin. 1996. Transcriptional activation and chromatin remodeling of the HIV-1 promoter in response to histone acetylation. EMBO J. 15:1112-1120. [PMC free article] [PubMed] [Google Scholar]
- 35.Verdin, E., P. Paras, Jr., and C. Van Lint. 1993. Chromatin disruption in the promoter of human immunodeficiency virus type 1 during transcriptional activation. EMBO J. 12:3249-3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yao, X. J., A. J. Mouland, R. A. Subbramanian, J. Forget, N. Rougeau, D. Bergeron, and E. A. Cohen. 1998. Vpr stimulates viral expression and induces cell killing in human immunodeficiency virus type 1-infected dividing Jurkat T cells. J. Virol. 72:4686-4693. [DOI] [PMC free article] [PubMed] [Google Scholar]