

Abstract
Applications of hydrophobic drug-based nanocarriers (NCs) remain largely limited because of their low loading capacity. Here, we report development of a multifunctional hybrid NC made of a magnetic Fe3O4 nanoparticle core and a mesoporous silica shell embedded with fluorescent carbon dots (CDs) and paclitaxel (PTX), and covered by another layer of silica. The NC is prepared via a one-pot process under mild condition. The PTX loading method introduced in this study simplifies drug loading process and demonstrates a high loading capacity as a result of the mesoporous silica dual-shell structure, supramolecular π-stacking between the conjugated rings of PTX molecules and aromatic rings of the CDs in the hybrid NC. The CDs also serve as both confocal and two-photon fluorescence imaging probes, while the Fe3O4 core serves as an MRI contrast agent. Significantly, NC releases PTX in response to NIR irradiation as a result of local heating of the embedded CDs and the heating of CDs also provides an additional therapeutic effect by thermally killing cancer cells in tumor in addition to the chemotherapeutic effect of released PTX. Both in vitro and in vivo results show that NC demonstrates high therapeutic efficacy through a synergistic effect from the combined chemo-photothermal treatments.
Keywords: hydrophobic drug, preloading, mesoporous silica, multifunctional imaging, synergistic effect
Graphical abstract

The manuscript reports a new nanocarrier comprised of a magnetic Fe3O4 core, fluorescent carbon dots, preloaded hydrophobic drug paclitaxel, and mesoporous silica for optical bioimaging, magnetic resonance imaging, and near infrared-responsive drug release. Results demonstrate that the hybrid NCs have high therapeutic efficacy through synergistic effect due to the combined chemo-photothermal treatments.
1. Introduction
Chemotherapy is a mainstay treatment for cancer.[1] However, the poor water-solubility and severe side effects of many hydrophobic anticancer drugs used in chemotherapy limit their overall therapeutic efficacies because of difficulties associated with their delivery through the intravenous route.[2, 3] Nanomaterial-based drug carriers, or nanocarriers (NC), have emerged as alternative delivery systems for intravenous delivery of hydrophobic drugs, which reduce the drug toxicity by encapsulation and improve the therapeutic efficacy through the enhanced permeability and retention effect.[4, 5] Among a wide variety of nanomaterials investigated, mesoporous silica-based NCs have attracted much attention due to their porous structure and large surface area enabling high loading capacity, and tailorable surface property, size and shape for targeted drug delivery.[6–9] Mesoporous silica-based NCs have also been made to release drugs in a controllable fashion or subjected in response to a stimulus such as a change in temperature, pH, or light intensity.[10–18] Furthermore, after functionalized with magnetic and fluorescent components, NCs can serve as multifunctional imaging agents for imaging-guided chemotherapy in cancer treatment,[19–22] to overcome the limitations of single-modal imaging technology such as the lower sensitivity of MRI and poor spatial resolution of fluorescence imaging.[4, 23–35]
In current nanotechnology, hydrophobic drugs are usually incorporated into mesoporous silica NCs after the NCs have been made, which often requires multi-step processes and long loading time, and yet exhibits a relatively lower loading efficiency. In addition, this post-loading of hydrophobic drugs was strictly preformed because drug efficacy was often affected during the synthesis of hybrid NCs. For example, the synthesis of mesoporous silica NCs involves harsh chemical corrosion, high temperature calcination, and use of organic solvent and reactive metals, which all can compromise the therapeutic effect of incorporated drugs.[36] An alternative approach is to encapsulate drugs into NCs during NC synthesis, where drugs are first preloaded into template nanoparticles that are then coated with a layer of polymer or silica.[5, 37–39] The template nanoparticles are then dissolved to yield drug-loaded NCs.[5, 37, 40] However, applications of the NCs made by this approach are often limited by their large hydrodynamic size (>100 nm) and lack of imaging components. Importantly, the step to dissolve the template nanoparticles in the fabrication process involves use of requirement of extra dissolution process involving acid or basic solvents, which may compromise the therapeutic effects of drugs.[19, 41, 42]
Here, we introduce a new synthesis approach to produce multifunctional mesoporous silica NCs that can preload drugs during the synthesis process under mild conditions. The resultant NC contains fluorescent carbon dots (CDs) and a magnetite component and thus has multi-modal imaging capacities in addition to the therapeutic function as a drug carrier. In this study a hydrophobic anticancer drug, paclitaxel (PTX), is used as the model anti-cancer drug. The preparation of this hybrid NC is a one-pot process that involves no template dissolution step mentioned above. It starts with the synthesis of mesoporous silica shell protected CDs and Fe3O4 NPs (Fe3O4@CDs@mSiO2) as template, followed by PTX loading and surface coating with mesoporous silica shell. The fluorescent CDs and magnetic Fe3O4 in the NCs serve to enable confocal and two-photon fluorescence imaging and MRI capabilities, respectively. Furthermore, the CDs in hybrid NCs can absorb and convert NIR light to heat which breaks the bonds between PTX and mSiO2 to release PTX, thus, demonstrating the ability of NIR-responsive drug release and combined photothermal/chemo-therapy for improved therapeutic efficacy.
2. Results and Discussion
Figure 1a illustrates the process for synthesis of Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs, which includes two steps. First, the hybrid NC (Fe3O4@CDs@mSiO2) was prepared by coating the Fe3O4 nanoparticles (NPs) with mesoporous silica (mSiO2), followed by encapsulation of fluorescent CDs into the mesoporous silica shell. The organic–inorganic mesoporous silica shell was prepared by hydrolysis and subsequent condensation of tetraethyl orthosilicate (TEOS) and octadecyltrimethoxysilane (C18TMS) in a mixture of ethanol and ammonia solution.[43] Ethanol serves as a solvent to form a homogeneous solution of TEOS, Fe3O4, CDs and PTX, ammonia acts as a catalyst to form silica nanosphere, and C18TMS was applied to a porogen to generate the porous structure.[44] The TEM image of Fe3O4 NPs in Figure 1b shows that the Fe3O4 NPs are well dispersed, ~10 nm in diameter, and have a uniform size distribution. The good monodispersity and stability of these NPs are attributable to the presence of abundant hydrophilic PEG coatings on Fe3O4.[45] Due to the presence of hydrophilic carboxyl groups on the CDs, the CDs can complexe with Fe3O4 NPs via hydrogen bonding, which facilitates the formation of stable Fe3O4@CDs@mSiO2 NCs. Figure 1c shows the TEM image of Fe3O4@CDs@mSiO2 NCs, revealing the core-shell structure (inset) of Fe3O4 and silica, as well as CDs (~ 3 nm in diameter, marked with red circles) trapped in the mesoporous silica shell.
Figure 1.
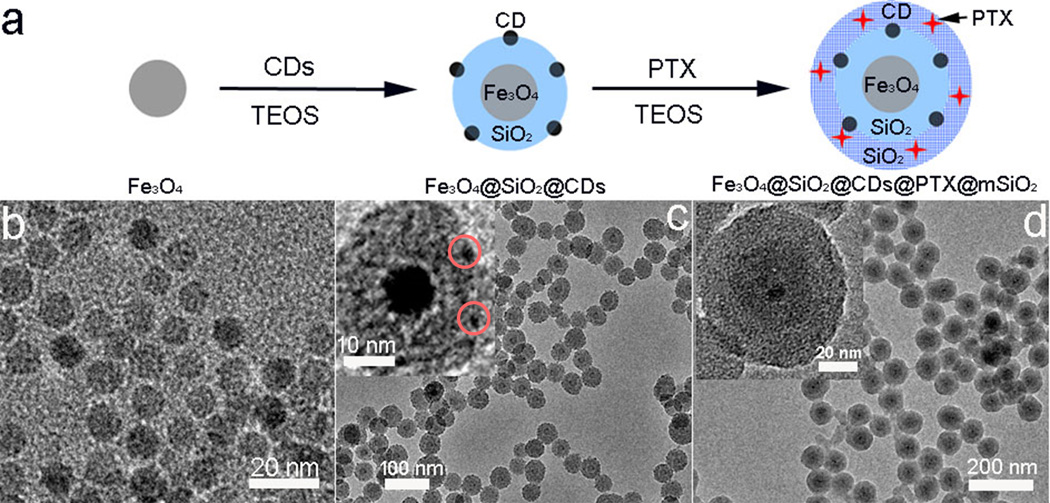
Synthesis and structure of Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs. (a) Schematic illustration of formation of Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs. Step I: surface coating and encapsulation of Fe3O4 and CDs; step II: preloading of PTX and surface coating of Fe3O4@CDs@mSiO2 with mesoporous silica. (b, c and d) TEM images of Fe3O4 core, Fe3O4@CDs@mSiO2 NCs and Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs. The insets in c and d are TEM images of a single Fe3O4@CDs@mSiO2 NC and Fe3O4@CDs@mSiO2@PTX@mSiO2 NC, respectively at a higher magnification.
Second, PTX was incorporated into the hybrid NC by mesoporous silica. Specifically, the reaction was allowed to proceed in ethanol so that hydrophobic PTX remained soluble. Due to the mesoporous structure and large surface area of the silica shell of Fe3O4@CDs@mSiO2, PTX can be readily loaded at high capacity. Figure 1d and Figure S1 (Supporting Information) shows the TEM and SEM images of as-prepared Fe3O4@CDs@mSiO2@PTX@mSiO2 hybrid NCs, respectively, which shows that the hybrid NCs are spherical in shape and ~70 nm in diameter. It also shows the dual-shell structure of of the Fe3O4@CDs@mSiO2@PTX@mSiO2 NC with the inner shell (~12 nm) composed of mesoporous silica that contains Fe3O4 and CDs and the outside shell (~20 nm) composed of mesoporous silica that are designed to load PTX.
The UV-vis absorption spectrospcopy was used to confirm the successful loading of CDs and PTX in the hybrid NCs. As shown in Figure S2 (Supporting Information), Fe3O4 cores and Fe3O4@mSiO2 NCs express no significant absorption at wavelength above 200 nm, whereas free CDs exhibit a clear broad peak centered at 240 nm due to the absorption of an aromatic Pi system.[46, 47] The hybrid NCs exhibit a weak absorption peak centered at 240 nm, suggesting that CDs were successfully incorporated into the NCs. In addition, the UV-vis absorption at 229 nm of Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs (Figure 2a) suggests that PTX was successfully loaded in the NCs when comparing the spectrum of free PTX. Dynamic light scattering was performed to obtain the size variation during synthesis. The increase in hydrodynamic size from 28 nm (Fe3O4) to 55 nm (Fe3O4@CDs@mSiO2) to 91 nm (Fe3O4@CDs@mSiO2@PTX@mSiO2) suggests the successful surface coating at both steps of the synthesis (Figure 2b). In addition, the distribution of hydrodynamic diameter (Dh) of Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs, with a zeta potential of −15.7 mV, is narrow, indicating that Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs have good size uniformity. Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs also have excellent stability in a cell culture medium containing 10% serum for up to 7 days (Figure S3, Supporting Information).
Figure 2.

Optical properties and size distribution of Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs. (a) UV-vis absorption spectra of free PTX, Fe3O4@CDs@mSiO2@mSiO2 NCs and Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs. (b) Hydrodynamic size distributions of Fe3O4, Fe3O4@CDs@mSiO2 NCs and Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs in distilled water at 25°C.
Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs containing both fluorescent CDs and magnetic Fe3O4 can serve as a contrast agent for multi-modal imaging. We first evaluated the optical imaging function of Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs in human SF-763 glioblastoma cells. As shown in the laser scanning confocal images of SF-763 cells incubated with Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs (Figure 3a–c), the hybrid NCs produced bright fluorescence and illuminated SF-763 cells in multicolor forms at wavelengths of 405, 488, and 546 nm. A comparison of the photoluminescence (PL) spectra (Figure S4, Supporting Information) and confocal images (Figure S5, Supporting Information) of free PTX and CDs with those of Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs further confirms that such optical property was generated by CDs embedded in the hybrid NCs. In addition, the confocal fluorescence signal did not show apparent attenuation after continuous irradiation at 488 nm for 60 min (Figure S6, Supporting Information), suggesting that Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs have excellent photostability and can be used for prolonged cellular imaging.
Figure 3.

Optical property of Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs. Laser scanning confocal microscopy images of SF-763 cells incubated with Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs for 2 h under different excitation wavelengths: (a) 405 nm, (b) 488 nm, (c) 546 nm. (d) DAPI nuclear stain, (e) two-photon fluorescence, and (f) overlaid images of SF-763 cells incubated with Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs. Excitation laser wavelength is 900 nm.
Two-photon fluorescence microscopy has attracted much attention as it provides direct cellular structure observation, deep penetration in biological tissues, low photobleaching and low autofluorescence.[48–50] Figure S7 (Supporting Information) shows the PL spectra of the free CDs obtained with a wide NIR excitation wavelength range from 980 nm to 700 nm, which demonstrates the upconverted emission ranging from 544 nm to 454 nm. Therefore, it is expected that Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs also have two-photon fluorescence cellular imaging capability. Two-photon fluorescence images of SF-763 cells incubated with Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs for 2 h in Figure 3d–f shows that the cells can be illuminated by the upconverted fluorescence emitted from Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs upon excitation by an NIR laser. In addition Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs were mainly located in the cytoplasm but not in the nuclei of the cells. Cellular uptake of this fluorescent marker was further evaluated using flow cytometry. As shown in Figure S8a (Supporting Information), an increase in PL intensity (from 57 to 174) in SF-763 cells demonstrates the nonspecific uptake of these NCs. Additioanlly, the release of PTX from NCs can be detected in SF-763 cells (Figure S8b, Supporting Information), further demonstrating that Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs can be potentially used as a transmembrane carrier for PTX.
The magnetic hysteresis behavior of Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs was examined at 300 K with an applied magnetic field of up to 40,000 Oe. As shown in Figure 4a, no apparent hysteresis loop was observed, suggesting the superparamagnetic nature of the hybrid NCs at room temperature. The saturation magnetization (Ms) of the hybrid NCs is 0.56 emu g−1, which is considerably lower than the Fe3O4 (~18 emu g−1) because the hybrid NCs contain only about 3.19 % of magnetic Fe3O4.[51] The temperature-dependent magnetization of Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs was measured after zero field cooling (ZFC) and field cooling (FC) under an applied magnetic field of 50 Oe between 4 and 300 K (Figure S9, Supporting Information). The ZFC curve indicates a blocking temperature (TB) of the hybrid NCs at 92 K. Above TB, the hybrid NCs are superparamagnetic, whereas below TB, they are ferromagnetic, which further confirms the superparamagnetism of Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs at room temperature.
Figure 4.

Magnetic properties and MRI of Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs. (a) The hysteresis behavior of Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs measured at room temperature. (b) R2 (1/T2) as a function of the concentration of Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs. (c) T2-weighted MR images and R2 maps of MRI phantoms of Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs at different NC concentrations.
The relaxation R2 of Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs was evaluated at 14 T by acquiring their phantom MR images at different iron concentrations. A linear correlation between the R2 (1/T2) and iron concentration was established (Figure 4b). The r2 relaxivity (the slop of R2 vs. iron concentration) of Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs was determined as 86.702 s−1 mM−1, which is lower than that of the Fe3O4 magnetic core (391.86 s−1 mM−1, Figure S10, Supporting Information) because of their relatively weak accessibility of water molecules.[52] The T2-weighted MR images and R2 maps (Figure 4c) shows that the T2 MRI signal intensity declines with the increase in hybrid NC concentration (corresponding to an increase in iron concentration). The incremental decrease in signal intensity is indicated by the enhanced darkness at increased Fe concentration from 0 to 1.0 mM. These results indicate that Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs can potentially serve as T2-weighted contrast agent for MR imaging..
Figure 5a–b shows the nitrogen adsorption/desorption isotherms and pore size distribution of Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs. The Brunauer–Emmett–Teller surface area and total pore volume of the hybrid NCs are 162.23 m2 g−1 and 0.27 cm3 g−1, respectively. The average Barrett–Joyner–Halenda pore diameter of the hybrid NCs calculated from the desorption branch of the isotherm is ~2.9 nm. These pores in the hybrid NCs could provide channels for PTX release. The drug loading capacity of the NCs was evaluated by quantifying the unloaded residual PTX separated from the reaction condition (Figure 5c). The HPLC-MS measurement displays the PTX drug loading of 27.8% and 30.9% for Fe3O4@mSiO2@mSiO2 and Fe3O4@CDs@mSiO2@mSiO2 NCs, respectively. This high drug loading can be attributed primarily to the porous structure and large surface area of the NCs. In addition, PTX could interact with CDs through the supramolecular π-stacking between the conjugated rings of PTX molecules and aromatic rings of CDs, which could increase the drug loading capacity.[53] The characteristic absorption peak of PTX displays a red-shift from 229 nm to 235 nm (Figure S11, Supporting Information) after PTX was loaded onto CDs, which confirms the supramolecular π-stacking between the conjugated rings of PTX and aromatic rings of CDs.
Figure 5.

Porous structure and drug release behavior of the hybrid NCs. (a) N2 adsorption-desorption isotherm and (b) pore diameter distribution of Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs. (c) HPLC analysis of initial PTX solution and residual PTX solution from the preparation of Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs. (d) Release profiles of PTX from Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs in PBS at 37°C, with or without of irradiation of NIR light. NIR was irradiated at 1.5 W cm−2 for 5 min each at predefined time points (6, 22, and 32 h). The inset in (d) is a schematic illustration of the release of PTX from Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs.
The PTX release under NIR irradiation was then investigated. Without NIR radiation, the PTX release from Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs was relatively slow and assumed a nearly steady rate after 12 h (Figure 5d). In contrast, a 5-min exposure to NIR irradiation markedly speeded up the release of PTX and the drug release rate returned to its pre-irradiation level upon the removal of radiation (Figure 5d). This irradiation-enabled drug release is attributable to the local heating produced by photothermal conversion by the CDs in the inner shell of hybrid NCs. Figure S12 (Supporting Information) confirms the highly efficient photothermal effect of the CDs (50 mg L−1), which increases water temperature by ~26°C in 5 min under NIR irradiation at an intensity of 1.5 W cm−2. This local heating by CDs not only weakens the drug-host interaction between PTX and CDs, but also increases the mobility of PTX as illustrated in the inset of Figure 5d.
Given that Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs were synthesized at 45°C, the stability of PTX in the reaction system was studied by incubating PTX at 45°C. As shown in Figure S13a (Supporting Information), the UV-vis spectrum of PTX incubated at 45°C was similar with that at 25°C. Also, they exhibited comparable cytotoxicity to SF-763 cells in vitro (Figure S13b, Supporting Information), indicating that a temperature as high as 45°C does not compromise the therapeutic efficacy of PTX.
The photothermal, and chemophotothermal efficacy of Fe3O4@CDs@mSiO2@mSiO2 NCs and Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs, respectively, were investigated in vitro. It was shown that Fe3O4@CDs@mSiO2@mSiO2 NCs (carrying no PTX) at a concentration of up to 50 µg/mL had negligible cytotoxicity against SF-763 cells after 72 h incubation (Figure 6a). In contrast, Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs significantly reduced the cell viability under the same conditions (Figure 6a), as a result of release of PTX molecules from the hybrid NCs. In addition, Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs can induce significantly higher cytotoxicity than free PTX in SF-763 cells, suggesting that Fe3O4@CDs@mSiO2@mSiO2 NCs can sustain PTX release and reduce the side-effect of free PTX (Figure S14, Supporting Information).
Figure 6.

In vitro chemotherapy and phototherapy using hybrid NCs. (a) In vitro cytotoxicity of Fe3O4@CDs@mSiO2@mSiO2 NCs and Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs without and with NIR irradiation at 1.5 W cm−2 for 5 min, as measured in terms of cell viability. (b) Therapeutic efficiencies of Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs as a drug carrier and photothermal therapy agent for chemo, photothermal, and their combined treatments. The t-test was used to compare the therapeutic efficiencies of combined treatment with the additive efficiencies of chemo and photothermal treatments alone. All p-values are smaller than 0.01.
Without NCs treatment, 5-min NIR irradiation has minimal effect on SF-763 cell viability (Figure S15, Supporting Information). However, Fe3O4@CDs@mSiO2@mSiO2 NCs exhibited some cytotoxic effect towards cells under 5 min NIR irradiation (Figure 6a), suggesting that they can serve as potential photothermal therapeutic agent in treating cancer cells. Notably, the combinatory treatment with NIR irradiation and PTX release resulted in significantly higher reduction in cell viability than the treatment with either Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs (chemotherapy) or 5-min NIR irradiation alone (photothermal therapy using Fe3O4@CDs@mSiO2@mSiO2 NCs) (Figure 6a).
Figure 6b compares the therapeutic efficacies of Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs as a PTX carrier and photothermal therapy agent. The therapeutic efficiencies were calculated by subtracting the cell viability from 100% from the combined chemo-photothermal treatment with the additive therapeutic efficiencies of independent chemo- and photothermal treatments, which were estimated using the relation of Tadditive = 100 − (fchemo × fphotothermal) × 100, where f is the fraction of surviving cells after each individual treatment.[54, 55] As expected, both the combinatory therapeutic efficiency and additive therapeutic efficiency are greater than the efficiency of the either chemo- or photothermal- therapy alone. Notably, the combinatory therapeutic efficiency of chemo- and photothermal- therapy with Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs is markedly greater than the additive therapeutic efficiency of chemo- and photothermal therapy, suggesting a therapeutic contribution from the synergetic effect of the combinatory treatment. t-test is used to compare the therapeutic efficiency of combined chemo-photothermal treatment with the additive therapeutic efficiency of independent chemo- and photothermal treatments, and all p-values are smaller than 0.01, indicating a significant difference. Clearly, Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs as a drug carrier demonstrated a highest therapeutic effect due to of the combined synergetic chemo-photothermal treatment.
Potential toxicity has been a major safety concern for nanoscaled materials in biomedical applications. In order to test the biocompatibility of the NCs in vivo, which is essential for the future use of this nanocarrier, we performed histological analysis on various tissues (heart, kidney, lung, spleen and liver) from mice injected with Fe3O4@CDs@mSiO2@mSiO2 and Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs at 0.05 mg mL−1 to identify signs of acute toxicity. Tissues were harvested from mice 120 h after NC injection, fixed in 10% formalin, embedded in paraffin, sectioned, and stained with hematoxylin and eosin (H&E). Images (Figure 7) were reviewed by a pathologist, and no discernible toxicity was observed in Fe3O4@CDs@mSiO2@mSiO2 treated animals. These results demonstrate that Fe3O4@CDs@mSiO2@mSiO2 NCs are nontoxic and safe. Also, these biocompatible hybrid NCs can be potentially used as a drug carrier to release PTX molecules for impoved chemotherapy.
Figure 7.

H&E stained tissue sections of mouse heart, kidney, liver, lung and spleen obtained from noninjected animals, and those injected with Fe3O4@CDs@mSiO2@mSiO2 (NO PTX NCs) or with Fe3O4@CDs@mSiO2@PTX@mSiO2 (PTX NCs) at a concentration of 0.05 mg mL−1. The scale bar 100 µm.
More importantly, Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs could efficiently kill 4T1-luc tumor cells under NIR irriadiation as demonstrated by the in vitro cell viability assay (Figure S16, Supporting Information). To further examine the efficacy of Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs in vivo, mice bearing 4T1-luc tumors were treated with NCs through intratumoral injection, and tumors were then subjected or not subjected (irradiation control) to NIR irradiation (808 nm, 1.5 W/cm2) for 5 min post-injection. Tumor-bearing mice were randomly divided into 4 groups with four mice per group, and treated with PBS (control), Fe3O4@CDs@mSiO2@mSiO2 NCs with NIR irradiation (photothermal therapy), Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs without NIR irradiation (chemotherapy), or Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs with NIR irradiation (combined therapy). Tumor sizes in mice were measured every two days for 10 days post-treatment. As shown in Figure S17a and b (Supporting Information), tumors in control mice and mice treated with photohtermal therapy continued to grow rapidaly, whereas tumors in mice received chemotheray showed a reduced growth rate. Notably, the combined treatement resulted in a dramatically-decreased tumor growth and yet no weight loss was oberved in these mice as compared to other groups (Figure S17c, Supporting Information). Such significant therapeutic effect of the combined treatment might be attributed to the increased drug release, cell membrane permeability, and internalization under NIR irradiation.
3. Conclusions
We developed a multifunctional hybrid NC containing Fe3O4, CDs, and preloaded hydrophobic PTX using mesoporous silica as scaffolds. The preloading of drug was accomplished in a one-pot process without extra dissolution of template cores under harsh conditions. The in situ preloading of PTX simplifies drug loading and enhances the loading capacity owing to the mesoporous dual-shell structures and supramolecular π-stacking between the conjugated rings of PTX molecules and aromatic rings of the embedded CDs. The hybrid NCs can also serve as a confocal and two-photon fluorescence imaging probe and T2-weighted MRI contrast agent. In addition, NIR-responsive PTX release from Fe3O4@CDs@mSiO2@PTX@mSiO2 can be realized through the photothermal effect generated by the embedded CDs under NIR irradiation. Importantly, this hybrid NC demonstrates good biocompatibility and can enable combined chemo-photothermal treatment.
4. Experimental Section
Materials
Paclitaxel (PTX) was purchased from LC Laboratories (Woburn, MA). All other chemicals were purchased from Sigma-Aldrich (St. Louis, MO). Glucose, hydrochloric acid (HCl, 37%), ethanol (C2H6O, 99%), tetraethyl silicate (TEOS, 98%), ammonia water (NH3·H2O, 27%) and octadecyltrimethoxysilane (C18TMS) were used as received.
Synthesis of PEG2000-NH2 monolayer-coated Fe3O4 NPs and fluorescent CDs
Monodispersed PEG2000-NH2 coated Fe3O4 NPs were synthesized following the our previously reported method.[56, 57] First, 50 mg of oleic acid-coated Fe3O4 was synthesized using the reported method [58] and suspended in 43 ml of anhydrous toluene followed by addition of 50 µL of trimethylamine in a three-neck round-bottom flask fitted with a Graham condenser. The flask was sealed with a rubber septum and purged with nitrogen. The solution was heated to 100°C and 0.10 ml SATES was added to the flask. 187.5 mg of mPEG2K-NH2 dissolved in 7 ml of anhydrous toluene was added to the flask 15 min after the addition of SATES. An additional 50 µL of SATES was injected 1h after the mPEG2K-NH2 injection, and the solution was allowed to react for another 6 h and 45 min. The solution was transferred to a single-neck round-bottom flask and NPs were precipitated with hexane. The NP precipitate was dispersed in THF, sonicated for 10 min, and precipitated with hexane. The resultant NP pellet was suspended in 10 ml anhydrous THF and sonicated for 10 min. 62.5 mg of mPEG2K-NH2 and 187.5 mg of bis(amine) functionalized PEG (PEG2Kbis(amine), MW 2000) were dissolved in 12 ml anhydrous THF and added to the NP solution. The flask was then sealed with a septum and purged with nitrogen. 12.5 mg of N,N1-dicyclohexylcarbodiimide (DCC) was dissolved in 2 ml anhydrous THF and added to the flask, and the reaction solution was placed in a sonication bath at 25°C and allowed to react for 16 h. Fully PEGylated NPs were precipitated with hexane, redispersed in 20 ml ethanol, sonicated for 10 min, and precipitated again with hexane. The pellet was fully dried and dispersed in distilled water with sonication for 10 min. The particles were purified through Sephacryl S-200 size exclusion gel chromatography.
Fluorescent CDs were obtained by acid-assisted ultrasonic and thermal treatment of glucose.[59, 60] 2.70 g of glucose was dissolved in 10 mL of deionized water. After intense sonication for 20 min, HCl (30.0 mL, 37 wt %) was slowly added into the above solution. The mixture solution was then treated ultrasonically for 8 h and transferred into a 50 mL Teflon-lined stainless autoclave. The precursor solution was heated to, and maintained at 200°C for 24 h. The CD solution was cooled naturally to room temperature and purified with repeated centrifugation and redispersion in water for three cycles. Finally, the CD solution was dialyzed for 7 days (Spectra/Por molecular porous membrane tubing, cutoff 3,000) at room temperature. The aqueous dispersion of CDs was then collected and dried to get solid CDs.
Synthesis of Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs
To synthesize the NCs, Fe3O4 (1.6 mg) and CDs (1 mg) were ultrasonic-dispersed in ethanol (20 mL) for 5 min. 1.6 mL NH3·H2O was then added into the mixture solution. After stirring for 10 min, 15 µL TEOS and 5 µL C18TMS were added into the above solution. The reaction was maintained at 45°C for 30 min. 30 µL TEOS, 20 µL C18TMS and 30 mg PTX dissolved in 1 mL ethanol were then injected into the flask at the speed of 1 mL/min under magnetic stirring at 45°C. The reaction solution was then stirred for another 1.5 h. NCs were collected by centrifugation of the mixture solution at 8,000 rmp for 10 min to remove unloaded CDs, PTX molecules and Fe3O4 NPs. The separated NCs were then re-dispersed in 10 mL ethanol with strong ultrasonication and washed three times to remove hydrophobic PTX molecules adsorbed on the NCs. The C18TMS was removed at the room temperature by repeated redispersion, centrifugation and washing of NCs using deionized water (10 mL) and ethanol (10 mL) for three times each. The NCs were purified by redispersing them in 10 mL distilled water with strong ultrasonication and washed three times to remove CDs adsorbed on the NCs. The amounts of as-synthesized Fe3O4@CDs@mSiO2@mSiO2 (without PTX) and Fe3O4@CDs@mSiO2@PTX@mSiO2 (with PTX) dried in vacuum were determined to be 67.08 and 96.91 mg, respectively. The unloaded PTX in the supernatant was quantified by HPLC-MS. The drug loading of Fe3O4@CDs@mSiO2@mSiO2 was calculated by (M0 − Mt)/MN ×100%, where M0 and Mt are the total mass of PTX dissolved in the initial solution and remained in the supernatant solution, respectively. MN is the mass of the Fe3O4@CDs@mSiO2@PTX@mSiO2 used in the loading process.
PTX released from Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs
The in vitro NIR-responsive release of PTX from Fe3O4@CDs@mSiO2@PTX@mSiO2 was assessed by a dialysis process. Purified Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs were re-dispersed in PBS solution (96.91 mL, 0.005 M, pH = 7.4). Two dialysis bags filled with 5 mL diluted Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs (1mg/mL) were immersed in 50 mL PBS and maintained at 37°C. One of them was exposed to the irradiation of NIR light (808 nm) with an output power of 1.5 W/cm2 for 5 min. The PTX released outside of the dialysis bag was sampled at defined time points and assayed by HPLC-MS. Cumulative release is expressed as the total percentage of drug released through the dialysis membrane over time.
Magnetic resonance imaging of Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs
R2 relaxtion (1/T2) values of Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs in PBS at different concentrations were evaluated at 14 T using a multi-spin echo acquisition technique. Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs in solution were pipetted into glass vials (3.25 mm I.D., 5 mm O.D., 200 µL volume). The vials were fastened together and placed into a water reservoir which served as a homogeneous background signal to minimize magnetic susceptibility variations near the samples. The secured vials were placed in a 25-mm single-channel 1H radiofrequency coil (PB Micro 2.5). Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs were imaged with a quantitative T2 multi-spin multi echo scan sequence (MSME) (TR = 2,500 ms, TE = 6.7 + 6n ms, [n = 0–16], in-plane resolution 78 × 156 µm2, matrix 256 × 128) with 0.5 mm slice thickness for 14 slices. Analysis of MRI data was accomplished with the FMRIB software library (FSL), Paravision 5.1 analysis package (Bruker), Osirix (Pixmeo) and ImageJ (NIH). T2 values were determined within a circular, 100-voxel region of interest.
Cell culture
Human SF-763 GBM cells were kindly provided by Prof. John R. Silber (Department of Neurological Surgery, University of Washington) and grown in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum and 1% antibiotic-antimycotic (Life technologies, Grand Island, NY). Mouse 4T1 breast cancer celles were purchased from American Type Culture Collection. Luciferanse-expressing 4T1 cells (4T1-luc) were generated in lab and grown in DMEM supplemented with 10% fetal bovine serum and 1% antibiotic-antimycotic. Cells were cultured in an incubator maintained at 37°C and 5% CO2 with 95% humidity.
Confocal imaging of Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs
SF-763 cells were seeded onto glass cover slips in a 6-well plate. After overnight incubation, cells were incubated with Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs (50 µg/mL), free PTX (15 µg/mL) or CDs (1 µg/mL) for 2 h. Cells were then washed with cold PBS 3 times, fixed with 4% paraformaldehyde for 15 min at 37°C, and mounted onto glass slides with ProLong® Gold Antifade Mountant (Life Technologies Inc., Gaithersburg, MD). The images of cells were acquired using a Laser Scanning Microscope Leica SP8X (Leica Microsystems GmbH, Germany). Three excitation wavelengths were used (405, 488 and 546 nm).
Two-photon fluorescent imaging of Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs
SF-763 cells were seeded onto glass cover slips in a 24-well plate. Twenty four hours after seeding, cells were incubated with Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs (50 µg/mL) for 2 h. Cells were then washed with PBS 3× and fixed with 4% paraformaldehyde for 10 min. Nuclei were stained with DAPI before cells were mounted onto glass slides with ProLong® Gold Antifade Mountant (Life Technologies Inc., Gaithersburg, MD). Two-photon imaging was performed using an Olympus FV1000 MPE BX61 multi-photon microscope with excitation wavelength at 900 nm.
Viability of Cells treated with Fe3O4@CDs@mSiO2@mSiO2 NCs or Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs with or without NIR irradiation
SF-763 and 4T1-luc cells were seeded in a 96-well plate and incubated overnight in the aforementioned growth conditions. On the following day, the medium was replaced with a medium containing Fe3O4@CDs@mSiO2@mSiO2 NCs or Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs or with medium control. Three different drug concentrations (50, 25 and 12.5 µg/mL) were used, and samples at each concentration were ran in sextuplicate. The cells were incubated with Fe3O4@CDs@mSiO2@mSiO2 NCs or Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs for 72 h. Wells containing the normal medium without samples were used as the control. For photothermal treatments, cells in wells were irradiated with 1.5 W cm−2 NIR light for 5 min. Cell viability was assessed using the alamar blue assay. Briefly, the medium was replaced with cell culture medium containing reagent and incubated for 2 h. Following the incubation, a microplate reader (SpectraMax i3, Molecular Devices, Sunnyvale, CA) was used to determine the fluorescence intensity of the dye (550ex/590em). The fluorescence intensity from Fe3O4@CDs@mSiO2@mSiO2 NCs and Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs was compared to those from untreated control cells to determine percent viability.
Cellular uptake of Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs by flow cytometry analysis
SF-763 cells were seeded onto a 12-well plate the day before Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs (50 µg/mL) addition. Cells were incubated with Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs for 1 h, followed by washing with cold PBS for 3 times. Cells were then digested by Trypsin-EDTA and washed with cold PBS. Cells were then fixed in 2 % paraformaldehyde at 4 °C overnight. After that, cells were centrifuged (500 g, 3 min) to remove fixative, washed once with PBS and resuspended in PBS for flow cytometry analysis. Data acquisition was performed with a FACSCanto II flow cytometer (BD Biosciences). The signals were collected and their excitation wavelengths were set to be 405 nm, and emission wavelengths were 450 nm, respectively.
HPLC detection of PTX uptake into cells
SF-763 cells were seeded into a 6-well plate (200,000 per well) and were allowed to grow overnight. On the following day, free PTX or Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs was added into cells at a dose of 50 µg/mL and incubated for 4 h. Cells were then washed 3 times with cold PBS and trypsinized. Cells were collected by centrifugation and freeze-dried. To extract PTX, 100 µL methanol was added into cell pellet and ultrasonicated for 10 min. Samples were centrifuged for 2 min at 10,000 g and supernatants were collected. The amount of PTX was determined using an HPLC system (Hitachi, Ltd., Tokyo, Japan, L-7100 intelligent pump, equipped with a L-7400 ultraviolet detector). XBridge BEH C18 column was used (130 Å, 3.5 µm, 4.6 mm × 100 mm) (Waters Corporation, Milford, MA). The solvent A was acetonitrile containing 0.1% acetic acid and the B solvent was DI water containing 0.1% acetic acid. The gradient was 60–100% solvent A in 6.5 min. The column temperature was 25°C and the flow rate was 0.6 mL/min. The injection volume was 20 µL and 254 nm was used as a detection wavelength.
Histopathological Evaluation
14 days after intravenous administration of Fe3O4@CDs@mSiO2@mSiO2 NCs or Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs at a concentration of 50 µg/mL and a volume of 200 µL, whole organs (heart, kidney, liver, lung and spleen) of C57BL/6 mice were removed through necropsy and preserved in 10% formalin for 48 h. Tissues were then embedded in paraffin, sliced into 5 µm sections, and stained with hematoxylin and eosin. Microscopic images of tissues were acquired using a Nikon ECLIPSE TE 2000-S microscope.
In vivo therapy of Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs
All animal studies were conducted in accordance with University of Washington Institute of Animal Care and Use Committee (IACUC) approved protocols as well as with federal guidelines. Female BALB/c mice of 5 weeks old were used in this study. 4T1-luc cells were trypsinized, and suspended in PBS (2 × 106 cells/mL), and were injected subcutaneously into the right flanks of mice (0.1 ml per mouse). Four days after tumor inoculation, mice were administered intratumorally 3 times for every two days with PBS, Fe3O4@CDs@mSiO2@mSiO2 or Fe3O4@CDs@mSiO2@PTX@mSiO2 NCs (200 µl, 0.5 mg/mL per mouse per injection). After every injection, a subset of mice were subjected to NIR irradiation (808 nm, 1.5 W/cm2) for 5 min. The length and width of the tumors were measured by a digital caliper. The tumor volume was calculated based on the following formula: width2 × length/2.
Characterization
TEM images were taken on a FEI TECNAI transmission electron microscope at an accelerating voltage of 100 kV. Field emission scanning electron microscopy (FE-SEM) images were acquired on an AMRAY 1910 scanning electron microscope. UV-vis absorption spectra were obtained on a Thermo Electron Co. Helios β UV-vis Spectrometer. PL spectra were obtained on a JOBIN YVON Co. FluoroMax®-3 Spectrofluorometer equipped with a Hamamatsu R928P photomultiplier tube. Magnetic properties were measured using a superconducting quantum interference device magnetometer (Quantum Design MPMS XL-7). The photothermal experiments were conducted using a near infrared lamp (808 nm) with a power density of 1.5 W/cm2 and a filter to block the ultraviolet-visible light. The hydrodynamic size of NCs was determined using Zetasizer Nano-ZS (Malvern Instruments, Worcestershire, UK) at room temperature. Nitrogen adsorption-desorption measurements were carried out on a Micromeritics ASAP 2020 instrument. HPLC-MS spectra were collected on a Bruker Esquire ion trap mass spectrometer (Bruker Daltonics, Billerica, MA) using the positive ion mode.
Supplementary Material
Acknowledgments
This work is supported by NIH grant R01CA161953. Two photon fluorescence imaging study is supported in part by a gift to the Institute for Stem Cell and Regenerative Medicine at the University of Washington. K. W. acknowledges support from the College of Engineering Dean's Fellowship (the Scott Fellowship and the Marsh Fellowship) at University of Washington.
Footnotes
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Dr. Hui Wang, Department of Materials Science and Engineering, University of Washington, Seattle, Washington 98195 (United States)
Dr. Kui Wang, Department of Materials Science and Engineering, University of Washington, Seattle, Washington 98195 (United States)
Mr. Bowei Tian, Department of Applied Mathematics, University of Washington, Seattle, Washington 98195 (United States)
Mr. Richard Revia, Department of Materials Science and Engineering, University of Washington, Seattle, Washington 98195 (United States)
Dr. Qingxin Mu, Department of Materials Science and Engineering, University of Washington, Seattle, Washington 98195 (United States)
Mr. Mike Jeon, Department of Materials Science and Engineering, University of Washington, Seattle, Washington 98195 (United States)
Ms. Fei-Chien Chang, Department of Materials Science and Engineering, University of Washington, Seattle, Washington 98195 (United States)
Prof. Miqin Zhang, Department of Materials Science and Engineering, University of Washington, Seattle, Washington 98195 (United States)
References
- 1.Chabner BA, Roberts TG. Nat. Rev. Cancer. 2005;5(1):65–72. doi: 10.1038/nrc1529. [DOI] [PubMed] [Google Scholar]
- 2.Lipinski CA. J. Pharmacol. Toxicol. Methods. 2000;44(1):235–249. doi: 10.1016/s1056-8719(00)00107-6. DOI http://dx.doi.org/10.1016/S1056-8719(00)00107-6. [DOI] [PubMed] [Google Scholar]
- 3.Merisko-Liversidge EM, Liversidge GG. Toxicol. Pathol. 2008;36(1):43–48. doi: 10.1177/0192623307310946. [DOI] [PubMed] [Google Scholar]
- 4.Lu J, Liong M, Zink JI, Tamanoi F. Small. 2007;3(8):1341–1346. doi: 10.1002/smll.200700005. [DOI] [PubMed] [Google Scholar]
- 5.Wang Y, Yan Y, Cui J, Hosta-Rigau L, Heath JK, Nice EC, Caruso F. Adv. Mater. 2010;22(38):4293–4297. doi: 10.1002/adma.201001497. [DOI] [PubMed] [Google Scholar]
- 6.Piao Y, Burns A, Kim J, Wiesner U, Hyeon T. Adv. Funct. Mater. 2008;18(23):3745–3758. [Google Scholar]
- 7.Slowing II, Vivero-Escoto JL, Wu C-W, Lin VSY. Adv. Drug Del. Rev. 2008;60(11):1278–1288. doi: 10.1016/j.addr.2008.03.012. DOI http://dx.doi.org/10.1016/j.addr.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 8.Chen Y, Chen H, Zeng D, Tian Y, Chen F, Feng J, Shi J. ACS Nano. 2010;4(10):6001–6013. doi: 10.1021/nn1015117. [DOI] [PubMed] [Google Scholar]
- 9.Stein A, Melde BJ, Schroden RC. Adv. Mater. 2000;12(19):1403–1419. DOI 10.1002/1521-4095(200010)12:19<1403::AID-ADMA1403>3.0.CO;2-X. [Google Scholar]
- 10.Meng H, Wang M, Liu H, Liu X, Situ A, Wu B, Ji Z, Chang CH, Nel AE. ACS Nano. 2015;9(4):3540–3557. doi: 10.1021/acsnano.5b00510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cao M, Wang P, Kou Y, Wang J, Liu J, Li Y, Li J, Wang L, Chen C. ACS Appl Mater Interfaces. 2015;7(45):25014–25023. doi: 10.1021/acsami.5b06938. [DOI] [PubMed] [Google Scholar]
- 12.Hao X, Hu X, Zhang C, Chen S, Li Z, Yang X, Liu H, Jia G, Liu D, Ge K, Liang X-J, Zhang J. ACS Nano. 2015;9(10):9614–9625. doi: 10.1021/nn507485j. [DOI] [PubMed] [Google Scholar]
- 13.Tian B, Liu S, Lu W, Jin L, Li Q, Shi Y, Li C, Wang Z, Du Y. Sci. Rep. 2016;6:21335. doi: 10.1038/srep21335. http://www.nature.com/articles/srep21335#supplementary-information. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alonso-Cristobal P, Oton-Fernandez O, Mendez-Gonzalez D, Díaz JF, Lopez-Cabarcos E, Barasoain I, Rubio-Retama J. ACS Applied Materials & Interfaces. 2015;7(27):14992–14999. doi: 10.1021/acsami.5b03881. [DOI] [PubMed] [Google Scholar]
- 15.Li M, Yan H, Teh C, Korzh V, Zhao Y. Chem. Commun. 2014;50(68):9745–9748. doi: 10.1039/c4cc02966f. [DOI] [PubMed] [Google Scholar]
- 16.Lai J, Shah BP, Zhang Y, Yang L, Lee K-B. ACS Nano. 2015;9(5):5234–5245. doi: 10.1021/acsnano.5b00641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Y, Hou Z, Ge Y, Deng K, Liu B, Li X, Li Q, Cheng Z, Ma Pa, Li C, Lin J. ACS Applied Materials & Interfaces. 2015;7(37):20696–20706. doi: 10.1021/acsami.5b05522. [DOI] [PubMed] [Google Scholar]
- 18.Liu J, Wang C, Wang X, Wang X, Cheng L, Li Y, Liu Z. Adv. Funct. Mater. 2015;25(3):384–392. [Google Scholar]
- 19.Louie A. Chem. Rev. 2010;110(5):3146–3195. doi: 10.1021/cr9003538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chapman S, Dobrovolskaia M, Farahani K, Goodwin A, Joshi A, Lee H, Meade T, Pomper M, Ptak K, Rao J, Singh R, Sridhar S, Stern S, Wang A, Weaver JB, Woloschak G, Yang L. Nano Today. 2013;8(5):454–460. doi: 10.1016/j.nantod.2013.06.001. DOI http://dx.doi.org/10.1016/j.nantod.2013.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bogart LK, Pourroy G, Murphy CJ, Puntes V, Pellegrino T, Rosenblum D, Peer D, Lévy R. ACS Nano. 2014;8(4):3107–3122. doi: 10.1021/nn500962q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liong M, Lu J, Kovochich M, Xia T, Ruehm SG, Nel AE, Tamanoi F, Zink JI. ACS Nano. 2008;2(5):889–896. doi: 10.1021/nn800072t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee D-E, Koo H, Sun I-C, Ryu JH, Kim K, Kwon IC. Chem. Soc. Rev. 2012;41(7):2656–2672. doi: 10.1039/c2cs15261d. [DOI] [PubMed] [Google Scholar]
- 24.Cai W, Chen X. J. Nucl. Med. 2008;49(Suppl 2):113S–128S. doi: 10.2967/jnumed.107.045922. [DOI] [PubMed] [Google Scholar]
- 25.Ellmann S, Beck M, Kuwert T, Uder M, Bäuerle T. Journal of Orthopaedic Translation. 2015;3(4):166–177. doi: 10.1016/j.jot.2015.07.004. DOI http://dx.doi.org/10.1016/j.jot.2015.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang P, Cheetham AG, Lin Y-a, Cui H. ACS Nano. 2013;7(7):5965–5977. doi: 10.1021/nn401667z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cuenca AG, Jiang H, Hochwald SN, Delano M, Cance WG, Grobmyer SR. Cancer. 2006;107(3):459–466. doi: 10.1002/cncr.22035. [DOI] [PubMed] [Google Scholar]
- 28.Farokhzad OC, Karp JM, Langer R. Expert Opin Drug Deliv. 2006;3(3):311–324. doi: 10.1517/17425247.3.3.311. [DOI] [PubMed] [Google Scholar]
- 29.Zhang P, Cheetham AG, Lin YA, Cui H. ACS Nano. 2013;7(7):5965–5977. doi: 10.1021/nn401667z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Palanikumar L, Kim HY, Oh JY, Thomas AP, Choi ES, Jeena MT, Joo SH, Ryu J-H. Biomacromolecules. 2015;16(9):2701–2714. doi: 10.1021/acs.biomac.5b00589. [DOI] [PubMed] [Google Scholar]
- 31.Chen Y, Chen H, Ma M, Chen F, Guo L, Zhang L, Shi J. J. Mater. Chem. 2011;21(14):5290–5298. [Google Scholar]
- 32.Xu B, Ju Y, Cui Y, Song G, Iwase Y, Hosoi A, Morita Y. Langmuir. 2014;30(26):7789–7797. doi: 10.1021/la500595b. [DOI] [PubMed] [Google Scholar]
- 33.Hudson SP, Padera RF, Langer R, Kohane DS. Biomaterials. 2008;29(30):4045–4055. doi: 10.1016/j.biomaterials.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fadeel B, Garcia-Bennett AE. Adv. Drug Del. Rev. 2010;62(3):362–374. doi: 10.1016/j.addr.2009.11.008. DOI http://dx.doi.org/10.1016/j.addr.2009.11.008. [DOI] [PubMed] [Google Scholar]
- 35.Wang H, Zhou S. Biomaterials Science. 2016;4(7):1062–1073. doi: 10.1039/c6bm00262e. [DOI] [PubMed] [Google Scholar]
- 36.Zhang Q, Wang W, Goebl J, Yin Y. Nano Today. 2009;4(6):494–507. DOI http://dx.doi.org/10.1016/j.nantod.2009.10.008. [Google Scholar]
- 37.Zhao Y, Lin LN, Lu Y, Chen SF, Dong L, Yu SH. Adv. Mater. 2010;22(46):5255–5259. doi: 10.1002/adma.201002395. [DOI] [PubMed] [Google Scholar]
- 38.Zhao Y, Luo Z, Li M, Qu Q, Ma X, Yu SH, Zhao Y. Angew. Chem. Int. Ed. Engl. 2015;54(3):919–922. doi: 10.1002/anie.201408510. [DOI] [PubMed] [Google Scholar]
- 39.Sur S, Fries AC, Kinzler KW, Zhou S, Vogelstein B. Proc. Natl. Acad. Sci. U. S. A. 2014;111(6):2283–2288. doi: 10.1073/pnas.1324135111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ping Y, Guo J, Ejima H, Chen X, Richardson JJ, Sun H, Caruso F. Small. 2015;11(17):2032–2036. doi: 10.1002/smll.201403343. [DOI] [PubMed] [Google Scholar]
- 41.Byrne JD, Betancourt T, Brannon-Peppas L. Adv. Drug Del. Rev. 2008;60(15):1615–1626. doi: 10.1016/j.addr.2008.08.005. DOI http://dx.doi.org/10.1016/j.addr.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 42.Jewell CM, Lynn DM. Adv. Drug Del. Rev. 2008;60(9):979–999. doi: 10.1016/j.addr.2008.02.010. DOI http://dx.doi.org/10.1016/j.addr.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.He Q, Cui X, Cui F, Guo L, Shi J. Microporous Mesoporous Mater. 2009;117(3):609–616. [Google Scholar]
- 44.Büchel G, Unger KK, Matsumoto A, Tsutsumi K. Adv. Mater. 1998;10(13):1036–1038. DOI 10.1002/(SICI)1521-4095(199809)10:13<1036::AID-ADMA1036>3.0.CO;2-Z. [Google Scholar]
- 45.Mu Q, Jeon M, Hsiao MH, Patton VK, Wang K, Press OW, Zhang M. Adv Healthc Mater. 2015;4(8):1236–1245. doi: 10.1002/adhm.201500034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chang Y-R, Lee H-Y, Chen K, Chang C-C, Tsai D-S, Fu C-C, Lim T-S, Tzeng Y-K, Fang C-Y, Han C-C, Chang H-C, Fann W. Nat Nano. 2008;3(5):284–288. doi: 10.1038/nnano.2008.99. DOI http://www.nature.com/nnano/journal/v3/n5/suppinfo/nnano.2008.99_S1.html. [DOI] [PubMed] [Google Scholar]
- 47.Wang H, Wei Z, Matsui H, Zhou S. Journal of Materials Chemistry A. 2014;2(38):15740–15745. [Google Scholar]
- 48.Helmchen F, Denk W. Nat Meth. 2005;2(12):932–940. doi: 10.1038/nmeth818. [DOI] [PubMed] [Google Scholar]
- 49.Denk W, Strickler J, Webb W. Science. 1990;248(4951):73–76. doi: 10.1126/science.2321027. [DOI] [PubMed] [Google Scholar]
- 50.Wang H, Sun Y, Yi J, Fu J, Di J, del Carmen Alonso A, Zhou S. Biomaterials. 2015;53:117–126. doi: 10.1016/j.biomaterials.2015.02.087. [DOI] [PubMed] [Google Scholar]
- 51.Park J, An K, Hwang Y, Park J-G, Noh H-J, Kim J-Y, Park J-H, Hwang N-M, Hyeon T. Nat Mater. 2004;3(12):891–895. doi: 10.1038/nmat1251. DOI http://www.nature.com/nmat/journal/v3/n12/suppinfo/nmat1251_S1.html. [DOI] [PubMed] [Google Scholar]
- 52.Chen F, Bu W, Zhang S, Liu J, Fan W, Zhou L, Peng W, Shi J. Adv. Funct. Mater. 2013;23(3):298–307. [Google Scholar]
- 53.Wang H, Ke F, Mararenko A, Wei Z, Banerjee P, Zhou S. Nanoscale. 2014;6(13):7443–7452. doi: 10.1039/c4nr01030b. [DOI] [PubMed] [Google Scholar]
- 54.Hahn GM, Braun J, Har-Kedar I. Proc. Natl. Acad. Sci. U. S. A. 1975;72(3):937–940. doi: 10.1073/pnas.72.3.937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu W, Shen J, Banerjee P, Zhou S. Adv. Funct. Mater. 2011;21(15):2830–2839. [Google Scholar]
- 56.Fang C, Bhattarai N, Sun C, Zhang M. Small. 2009;5(14):1637–1641. doi: 10.1002/smll.200801647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mu Q, Lin G, Patton VK, Wang K, Press OW, Zhang M. Journal of Materials Chemistry B. 2016;4(1):32–36. doi: 10.1039/C5TB02123E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Park J, An K, Hwang Y, Park JG, Noh HJ, Kim JY, Park JH, Hwang NM, Hyeon T. Nat Mater. 2004;3(12):891–895. doi: 10.1038/nmat1251. [DOI] [PubMed] [Google Scholar]
- 59.Wang H, Zhuang J, Velado D, Wei Z, Matsui H, Zhou S. ACS Appl Mater Interfaces. 2015;7(50):27703–27712. doi: 10.1021/acsami.5b08443. [DOI] [PubMed] [Google Scholar]
- 60.Wang H, Yi J, Velado D, Yu Y, Zhou S. ACS Applied Materials & Interfaces. 2015;7(29):15735–15745. doi: 10.1021/acsami.5b04744. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.