

Abstract
Background
Haploidentical donor (HID) hematopoietic stem cell transplantation (HSCT) is an alternative treatment method for severe aplastic anemia (SAA) patients lacking suitable identical donors and those who are refractory to immunosuppressive therapy (IST). The current study evaluated the feasibility of upfront haploidentical HSCT in SAA patients.
Methods
We conducted a multicenter study based on a registry database. One hundred fifty-eight SAA patients who underwent upfront transplantation between June 2012 and September 2015 were enrolled.
Results
Eighty-nine patients had haploidentical donors (HIDs), and 69 had matched related donors (MRDs) for HSCT. The median times for myeloid engraftment in the HID and MRD cohorts were 12 (range, 9–20) and 11 (range, 8–19) days, with a cumulative incidence of 97.8 and 97.1% (P = 0.528), respectively. HID recipients had an increased cumulative incidence of grades II–IV acute graft-versus-host disease (aGVHD) (30.3 vs. 1.5%, P < 0.001), grades III–IV aGVHD (10.1 vs. 1.5%, P = 0.026), and chronic GVHD (cGVHD) (30.6 vs. 4.4%, P < 0.001) at 1 year but similar extensive cGVHD (3.4 vs. 0%, P = 0.426). The three-year estimated overall survival (OS) rates were 86.1 and 91.3% (P = 0.358), while the three-year estimated failure-free survival (FFS) rates were 85.0 and 89.8% (P = 0.413) in the HID and MRD cohorts, respectively. In multivariate analysis, survival outcome for the entire population was significantly adversely associated with increased transfusions and poor performance status pre-SCT. We did not observe differences in primary engraftment and survival outcomes by donor type.
Conclusions
Haploidentical SCT as upfront therapy was an effective and safe option for SAA patients, with favorable outcomes in experienced centers.
Electronic supplementary material
The online version of this article (doi:10.1186/s13045-017-0398-y) contains supplementary material, which is available to authorized users.
Keywords: Upfront, Haploidentical transplantation, Acquired severe aplastic anemia
Background
Aplastic anemia (AA) is a rare but potentially fatal disorder characterized by hypocellular bone marrow and pancytopenia. Severe aplastic anemia (SAA) is a life-threatening type for which allogeneic hematopoietic stem cell transplantation (HSCT) and immunosuppressive therapy (IST) are the principle treatment modalities. Upfront matched related donor (MRD) HSCT is recommended for patients with SAA younger than 35 years of age [1] and, reportedly, results in long-term survival rates over 80% [2–5]. Unfortunately, only about 30% of patients have a matched sibling. According to current therapeutic algorithms, IST with a combination of horse antithymocyte globulin (ATG) and cyclosporin A (CsA) is the preferred first-line treatment for patients without a MRD, and HSCT from a haploidentical donor (HID) should be delayed until one or two courses of IST have failed [1]. However, approximately one third of patients did not respond to IST and one third of responders relapsed after initial therapy [5, 6]. In addition, clonal evolution was also observed in 15% of cases [7]. Furthermore, only rabbit and porcine ATG are available in China and are much less effective than horse ATG [8]. As such, IST has a high failure rate, which may impact patient quality of life due to the necessity for further treatment.
A study in the Asian Pacific observed that transplantation from alternative donors achieved comparable outcome to that from MRD, with a 5-year OS of 83.7 and 90.6% in pediatric patients [9]. Another study from European Cooperative Group for Bone Marrow Transplantation (EBMT) also demonstrated survival outcomes for upfront-unrelated donor HSCT similar to those of MRD HSCT in pediatric SAA (2-year OS of 96 and 91%) [10]. In recent years, remarkable improvement has also been made in HID SCT [11–13], primarily due to optimal conditioning regimens, improved supportive care, and advances in medications. However, published data on HID SCT for SAA are limited and mostly restricted in mixed variety line of therapy. In addition, the option of upfront HID SCT has never been investigated in a comparative manner. We previously developed a novel protocol and successfully used a salvage therapy for SAA patients [14]. To further assess the therapeutic effect and safety of upfront HID SCT for the treatment of SAA, we conducted a multicenter retrospective study based on data from a registry database.
Methods
Patients
Based on the Chinese Bone Marrow Transplantation Registry (CBMTR) registry database, 11 transplant centers in China that used the same protocol for HID HSCT in SAA patients were invited to join this study. Between June 2012 and September 2015, a total of 158 consecutive patients with acquired SAA underwent HSCT from a HID (n = 89) or MRD (n = 69) as upfront treatment. Disease severity was defined as previously described [15]. Patients with congenital bone marrow disorders (Fanconi anemia, Diamond-Blackfan anemia, and dyskeratosis congenital (DKC)) were excluded clinically and by laboratory assays. Trephine biopsy and chromosome tests were routinely performed. The chromosome breakage and gene test was used to exclude Fanconi anemia. Telomerase RNA component (TERC) mutation analysis was used to detect hidden forms of DKC, which was performed when congenital forms were suspected based on patient history and clinical analysis. The study was approved by the institutional review board of each of the eleven participating institutions, and written informed consent was obtained from all subjects in accordance with the principles of the Declaration of Helsinki.
Conditioning regimen
Conditioning therapy in haploidentical transplantation consisted of the following: 0.8 mg/kg intravenous (i.v.) busulfan (BU) four times daily on days −7 and −6; 50 mg/kg i.v. cyclophosphamide (CY) once daily on days −5, −4, −3, and −2; and 2.5 mg/kg i.v. ATG (rabbit, Sangstat product Lyons, France) once daily for four consecutive days from days 5 to 2.
In matched related SCT, patients received either CY + ATG (CY: 50 mg/kg/day and ATG 2.5 mg/kg/day (rabbit)) or CY + ATG+ fludarabine (Flu) regimen. The three-drug conditioning consisted of 120–200 mg/m2 total Flu combined with 100–200 mg/kg total modified CY and 10–12.5 mg/kg total ATG.
Stem cell mobilization and collection
The majority of patients received granulocyte colony-stimulating factor (G-CSF)-primed bone marrow (BM) combined with G-CSF-primed peripheral blood (PB) hematopoietic stem cell (HSC). Donor stem cell mobilization was performed using subcutaneous G-CSF (Filgrastim, Kirin, Japan or Granocyte, Chugai, Japan) at 5 μg/kg/day from day -3 until the last day of collection. BM grafts were harvested on day 1. The target mononuclear cell (MNC) count from BM was 2–4 × 108/kg recipient weight. On day 2, peripheral blood stem cells (PBSCs) were collected by apheresis using a COBE Blood Cell Separator (Gambro BCT, Lakewood, CO, USA). The target MNC count from BM and PB was 6–8 × 108/kg recipient weight. An additional harvest of PBSCs was performed on day 3 if the target MNC count was not achieved.
GVHD prophylaxis, management, and supportive care
In the HID cohort, all patients received CsA, MMF, and short-term MTX as acute graft-versus-host disease (aGVHD) prophylaxis [16]. CsA (1.5 mg/kg, q 12 h, i.v.) was administered from day −9 with a target trough concentration of 150–250 ng/mL. It was switched to oral administration when recipient bowel function returned to normal. From day −9, MMF (0.5 g q 12 h, 0.25 g q 12 h in pediatric patients) was administered orally, which was tapered to half on day +30 and discontinued on day +60. MTX (15 mg/m2) was administered intravenously on day +1, followed by a dose of 10 mg/m2 on days +3, +6, and +11 (in MRD HSCT, MTX +1, +3, and +6). In the MRD cohort, MMF dosage was tapered upon engraftment.
Acute and chronic GVHD (aGVHD and cGVHD) were diagnosed and graded according to international criteria [17, 18]. With regards to the treatment of aGVHD, the effective concentration of CsA was resumed, and 1–2 mg/kg/day methylprednisolone equivalents were administered as first-line therapy. For steroid-refractory aGVHD, second-line treatments such as tacrolimus (FK506), MMF, and CD25 monoclonal antibody (CD25 mAb) or MTX were administered.
G-CSF (5 μg/kg/day) was administered subcutaneously from day +6 until myeloid engraftment in the HID but not in the MRD cohort. Viral surveillance included screening for cytomegalovirus (CMV) and Epstein-Barr virus (EBV) by polymerase chain reaction twice weekly. Preemptive ganciclovir (DHPG) or foscarnet therapy was initiated for positive CMV polymerase chain reaction findings. Other infection prevention and supportive care were provided in accordance with previous articles [19, 20].
Definitions and evaluation
Myeloid and platelet engraftment were defined as previously described [21]. Full donor chimerism (FDC) was defined as the presence of >95% donor hematopoietic cells. Patients who did not reach neutrophil counts >0.5 × 109/L for three consecutive days after transplantation were considered to have had primary graft failure. Patients with initial engraftment in whom recurrent pancytopenia with obviously hypocellular BM and without moderate to severe acute GVHD were considered to have had secondary graft failure [22]. OS was defined as the time from the date of HSCT to death from any cause or last follow-up. Failure-free survival (FFS) was defined as survival with response. Death, primary or secondary graft failure, and relapse were considered treatment failures. GFFS (GVHD-free, failure-free survival) was defined as grades III–IV acute GVHD, extensive chronic GVHD, and treatment failures as the above. Transplantation-related mortality (TRM) was defined as death without relapse. Regimen-related toxicity was evaluated according to Seattle Toxicity Criteria [23]. Performance status was graded according to Eastern Cooperative Oncology Group (ECOG) scoring.
Statistical analysis
The last follow-up for all surviving patients was April 30, 2016. Patients lost to follow-up were censored at the time of their withdrawal. Differences in the distribution of various parameters for the two groups were compared using chi-square or Student’s t tests as appropriate. Analyses of OS, FFS, and GFFS were performed using the Kaplan–Meier method, with differences compared by log-rank tests. Cumulative incidences of engraftment and GVHD were estimated in the competing risk model, with early death as the competing event. Univariate and multivariate analyses were performed to determine whether any of the selected factors were predictive of the endpoints. In multivariate analysis, all factors with P < 0.1 in univariate analysis were evaluated in the Cox regression model with a backward stepwise model selection approach. Significant factors (P < 0.05) were considered to be independently predictive of the outcomes. All statistical analyses were performed using SPSS Version 13.0 and R software package (version 2.6.1; http://www.r-project.org).
Results
Basic characteristics
Table 1 shows the patient and transplant characteristics of the 158 patients. All patients had no IST or had only CsA as IST for a limited time before transplantation. All cases underwent transplantation within 4 months after definite diagnosis. The two cohorts were similar with regard to male/female ratio, interval between diagnosis and transplant, red blood cell (RBC) transfusions, and ECOG scores prior to transplant. HID patients were younger than patients in the MRD cohort (median age 22 years [range, 4–51 years] vs. 33 years [range, 7–61 years], respectively; P < 0.001). Recipients of mismatched transplants had more male donors than did the recipients of matched transplants. Approximately 90% of patients in the HID group received grafts from BM and PB, compared to 63.2% in the MRD cohort. The graft compositions in the two groups were similar except that the matched patients had higher CD34 cell counts (4.5 [range, 1.5–30.2] × 106/kg vs. 3.6 [range, 0.5–18.8] × 106/kg, respectively; P = 0.001).
Table 1.
Patient and graft characteristics
| Variable | Haploidentical N = 89 | Matched related N = 69 | P |
|---|---|---|---|
| Age (years), median (range) | 22 (4–51) | 33 (7–61) | <0.001 |
| Children, no. (%) | 33 (37.1%) | 7 (10.1%) | <0.001 |
| Adult, no. (%) | 56 (62.9%) | 62 (89.9%) | |
| Male/female, no. | 57/32 | 39/30 | 0.337 |
| SAA/VSAA, no. | 69/20 | 58/11 | 0.305 |
| Disease course (months), median (range) | 0.507 | ||
| Interval from SAA diagnosis to SCT | 1.5 (1–4.0) | 1.0 (1–3.0) | |
| Previous transfusion | |||
| Median units of RBC (range) | 7.0 (0–30) | 8.0 (2–34) | 0.115 |
| Median units of PLT (range) | 13.5 (0–90) | 10.0 (2–90) | 0.036 |
| aECOG pre-SCT, median, (range) | 1 (0–3) | 1 (0–2) | 0.568 |
| Donor-patient sex match, no. (%) | 0.042 | ||
| Male-male | 40 (44.9%) | 17 (24.6%) | |
| Male-female | 19 (21.3%) | 15 (21.7%) | |
| Female-male | 19 (21.3%) | 22 (31.9%) | |
| Female-female | 11 (12.4%) | 15 (21.7%) | |
| Donor type, no. (%) | <0.001 | ||
| Sibling | 25 (28.1%) | 69 (100%) | |
| Parent | 57 (64.0%) | – | |
| Child | 5 (5.6%) | – | |
| bOthers | 2 (2.2%) | – | |
| HLA type, no (%) | <0.001 | ||
| 6/6 | 4 (4.5%) | 69 (100%) | |
| 5/6 | 6 (6.7%) | – | |
| 4/6 | 21 (23.6%) | – | |
| 3/6 | 58 (65.2%) | – | |
| ABO matched, no. (%) | 0.414 | ||
| Matched | 49 (55.1%) | 44 (63.8%) | |
| Minor mismatched | 18 (20.2%) | 13 (18.8%) | |
| Major mismatched | 14 (15.7%) | 10 (14.5%) | |
| Different | 8 (9.0%) | 2 (2.9%) | |
| Graft type, no. (%) | <0.001 | ||
| BM + PB | 78 (87.6%) | 43 (63.2%) | |
| BM only | 9 (10.1%) | 3 (4.4%) | |
| PB only | 2 (2.2%) | 22 (32.4%) | |
| Median MNCs, ×10^8/kg (range) | 9.9 (3.4–32.0) | 10.5 (4.6–26.4) | 0.817 |
| Median CD34+ count, ×10^6/kg (range) | 3.6 (0.5–18.8) | 4.5 (1.5–30.2) | 0.001 |
| Median CD3+ count, ×10^8/kg (range) | 1.8 (0.1–6.7) | 2.1 (0.1–7.7) | 0.530 |
| Median CD4+ count, ×10^8/kg (range) | 1.0 (0.1–3.5) | 1.1 (0.1–5.0) | 0.337 |
| Median CD8+ count, ×10^8/kg (range) | 0.7 (0.1–3.0) | 0.8 (0.1–2.7) | 0.147 |
| Median follow-up among alive patients, mo. (range) | 21.4 (7.1–47.6) | 26.0 (7.5–47.6) | 0.258 |
| Neutrophil engraftment, median (range) | 12 (9–20) | 11 (8–19) | 0.151 |
| Platelet engraftment, median (range) | 15 (6–91) | 14 (7–36) | 0.484 |
BM bone marrow, PB peripheral blood, MNC mononuclear cell
*Patient age, previous transfusion of platelet, donor-recipient sex match, graft type, and infused CD34 cells differed significantly between the two groups (P < 0.05). There were no other significances between group differences
aECOG (Eastern Cooperative Oncology Group scale) is used to evaluate patients’ performance status
bOther donor types were from cousins
Engraftment
In the HID group, 87 of 88 (98.9%) cases who survived more than 28 days achieved myeloid engraftment at a median of 12 days (range, 9–20 days). In the MRD cohort, 67 evaluable patients engrafted at a median of 11 days (range, 8–19 days). The cumulative incidences of 28-day engraftment were 97.75 ± 0.03% and 97.10 ± 0.05% (P = 0.528, Additional file 1: Figure S1a) in the mismatched and matched groups, respectively. Eighty-six and 66 patients had platelet engraftments in the HID and MRD groups at 15 days (range, 6–91 days) vs. 14 days (range, 7–36 days), with cumulative incidences of platelet engraftment of 96.63 ± 0.05% and 95.65 ± 0.08%, respectively (P = 0.989, Additional file 1: Figure S1b). One HID patient failed to achieve primary engraftment and underwent a second transplant from the original donor; however, he experienced primary graft failure again. One patient in the MRD cohort experienced secondary graft failure on day +45 and refused further therapy.
GVHD
The cumulative incidence of grades II–IV aGVHD at 100 days were 30.34 ± 0.24% and 1.45 ± 0.02% after HID and MRD transplants, respectively (P < 0.001, Additional file 1: Figure S2a). The cumulative incidence of grades III–IV aGVHD at 100 days were 10.11 ± 0.10% and 1.45 ± 0.02% (P = 0.026, Additional file 1: Figure S2b). Multivariate analysis identified no significant factors in II–IV aGVHD, and HID was the only independent factor associated with III–IV aGVHD (Table 2).
Table 2.
Multivariate analysis of adverse factors associated with survival outcomes and GVHD
| Outcome | Hazard ratio (95% confidence interval) | P value |
|---|---|---|
| Overall survival | ||
| Previous RBC (>10) | 6.8 (1.9–23.8) | 0.003 |
| ECOG (>2) | 2.9 (1.1–7.9) | 0.032 |
| Failure-free survival | ||
| Previous RBC (>10) | 5.4 (1.8–16.3) | 0.003 |
| ECOG (>2) | 3.0 (1.2–7.5) | 0.022 |
| II–IV aGVHD | ||
| HID | 1.3 (0.9–1.8) | 0.181 |
| III–IV aGVHD | ||
| HID | 1.6 (1.2–2.2) | 0.006 |
HID haploidentical donor, aGVHD acute graft-versus-host disease, cGVHD chronic graft-versus-host disease
Eighty-three and 65 patients in the HID and MRD cohorts, respectively, with survival longer than 100 days after transplantation were evaluable for the incidence of cGVHD. HID patients had a higher three-year cumulative incidence of cGVHD than did the MRD patients (39.30 ± 0.54% vs. 8.35 ± 0.13%, P < 0.001, Additional file 1: Figure S3a). However, the two groups had similar three-year incidences of extensive cGVHD (3.42 ± 0.04% vs. 2.03 ± 0.04%, P = 0.426, Additional file 1: Figure S3b). During the follow-up, three mismatched and one matched patients with extensive cGVHD received systemic therapy.
Infectious complications and immune reconstitution
The most common infection was the reactivation of CMV, which occurred in 46 (51.7%) HID and 30 (43.5%) MRD patients (P = 0.306), at a median of 30 (range, 16-74) and 28 (range, 11-49) days post-transplantation. Only one HID patient developed CMV enteritis on day +33 and recovered after administration of antiviral drugs combined with an infusion of CMV-specific cytotoxic T lymphocytes (CMV-CTL). Twenty-five (28.1%) and 15 (21.7%) suffered EBV viremia in the HID and MRD-SCT groups (P = 0.363). The median times to EBV viremia in the two cohorts were 41 (range, 26–73) and 34 (range, 18–89) days, respectively. One HID and one MRD case developed EBV-associated post-transplant lymphoproliferative disorders (PTLD) on days +76 and +68, respectively.
The outcomes of immune reconstitution are shown in Fig. 1. CD3, CD4, and CD19 concentrations were comparable between the two cohorts from 6 months post-SCT. Furthermore, equivalent levels of immunoglobulins A, G, and M (IgA, IgG, IgM) were achieved at 1 year.
Fig. 1.
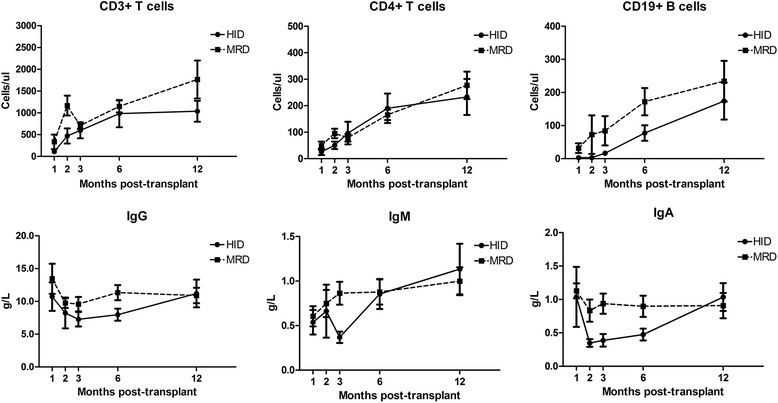
Immune reconstitution. Reconstitution of CD3, CD4, and CD19 lymphocytes were comparable from 6 months post-SCT. Equivalent levels of immunoglobulins A, G, and M (IgA, IgG, IgM) were achieved at 1 year between two cohorts
Transplantation-related mortality
During a median follow-up of 22.6 months (range, 7.1–47.6), 12 and 6 were in the HID and the MRD groups, respectively, with a median time to death of 96.5 (range, 2–345 days) and 51 days (2–244 days). Analyses of TRM revealed that GVHD and infection were the major causes of death in the two groups. In the HID cohort, six patients (6.74%) died of infection (two fungal, one EBV-associated PTLD, and three serious bacterial infections), four (4.49%) died of GVHD (three severe aGVHD and one extensive cGVHD), one (1.12%) of regimen-related toxicity (RRT), and one of primary graft failure. Six (8.70%) patients died of TRM in the MRD cohort, which included three (4.35%) of infection (two fungal and one bacterial), one (1.45%) due to severe aGVHD, one from RRT, and one from secondary graft failure.
Survival outcomes and follow-up
The three-year probabilities of overall survival (OS) were 86.1 ± 3.7% and 91.3 ± 3.4% after HID and MRD-related donor transplants, respectively (P = 0.358, Fig. 2). The three-year FFS was also not significantly different in the upfront HID HSCT cohort (85.0 ± 3.9%) vs. the MRD controls (89.8 ± 3.7%) (P = 0.413, Fig. 3). Increased RBC transfusions, longer SAA courses, and poorer performance scores significantly predicted survival outcomes in univariate analysis (Additional file 1: Table S1). In multivariate analysis, the risks of mortality did not differ significantly by donor type (Table 2), but mortality was significantly higher in patients receiving increased RBC transfusions and in those with poor performance scores. The estimated GFFS at 1 year was also similar (80.8 ± 4.2% and 88.4 ± 3.9%, P = 0.282, Fig. 4) in mismatched and matched patients.
Fig. 2.
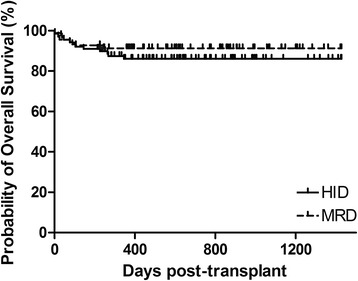
Overall survival of two cohorts: HID, 3-year OS of 86.1% ± 3.7%; MRD, 3-year OS of 91.3% ±3.4% (P = 0.358)
Fig. 3.
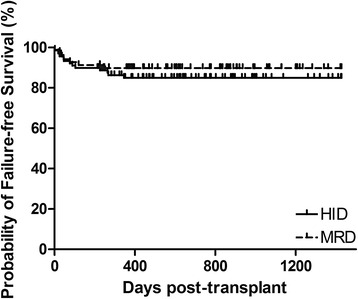
Failure-free survival of two cohorts: HID, 3-year FFS of 85.0% ± 3.9%; MRD, 3-year FFS of 89.8% ± 3.7% (P = 0.413)
Fig. 4.
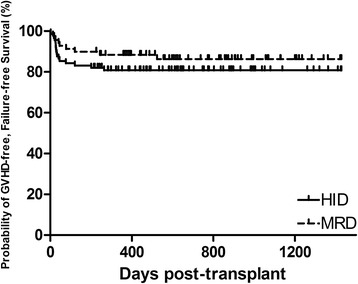
GVHD, failure-free survival of two cohorts: HID, 1-year GFFS of 80.8% ± 4.2%; MRD 1-year GFFS of 88.4% ± 3.9% (P = 0.282)
As of April 30, 2016, all of the 140 surviving patients (77 HID and 63 MRD patients) achieved transfusion independence. Among the 120 patients (63 HID and 57 MRD) who were followed up for more than 1 year, 100% alive patients break away from transfusion. 87.9 vs. 88.2% had normal WBC count, 90.9 vs. 88.2% had normal platelet (PLT) count, and 93.9 vs. 91.2% cases attained hemoglobin above 100 g/L in the HID and MRD groups, respectively. In addition, 86.8% of HID and 86.7% of MRD patients achieved Karnofsky Performance Status Scale (KPS) scores ≥90.
Discussion
Allo-HSCT from MRD leads to long-term survival in over 80% of patients, with most survivors having a normal performance status [24–27]. In general, HSCT with sustained engraftment restores bone marrow function and precludes the late complications such as relapse and clonal evolution observed with IST. More than two thirds of patients lack an MRD; however, a HID is a readily available for nearly all patients. Indeed, transplantation from HID has benefited from the same improvements as has transplantation from matched siblings, and great progress has been made in both engraftment and survival in HID [28]. Furthermore, rabbit ATG is markedly inferior to horse ATG as a first-line treatment for SAA [8]. Thus, both the unavailability of horse ATG and poor economic status which afforded either IST or SCT have prompted interest in the feasibility of upfront HID HSCT in developing countries such as China. Hence, we conducted a study based on registry data regarding the feasibility of upfront HID SCT. To the best of our knowledge, we are the first to compare outcomes of consecutive patients undergoing upfront HID SCT with upfront allo-HSCT using MRD. Data from our study demonstrated that comparable engraftment and survival outcomes were achieved in both cohorts.
Graft failure is a central problem in HSCT for SAA, occurring more frequently than in other hematological malignancies. In the initial attempts with MRD-SCT, using CY alone as conditioning regimen and MTX alone for GVHD prophylaxis, the rates of GF with MRD-SCT were over 30% [29]. Higher GF rates of 70% were also observed in early experience with HID SCT [30]. However, the current situation is considerably different. CY with ATG for conditioning followed by post-grafting CsA plus MTX resulted in an engraftment rate of 95% in matched siblings [24]. Similarly, several studies on HID transplants reported incidence of GF ranging from 0 to 25% due to recent advances in ex vivo depletion of T cells or unmanipulated in vivo regulation [21, 28, 31–35]. Our findings showed that the HID cohort without in vitro T cell depletion had myeloid (97.75 vs. 97.10%) and platelet (96.63 vs. 95.65%) engraftment rates comparable to those of the MRD cohort. As analyzed in our previous research [21], the reasons for these encouraging engraftment outcomes in mismatched transplants may be multifactorial, including adding BU to CY + ATG for intensified conditioning [36, 37], using G-CSF-mobilized grafts [38] and combining CsA, MTX, and MMF as GVHD prophylaxis.
As SAA is a non-neoplastic hematologic disorder, another goal of transplantation is to avoid acute or chronic GVHD after successful engraftment. Although higher proportions of patients with II–IV (30.34 vs. 1.45%) and III–IV aGVHD (10.11 vs. 1.45%) in the HID than in the MRD cohorts, the incidences of aGVHD were comparable to those of recent studies involving ex vivo T cell depletion haploidentical transplants, reported II–IV aGVHD rates of 30–33% [11, 12, 28, 31]. Unexpectedly, univariate analysis showed that older age (≥18 years) and PB graft were associated with a reduced incidence of II–IV aGVHD in our total cohort. However, it is important to realize that adult and PB graft were significantly more common in the matched group, and no significant factors were identified in multivariate analysis. Although we found that risks of severe aGVHD differed significantly by donor type, the incidence of severe aGVHD-related TRM was not obviously higher in the HID cohort. PTCY or CD25mAb may reduce the incidence of severe GVHD, which should be assessed in future studies.
Extensive cGVHD has a major impact on quality of life in HSCT recipients [39]. The incidence of extensive cGVHD was similar between the HID and MRD HSCT cohorts in our study. However, limited GVHD may also affect quality of life post-transplantation. We assessed quality of life during the follow-up period; most patients with limited cGVHD rated their quality of life as excellent and their symptoms as minimal or mild. Our study demonstrated that nearly 90% of patients in both cohorts who survived for more than a year achieved KFS scores above 90. Further long-term follow-up is necessary to confirm this positive performance status. Besides, similar 1-year GFFS in two cohorts also supported that patients alive had comparable survival rates without ongoing morbidity.
Infection was also a major barrier to the wider application of HID SCT. However, delayed immune reconstitution within 6 months post-transplantation was observed in our HID cohort. The possible reasons were as follows: first, an intensified immunological suppression conditioning regimen was used in the mismatched patient group to promote engraftment and prevent GVHD; second, additional immunological suppression therapy was administered because of higher incidences of aGVHD in the HID cohort. Encouragingly, the incidence of viral infection was similar between the two groups. In the HID and MRD cohorts, CMV reactivation occurred in 51.7 and 43.5% of patients, but only one mismatched patient developed disease. EBV viremia was detected in 28.1 and 21.7% of patients in each group and lymphoproliferative disorder in 1.1 and 1.4%, respectively. Ganciclovir was commonly administered as prophylaxis, intensive surveillance was performed twice weekly, and preemptive treatment was administered promptly once viremia was detected, which may have helped to decrease the incidence of lethal virus infection.
We have observed comparable survival (three-year OS of 86.1 vs. 91.3%, FFS of 85.0 vs. 89.8%) after HID and MRD transplantations, with increased RBC transfusions and poor ECOG being the adverse factors. Consistent with other reports [22, 40], heavily transfused patients have worse outcomes, including increased graft failure and poor survival. In our data, increased transfusions had no influence on either engraftment or GVHD. One possible explanation is that iron deposition brought by transfusions decreased organ function prior to SCT. Passweg et al. also observed that poor performance scores affected outcomes after transplantation [41]. All of these findings point to the need for reducing such delays pre-transplantation, which may reduce the need for multiple transfusions and the incidence of pre-treatment infections, thus improving performance status at transplantation and improving OS rates.
Our study has several limitations, including the potential for selection bias; the fact that the patients in the HID cohort were younger than those in the MRD cohort and that the donor-recipient sex match and graft source differed significantly between groups implied that comparisons between GVHD rates in these groups were not feasible. As age has been shown to influence outcomes [42], we also stratified our population into pediatric and adult patients and observed similar outcomes in pediatric HID vs. MRD and adult HID vs. MRD. Despite these limitations, our data offer compelling comparative evidence of the value of HID SCT as a front-line treatment in developing countries.
HID SCT has several obvious advantages compared to other alternative donors. First, donors are available for nearly all patients; second, SCT can be performed immediately, which is particularly crucial for patients with vSAA who need prompt therapy; and third, continued donor access is available for rescuing graft failure or for treating infections.
Conclusions
In summary, our findings show that upfront HID HSCT is a safe and feasible choice for patients without suitably matched donors. Our data indicates that there might be a change in the SAA treatment algorithm in developing countries, and upfront HID HSCT might be considered alongside IST for patients lacking matched donors in specialized centers. Further prospective multicenter research is needed to confirm that.
Acknowledgements
We give thanks to all colleagues for participating in the research.
Funding
This work was partly supported by the Collaborative Innovation Center of Hematology China, National Natural Science Foundation of China (Grant No. 81230013 and 81370666), and Beijing Municipal Science and Technology Program (No. Z141100000214011 and Z151100001615020).
Availability of data and materials
All the data generated or analyzed during this study are included in this published article (and its supplementary information files).
Authors’ contributions
X-JH designed the research; L-PX and X-JH analyzed the data and wrote the manuscript; and all authors provided the patients’ data and gave the final approval for the manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
The study was approved by each institutional review board at the eleven participating institutions, and written informed consent was obtained from all subjects in accordance with the Declaration of Helsinki.
Abbreviations
- AA
Aplastic anemia
- aGVHD
Acute graft-versus-host disease
- ATG
Antithymocyte globulin
- BM
Bone marrow
- BU
Busulfan
- CBMTR
Chinese Bone Marrow Transplantation Registry
- CD25 mAb
CD25 monoclonal antibody
- cGVHD
Chronic graft-versus-host disease
- CMV
Cytomegalovirus
- CMV-CTL
Cytomegalovirus-specific cytotoxic T lymphocytes
- CsA
Cyclosporin A
- CY
Cyclophosphamide
- EBV
Epstein-Barr virus
- ECOG
Eastern Cooperative Oncology Group
- FDC
Full donor chimerism
- FFS
Failure-free survival
- Flu
Fludarabine
- GFFS
GVHD-free, failure-free survival
- HID
Haploidentical donor
- HSCT
Hematopoietic stem cell transplantation
- IST
Immunosuppressive therapy
- KPS
Karnofsky Performance Status Scale
- MMF
Mycophenolate mofetil
- MNC
Mononuclear cell
- MRD
Matched related donor
- MTX
Methotrexate
- OS
Overall survival
- PB
Peripheral blood
- PTLD
Post-transplant lymphoproliferative disorders
- RBC
Red blood cell
- RRT
Regimen-related toxicity
- SAA
Severe aplastic anemia
- TRM
Transplantation-related mortality
Additional file
Univariate analysis of factors associated with survival outcomes. Table S2. Univariate analysis of factors associated with II–IV aGVHD and III–IV aGVHD. Figure S1a. The cumulative incidence of 28-day neutrophil engraftment:HID97.75 ± 0.03%, MRD97.10 ± 0.05% (P = 0.528). Figure S1b. The cumulative incidence of platelet engraftment: HID96.63 ± 0.05%, MRD 95.65 ± 0.08% (P = 0.989). Figure S2a. The cumulative incidence of II–IV aGVHD: HID 30.34 ± 0.24%, MRD1.45 ± 0.02% (P < 0.001). Figure S2b. The cumulative incidence of III–IV aGVHD: HID10.11 ± 0.10%, MRD1.45 ± 0.02% (P = 0.026). Figure S3a. The cumulative incidence of cGVHD (P < 0.001). Figure S3b. The cumulative incidence of extensive cGVHD (P = 0.426). (DOCX 42 kb)
Contributor Information
Lan-Ping Xu, Email: lpxu_0415@sina.com.
Song Jin, Email: jin088@sina.com.
Shun-Qing Wang, Email: drwangshq@medmail.com.cn.
Ling-Hui Xia, Email: linghuixia@hust.edu.cn.
Hai Bai, Email: baihai98@tom.com.
Su-Jun Gao, Email: sujung1963@sina.com.
Qi-Fa Liu, Email: liuqifa628@163.com.
Jian-Min Wang, Email: jmwang@medmail.com.cn.
Xin Wang, Email: xinw007@126.com.
Ming Jiang, Email: jiangmingyy@126.com.
Xi Zhang, Email: zhangxxi@sina.com.
De-Pei Wu, Email: wudepei@medmail.com.cn.
Xiao-Jun Huang, Phone: 8610-8832-6006, Email: huangxiaojun@bjmu.edu.cn.
References
- 1.Killick SB, Bown N, Cavenagh J, Dokal I, Foukaneli T, Hill A, Hillmen P, Ireland R, Kulasekararaj A, Mufti G, et al. Guidelines for the diagnosis and management of adult aplastic anaemia. Br J Haematol. 2016;172:187–207. doi: 10.1111/bjh.13853. [DOI] [PubMed] [Google Scholar]
- 2.Locasciulli A, Oneto R, Bacigalupo A, Socie G, Korthof E, Bekassy A, Schrezenmeier H, Passweg J, Fuhrer M. Outcome of patients with acquired aplastic anemia given first line bone marrow transplantation or immunosuppressive treatment in the last decade: a report from the European Group for Blood and Marrow Transplantation (EBMT) Haematologica. 2007;92:11–8. doi: 10.3324/haematol.10075. [DOI] [PubMed] [Google Scholar]
- 3.Kim HJ, Park CY, Park YH, Kim YJ, Kim DW, Min WS, Kim CC. Successful allogeneic hematopoietic stem cell transplantation using triple agent immunosuppression in severe aplastic anemia patients. Bone Marrow Transplant. 2003;31:79–86. doi: 10.1038/sj.bmt.1703786. [DOI] [PubMed] [Google Scholar]
- 4.Armand P, Antin JH. Allogeneic stem cell transplantation for aplastic anemia. Biol Blood Marrow Transplant. 2007;13:505–16. doi: 10.1016/j.bbmt.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 5.Young NS, Calado RT, Scheinberg P. Current concepts in the pathophysiology and treatment of aplastic anemia. Blood. 2006;108:2509–19. doi: 10.1182/blood-2006-03-010777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scheinberg P, Wu CO, Nunez O, Young NS. Long-term outcome of pediatric patients with severe aplastic anemia treated with antithymocyte globulin and cyclosporine. J Pediatr. 2008;153:814–9. doi: 10.1016/j.jpeds.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saracco P, Quarello P, Iori AP, Zecca M, Longoni D, Svahn J, Varotto S, Del Vecchio GC, Dufour C, Ramenghi U, et al. Cyclosporin A response and dependence in children with acquired aplastic anaemia: a multicentre retrospective study with long-term observation follow-up. Br J Haematol. 2008;140:197–205. doi: 10.1111/j.1365-2141.2007.06903.x. [DOI] [PubMed] [Google Scholar]
- 8.Scheinberg P, Nunez O, Weinstein B, Scheinberg P, Biancotto A, Wu CO, Young NS. Horse versus rabbit antithymocyte globulin in acquired aplastic anemia. N Engl J Med. 2011;365:430–8. doi: 10.1056/NEJMoa1103975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen J, Lee V, Luo CJ, Chiang AK, Hongeng S, Tan PL, Tan AM, Sanpakit K, Li CF, Lee AC, et al. Allogeneic stem cell transplantation for children with acquired severe aplastic anaemia: a retrospective study by the Viva-Asia Blood and Marrow Transplantation Group. Br J Haematol. 2013;162:383–91. doi: 10.1111/bjh.12405. [DOI] [PubMed] [Google Scholar]
- 10.Dufour C, Veys P, Carraro E, Bhatnagar N, Pillon M, Wynn R, Gibson B, Vora AJ, Steward CG, Ewins AM, et al. Similar outcome of upfront-unrelated and matched sibling stem cell transplantation in idiopathic paediatric aplastic anaemia. A study on behalf of the UK Paediatric BMT Working Party, Paediatric Diseases Working Party and Severe Aplastic Anaemia Working Party of EBMT. Br J Haematol. 2015;171:585–94. doi: 10.1111/bjh.13614. [DOI] [PubMed] [Google Scholar]
- 11.Wang Y, Liu QF, Xu LP, Liu KY, Zhang XH, Ma X, Fan ZP, Wu DP, Huang XJ. Haploidentical vs identical-sibling transplant for AML in remission: a multicenter, prospective study. Blood. 2015;125:3956–62. doi: 10.1182/blood-2015-02-627786. [DOI] [PubMed] [Google Scholar]
- 12.Wang Y, Liu DH, Liu KY, Xu LP, Zhang XH, Han W, Chen H, Chen YH, Wang FR, Wang JZ, et al. Long-term follow-up of haploidentical hematopoietic stem cell transplantation without in vitro T cell depletion for the treatment of leukemia: nine years of experience at a single center. Cancer. 2013;119:978–85. doi: 10.1002/cncr.27761. [DOI] [PubMed] [Google Scholar]
- 13.Gao L, Zhang C, Gao L, Liu Y, Su Y, Wang S, Li B, Yang T, Yuan Z, Zhang X. Favorable outcome of haploidentical hematopoietic stem cell transplantation in Philadelphia chromosome-positive acute lymphoblastic leukemia: a multicenter study in Southwest China. J Hematol Oncol. 2015;8:90. doi: 10.1186/s13045-015-0186-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu LP, Wang SQ, Wu DP, Wang JM, Gao SJ, Jiang M, Wang CB, Zhang X, Liu QF, Xia LH, et al. Haplo-identical transplantation for acquired severe aplastic anaemia in a multicentre prospective study. Br J Haematol. 2016;175:265–74. doi: 10.1111/bjh.14225. [DOI] [PubMed] [Google Scholar]
- 15.Camitta BM, Thomas ED, Nathan DG, Santos G, Gordon-Smith EC, Gale RP, Rappeport JM, Storb R. Severe aplastic anemia: a prospective study of the effect of early marrow transplantation on acute mortality. Blood. 1976;48:63–70. [PubMed] [Google Scholar]
- 16.Lai YR, Chen YH, Hu DM, Jiang M, Liu QF, Liu L, Hou J, Schwarzenberger P, Li QC, Zhang ZM, et al. Multicenter phase II study of a combination of cyclosporine a, methotrexate and mycophenolate mofetil for GVHD prophylaxis: results of the Chinese Bone Marrow Transplant Cooperative Group (CBMTCG) J Hematol Oncol. 2014;7:59. doi: 10.1186/s13045-014-0059-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Przepiorka D, Weisdorf D, Martin P, Klingemann HG, Beatty P, Hows J, Thomas ED. 1994 Consensus Conference on Acute GVHD Grading. Bone Marrow Transplant. 1995;15:825–8. [PubMed] [Google Scholar]
- 18.Shulman HM, Sullivan KM, Weiden PL, McDonald GB, Striker GE, Sale GE, Hackman R, Tsoi MS, Storb R, Thomas ED. Chronic graft-versus-host syndrome in man. A long-term clinicopathologic study of 20 Seattle patients. Am J Med. 1980;69:204–17. doi: 10.1016/0002-9343(80)90380-0. [DOI] [PubMed] [Google Scholar]
- 19.Huang XJ, Liu DH, Liu KY, Xu LP, Chen H, Han W, Chen YH, Wang JZ, Gao ZY, Zhang YC, et al. Haploidentical hematopoietic stem cell transplantation without in vitro T-cell depletion for the treatment of hematological malignancies. Bone Marrow Transplant. 2006;38:291–7. doi: 10.1038/sj.bmt.1705445. [DOI] [PubMed] [Google Scholar]
- 20.Lu DP, Dong L, Wu T, Huang XJ, Zhang MJ, Han W, Chen H, Liu DH, Gao ZY, Chen YH, et al. Conditioning including antithymocyte globulin followed by unmanipulated HLA-mismatched/haploidentical blood and marrow transplantation can achieve comparable outcomes with HLA-identical sibling transplantation. Blood. 2006;107:3065–73. doi: 10.1182/blood-2005-05-2146. [DOI] [PubMed] [Google Scholar]
- 21.Xu LP, Liu KY, Liu DH, Han W, Chen H, Chen YH, Zhang XH, Wang Y, Wang FR, Wang JZ, Huang XJ. A novel protocol for haploidentical hematopoietic SCT without in vitro T-cell depletion in the treatment of severe acquired aplastic anemia. Bone Marrow Transplant. 2012;47:1507–12. doi: 10.1038/bmt.2012.79. [DOI] [PubMed] [Google Scholar]
- 22.Champlin RE, Horowitz MM, van Bekkum DW, Camitta BM, Elfenbein GE, Gale RP, Gluckman E, Good RA, Rimm AA, Rozman C, et al. Graft failure following bone marrow transplantation for severe aplastic anemia: risk factors and treatment results. Blood. 1989;73:606–13. [PubMed] [Google Scholar]
- 23.Bearman SI, Appelbaum FR, Buckner CD, Petersen FB, Fisher LD, Clift RA, Thomas ED. Regimen-related toxicity in patients undergoing bone marrow transplantation. J Clin Oncol. 1988;6:1562–8. doi: 10.1200/JCO.1988.6.10.1562. [DOI] [PubMed] [Google Scholar]
- 24.Kahl C, Leisenring W, Deeg HJ, Chauncey TR, Flowers ME, Martin PJ, Sanders JE, Storb R. Cyclophosphamide and antithymocyte globulin as a conditioning regimen for allogeneic marrow transplantation in patients with aplastic anaemia: a long-term follow-up. Br J Haematol. 2005;130:747–51. doi: 10.1111/j.1365-2141.2005.05667.x. [DOI] [PubMed] [Google Scholar]
- 25.Anasetti C, Doney KC, Storb R, Meyers JD, Farewell VT, Buckner CD, Appelbaum FR, Sullivan KM, Clift RA, Deeg HJ, et al. Marrow transplantation for severe aplastic anemia. Long-term outcome in fifty “untransfused” patients. Ann Intern Med. 1986;104:461–6. doi: 10.7326/0003-4819-104-4-461. [DOI] [PubMed] [Google Scholar]
- 26.Kiem HP, McDonald GB, Myerson D, Spurgeon CL, Deeg HJ, Sanders JE, Doney K, Appelbaum FR, Sullivan KM, Witherspoon RP, Storb R. Marrow transplantation for hepatitis-associated aplastic anemia: a follow-up of long-term survivors. Biol Blood Marrow Transplant. 1996;2:93–9. [PubMed] [Google Scholar]
- 27.Eapen M, Ramsay NK, Mertens AC, Robison LL, DeFor T, Davies SM. Late outcomes after bone marrow transplant for aplastic anaemia. Br J Haematol. 2000;111:754–60. [PubMed] [Google Scholar]
- 28.Im HJ, Koh KN, Seo JJ. Haploidentical hematopoietic stem cell transplantation in children and adolescents with acquired severe aplastic anemia. Korean J Pediatr. 2015;58:199–205. doi: 10.3345/kjp.2015.58.6.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McCann SR, Bacigalupo A, Gluckman E, Hinterberger W, Hows J, Ljungman P, Marin P, Nissen C, van’t Veer Kerthof E, Raghavachar A, et al. Graft rejection and second bone marrow transplants for acquired aplastic anaemia: a report from the Aplastic Anaemia Working Party of the European Bone Marrow Transplant Group. Bone Marrow Transplant. 1994;13:233–7. [PubMed] [Google Scholar]
- 30.Wagner JL, Deeg HJ, Seidel K, Anasetti C, Doney K, Sanders J, Sullivan KM, Storb R. Bone marrow transplantation for severe aplastic anemia from genotypically HLA-nonidentical relatives. An update of the Seattle experience. Transplantation. 1996;61:54–61. doi: 10.1097/00007890-199601150-00012. [DOI] [PubMed] [Google Scholar]
- 31.Im HJ, Koh KN, Choi ES, Jang S, Kwon SW, Park CJ, Chi HS, Seo JJ. Excellent outcome of haploidentical hematopoietic stem cell transplantation in children and adolescents with acquired severe aplastic anemia. Biol Blood Marrow Transplant. 2013;19:754–9. doi: 10.1016/j.bbmt.2013.01.023. [DOI] [PubMed] [Google Scholar]
- 32.Wang Z, Zheng X, Yan H, Li D, Wang H. Good outcome of haploidentical hematopoietic SCT as a salvage therapy in children and adolescents with acquired severe aplastic anemia. Bone Marrow Transplant. 2014;49:1481–5. doi: 10.1038/bmt.2014.187. [DOI] [PubMed] [Google Scholar]
- 33.Gao L, Li Y, Zhang Y, Chen X, Gao L, Zhang C, Liu Y, Kong P, Wang Q, Su Y, et al. Long-term outcome of HLA-haploidentical hematopoietic SCT without in vitro T-cell depletion for adult severe aplastic anemia after modified conditioning and supportive therapy. Bone Marrow Transplant. 2014;49:519–24. doi: 10.1038/bmt.2013.224. [DOI] [PubMed] [Google Scholar]
- 34.Clay J, Kulasekararaj AG, Potter V, Grimaldi F, McLornan D, Raj K, de Lavallade H, Kenyon M, Pagliuca A, Mufti GJ, Marsh JC. Nonmyeloablative peripheral blood haploidentical stem cell transplantation for refractory severe aplastic anemia. Biol Blood Marrow Transplant. 2014;20:1711–6. doi: 10.1016/j.bbmt.2014.06.028. [DOI] [PubMed] [Google Scholar]
- 35.Esteves I, Bonfim C, Pasquini R, Funke V, Pereira NF, Rocha V, Novis Y, Arrais C, Colturato V, de Souza MP, et al. Haploidentical BMT and post-transplant Cy for severe aplastic anemia: a multicenter retrospective study. Bone Marrow Transplant. 2015;50:685–9. doi: 10.1038/bmt.2015.20. [DOI] [PubMed] [Google Scholar]
- 36.Dulley FL, Vigorito AC, Aranha FJ, Sturaro D, Ruiz MA, Saboya R, Macedo MC, Da Silva RL, Chamone DA, Mehta J, et al. Addition of low-dose busulfan to cyclophosphamide in aplastic anemia patients prior to allogeneic bone marrow transplantation to reduce rejection. Bone Marrow Transplant. 2004;33:9–13. doi: 10.1038/sj.bmt.1704325. [DOI] [PubMed] [Google Scholar]
- 37.Vo PT, Pantin J, Ramos C, Cook L, Cho E, Kurlander R, Khuu H, Barrett J, Leitman S, Childs RW. Conditioning with rabbit versus horse ATG dramatically alters clinical outcomes in identical twins with severe aplastic anemia transplanted with the same allogeneic donor. J Hematol Oncol. 2015;8:78. doi: 10.1186/s13045-015-0173-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu LP, Liu KY, Liu DH, Chen H, Han W, Chen YH, Wang Y, Huang XJ. The inferiority of G-PB to rhG-CSF-mobilized blood and marrow grafts as a stem cell source in patients with high-risk acute leukemia who underwent unmanipulated HLA-mismatched/haploidentical transplantation: a comparative analysis. Bone Marrow Transplant. 2010;45:985–92. doi: 10.1038/bmt.2009.311. [DOI] [PubMed] [Google Scholar]
- 39.Viollier R, Passweg J, Gregor M, Favre G, Kuhne T, Nissen C, Gratwohl A, Tichelli A. Quality-adjusted survival analysis shows differences in outcome after immunosuppression or bone marrow transplantation in aplastic anemia. Ann Hematol. 2005;84:47–55. doi: 10.1007/s00277-004-0930-3. [DOI] [PubMed] [Google Scholar]
- 40.Gluckman E, Horowitz MM, Champlin RE, Hows JM, Bacigalupo A, Biggs JC, Camitta BM, Gale RP, Gordon-Smith EC, Marmont AM, et al. Bone marrow transplantation for severe aplastic anemia: influence of conditioning and graft-versus-host disease prophylaxis regimens on outcome. Blood. 1992;79:269–75. [PubMed] [Google Scholar]
- 41.Passweg JR, Perez WS, Eapen M, Camitta BM, Gluckman E, Hinterberger W, Hows JM, Marsh JC, Pasquini R, Schrezenmeier H, et al. Bone marrow transplants from mismatched related and unrelated donors for severe aplastic anemia. Bone Marrow Transplant. 2006;37:641–9. doi: 10.1038/sj.bmt.1705299. [DOI] [PubMed] [Google Scholar]
- 42.Gupta V, Eapen M, Brazauskas R, Carreras J, Aljurf M, Gale RP, Hale GA, Ilhan O, Passweg JR, Ringden O, et al. Impact of age on outcomes after bone marrow transplantation for acquired aplastic anemia using HLA-matched sibling donors. Haematologica. 2010;95:2119–25. doi: 10.3324/haematol.2010.026682. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All the data generated or analyzed during this study are included in this published article (and its supplementary information files).