

Abstract
Astrocytes form borders (glia limitans) that separate neural from non-neural tissue along perivascular spaces, meninges and tissue lesions in the central nervous system (CNS). Transgenic loss-of-function studies reveal that astrocyte borders and scars serve as functional barriers that restrict the entry of inflammatory cells into CNS parenchyma in health and disease. Astrocytes also have powerful pro-inflammatory potential. Thus, astrocytes are emerging as pivotal regulators of CNS inflammatory responses. This article reviews evidence that astrocytes play critical roles in attracting and restricting CNS inflammation, with important implications for diverse CNS disorders.
Damage and disease in the central nervous system (CNS) trigger coordinated multicellular responses that involve glia, neurons and non-neural cells. Immune and inflammatory cells play critical roles in these responses, particularly as regards neutralizing microbial pathogens and clearing debris. Fine tuned regulation of inflammation is essential to equilibrate between debris accumulation and excessive phagocytosis, either of which can be harmful. Accordingly, diverse cellular and molecular regulatory mechanisms unfold in and around CNS tissue lesions that balance inflammatory cell-mediated debris clearance with maximizing the preservation of healthy tissue and restricting the spread of cytotoxic inflammation1.
Inflammation plays a role in most if not all CNS insults and there is intense interest in understanding the cellular and molecular mechanisms that regulate the onset and resolution of CNS inflammation. There is an equally intense search for causal mechanisms underlying the initiation and progression of CNS autoimmune inflammatory disorders2, 3. Although the principal effectors of CNS inflammation are circulating bone marrow-derived leukocytes, there is increasing recognition that CNS intrinsic cells play essential roles in recruiting, instructing and retaining leukocytes at sites of CNS insults. Among CNS intrinsic cells, myeloid lineage-derived microglia are well established as early sensors of damage and recruiters of multicellular inflammation4. In addition, astrocytes are now emerging as cells that can exert either potent pro-inflammatory functions or critically protective anti-inflammatory functions, as regulated by specific signaling inputs. This article reviews and examines astrocyte contributions to the recruitment, instruction and restriction of immune and inflammatory cells at sites of CNS injury and disease.
Astrocyte responses to CNS damage and disease
In addition to their many essential functions in healthy CNS, astrocytes respond to CNS insults through a process called astrogliosis5–7. Although astrogliosis is often referred to as if it were a single uniform entity, this is emphatically not the case. Evidence accumulated over the past twenty years provides compelling evidence that astrogliosis is a spectrum of heterogeneous potential changes (Box 1), and that these changes are determined in a context specific manner by diverse specific signaling events that vary with the nature and severity of different CNS insults7–11. Functions and effects of astrogliosis are being elucidated. Reactive astrocytes have the potential to release diverse molecules that impact in many ways on nearby cells of all types7–10.
Astrogliosis and scar formation have sometimes been regarded as responses that are predominantly harmful to outcome and should therefore be broadly inhibited. This notion is refuted by a large volume of in vivo experimental studies from many laboratories, which show that normally occurring astrogliosis and scar formation exert numerous essential beneficial functions that improve outcome, including wound closure, neuronal protection, blood-brain barrier repair and restriction of CNS inflammation6–8, 10, 12. Nevertheless, under specific circumstances, astrogliosis has the potential to lead to harmful effects such as exacerbating inflammation or interfering with synapse sprouting or axon growth6, 7, 13. Both the beneficial functions of astrogliosis and its potentially harmful effects are regulated by specific signaling mechanisms that occur in specific contexts and thus represent targets for potential therapeutic manipulation7, 14. In addition, there is accumulating clinical and experimental evidence that dysfunctions of astrocytes and astrogliosis, either through loss of beneficial functions or inappropriate gain of detrimental effects, have the potential to contribute to or be primary causes of CNS disorders, leading to the notion of astrocytopathies7, 15 as discussed below.
Restricted leukocyte trafficking in CNS
Regulation of leukocyte trafficking in CNS has similarities and differences with other tissues. As in other tissues, leukocyte responses to CNS damage, infection and disease involve two arms of the immune system referred to as innate and adaptive. Innate inflammatory responses are coordinated by receptors for conserved molecular patterns associated with pathogens (PAMPs) or with cell damage or danger (DAMPs)16, 17. Adaptive immune responses are coordinated by antigen receptors generated de novo and rearranged to achieve specificity18, 19.
In contrast with most other tissues, CNS tissues exhibit some degree of ‘immune privilege’ that is not absolute, but does restrict and regulate the access of blood-borne antibodies and leukocytes to CNS parenchyma20. This relative immune privilege is implemented by CNS intrinsic cells that (i) create a partial but not absolute blood-brain barrier to circulating antibodies and other immune effector molecules, and (ii) regulate the entry and trafficking of leukocytes in CNS tissue from the circulation, meninges and choroid plexus3, 20–22. The restricted access of leukocytes to CNS parenchyma from the circulation involves several levels of control that regulate the diapedesis and entry of leukocytes past endothelial barriers into perivascular spaces as well as their passage beyond perivascular spaces into CNS parenchyma21, 23. Diverse CNS cells take part in these activities including CNS endothelia, pericytes, other perivascular cells, microglia and astrocytes21, 23.
Astrocyte restriction of CNS inflammation
Astrocytes form structurally prominent borders (glia limitans) that line all interfaces between CNS neural parenchyma and non-neural cells (meningeal cells, endothelia, pericytes, fibroblast lineage cells etc.) along all blood vessels (Fig. 1a) and meningeal surfaces (Fig. 1b). In healthy CNS tissue, these astrocyte borders form functional barriers that present molecular cues that help to restrict leukocyte access into CNS parenchyma from adjacent non-neural tissues that exhibit high levels of leukocyte trafficking for immune surveillance21, 23, 24. At sites of CNS tissue damage, astrocytes form scars that share structural similarities with perivascular and meningeal astrocyte glia limitans borders (Fig. 1c)25. CNS lesions exhibit high levels of immune and inflammatory cell trafficking after traumatic injury, stroke, infection, neurodegenerative disease and autoimmune attack. Astrocyte scars are formed by newly proliferated astrocytes that line all interfaces between viable CNS neural parenchyma and non-neural cells at sites of tissue damage (Fig. 1c)26, 27. There is now compelling evidence that astrocyte scar borders serve as functional barriers that use contact- and diffusion-mediated molecular mechanisms to restrict and regulate the entry of inflammatory cells into CNS parenchyma from non-neural lesion cores, as demonstrated in particular by transgenically targeted loss-of-function experiments (Table 1).
Figure 1. Astrocytes form borders (glia limitans) that serve as functional barriers at interfaces between non-neural tissue and CNS neural parenchyma along blood vessels (a), meninges (b) and tissue lesions (c).
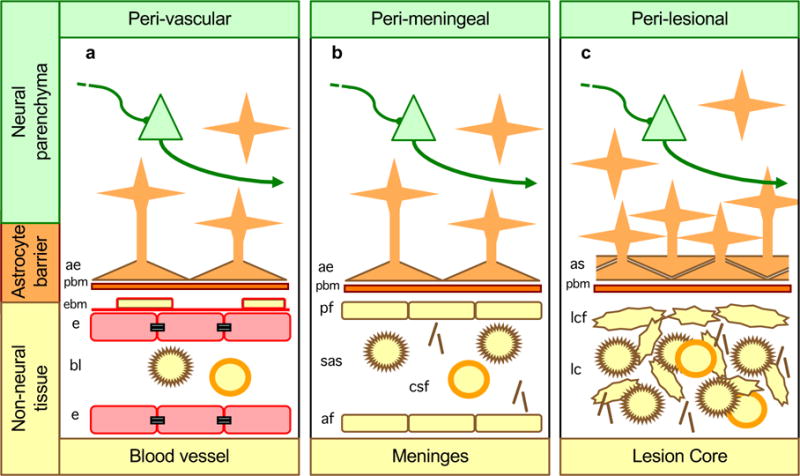
a| Along blood vessels, astrocyte endfeet and parenchymal basement membrane (PBM) present diverse molecular cues that constitute part of the multiple functional barriers, including endothelia with tight junctions and endothelial basement membrane (EBM), across which leukocytes must be actively recruited to pass from the bloodstream into CNS neural parenchyma21, 23, 24. b| Abutting the meninges, astrocyte endfeet and parenchymal basement membranes present multiple molecular cues that restrict leukocytes in the subarachnoid space (SAS) from passing freely into CNS parenchyma. c| Surrounding tissue lesion cores (which are comprised of non-neural cells including leukocytes), astrocyte scars and parenchymal basement membrane are similar in appearance, organization and function to astrocyte borders lining non-neural cells along meninges and blood vessels, and similarly restrict the entry of leukocytes into adjacent CNS parenchyma.
|
neuron |
|
macrophage |
|
|
T cell | ||

|
astrocyte |
|
collagen |
|
|
astrocyte |
|
tight junction |
Table 1.
Anti-inflammatory astrocyte functions and molecular mechanisms demonstrated in vivo by transgenic loss-of function studies in mice
| Disease Model | Deleted or downregulated astrocyte type or astrocyte molecule | Functions revealed | References |
|---|---|---|---|
| Stab brain injury | Newly proliferated scar astrocyte | Repairs blood-brain barrier; Restricts inflammation; Protects neurons. |
28 |
| Spinal cord injury | Newly proliferated scar astrocyte | Repairs blood-brain barrier; Restricts inflammation; Protects neurons; Preserves motor function. |
30 |
| Spinal cord injury | STAT3 | Required for scar formation; Restricts inflammation; Protects neurons; Preserves motor function. |
26, 41, 54 |
| Focal brain injury | Newly proliferated scar astrocyte | Restricts inflammation; Protects neurons. |
127 |
| Spinal cord injury | BMPR1a | Promotes scar formation; Restricts inflammation |
128 |
| Stroke | GFAP & Vim | Required for scar structure; Restricts inflammation. |
31, 34 |
| Stroke | Notch1 | Promotes proliferation and scar formation; Restricts inflammation. |
129, 130 |
| Stroke | TGFβ | Restricts inflammation. | 62 |
| EAE | GFAP | Critical for border structure; Restricts inflammation. |
58 |
| EAE | CRYAB | Restricts inflammation. | 70 |
| EAE | Newly proliferated scar astrocyte | Restricts inflammation. | 32, 33 |
| EAE | GP130 | Critical for barrier function; Restricts inflammation. |
56 |
| EAE | ERα | Restricts inflammation. | 74, 75 |
| EAE | A20 | Blocks NFκB signaling; Restricts inflammation. |
68 |
| EAE | SHH | Maintains blood-brain barrier. | 51 |
| Infection | GP130 | Critical for barrier formation; Inflammatory cell infiltration. |
55 |
| Infection | TGFβ | Inflammatory cell infiltration. Protects neurons |
63 |
| Liposomal storage disease | GFAP & Vim | Critical for border structure; Protects neurons. |
59 |
| Alzheimer’s disease model | GFAP & Vim | Critical for border structure; Reduces Aβ load; Restricts inflammation. |
60 |
| Alzheimer’s disease model | APOE | Maintains blood-brain barrier; Restricts inflammation. |
53 |
| Parkinson’s disease model | DRD2 | Restricts inflammation. | 69 |
| Parkinson’s disease model | CRYAB | Restricts inflammation. | 69 |
Astrocyte restriction of cytotoxic CNS inflammation is a recent discovery28. Astrocyte scar formation around CNS lesions has been recognized for over 125 years, but for much of this time, scar functions have been enigmatic and astrocyte scars have been studied primarily as potential inhibitors of axon regrowth after injury13. Over the past twenty years, transgenic technology revolutionized the ability to study CNS cellular and molecular mechanisms in vivo, including mechanisms of neural injury and repair. Early transgenic loss-of-function studies of astrogliosis unexpectedly revealed an essential function for proliferating reactive astroglia in restricting cytotoxic inflammation in gut and brain28, 29. These observations have been reinforced by numerous related findings (Tables 1,2), and there is now compelling evidence that scar-forming astrocytes exert critical functions that restrict the spread of inflammation into CNS parenchyma from inflamed non-neural tissue at perivascular spaces (Fig. 2Aa,Ab), meninges (Fig. 2Ba,Bb) and tissue lesions (Fig. 2Ca,Cb), after diverse CNS insults including trauma, ischemia, autoimmune attack and neurodegeneration28, 30–34. Nevertheless, in addition to the considerable evidence for astrocyte anti-inflammatory barrier functions, there is equally substantial evidence that astrocytes exert pro-inflammatory functions. Thus, astrocytes are emerging as cells that have complex roles in both recruiting and restricting inflammatory cells.
Table 2.
Selected examples of astrocyte-produced molecules that exert or regulate anti-inflammatory or pro-inflammatory functions.
| Astrocyte molecules | Anti-inflammatory functions | Reference |
|---|---|---|
| Cytokines and growth factors | ||
| TGFβ, IL6, IL10, IL11, IL19, IL27 | Activate diverse anti-inflammatory signaling mechanisms. | 37–39, 131–133 |
| SHH | Maintains blood-brain barrier by acting on endothelia Smo receptors; Induces proliferation of scar-forming astrocytes. |
51, 134 |
| Intracellular signaling factors | ||
| STAT3, A20, Gal9, CRYAB | Suppress pro-inflammatory signaling mechanisms. | 26, 41, 54, 68, 69, 71, 135 |
| Receptors | ||
| ERα, GP130, DRD2, | Suppress pro-inflammatory signaling mechanisms. | 55, 69, 74, 75 |
| Small inter-cellular effector molecules | ||
| RA | Reduces oxidative stress, protects blood-brain barrier function, reduces inflammation. | 66 |
| microRNAs | ||
| miR-181, Dicer1, miR-17-5p | Suppress pro-inflammatory signaling mechanisms; Required for scar-forming astrocyte proliferation via JAK/STAT signaling. |
72, 136 |
| Astrocyte molecules | Pro-inflammatory functions | Reference |
|---|---|---|
| Chemokines | ||
| CCL2, CCL5, CCL7, CCL8, CCL12 | Recruit diverse leukocytes including monocytes, macrophages, T cells, eosinophils, basophils, mast cells, dendritic cells. | 37–39, 43, 44, 132 |
| CXCL1, CXCL9, CXCL10, CXCL12, CXCL16 | Recruit diverse leukocytes including monocytes, macrophages, neutrophil, T cells, NKT cells, B cells, dendritic cells. | 37–39, 45, 132 |
| Cytokines and growth factors | ||
| IL1β, IL6, IL11, IL15, IL17, TNFα, BAFF | Activate diverse pro-inflammatory signaling mechanisms. | 37–39, 131, 132, 137 |
| VEGF | Opens endothelial blood-brain barrier and promotes leukocyte extravasation. | 46, 47 |
| Intracellular signaling factors | ||
| NFκB, SOCS3, | Activate diverse pro-inflammatory signaling mechanisms. | 40–42 |
| Act1 | Activates IL17 mediated pro-inflammatory signaling mechanisms. | |
| Small inter-cellular effector molecules | ||
| PGE, NO | Diverse pro-inflammatory effects. | 135 |
Figure 2. Dysfunction of astrocyte functional barriers lining inflamed non-neural tissue leads to increased spread of neurotoxic inflammation into adjacent neural parenchyma.
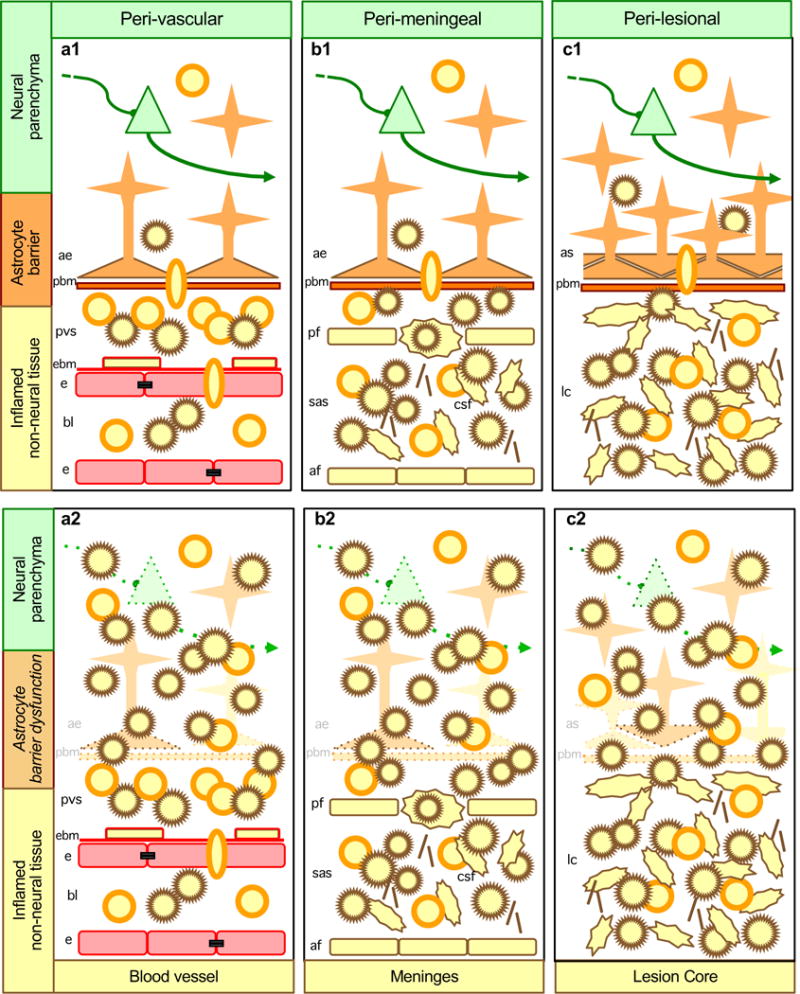
Aa| Along blood vessels, during autoimmune inflammation or certain infections, leukocytes are recruited across barriers of endothelia and endothelial basement membrane (EBM) and collect in perivascular spaces (PVS) but are restricted from passing freely into CNS parenchyma by molecular functional barriers presented by parenchymal basement membrane (PBM) and astrocyte endfeet. Ab| When astrocyte endfeet and parenchymal basement membrane are disrupted or rendered dysfunctional by experimental transgenic manipulations or by certain disease mechanisms, cytotoxic inflammation spreads into CNS parenchyma. Ba| Along meninges, during infections and autoimmune inflammation, leukocytes freely traffic and collect in the subarachnoid space (SAS) but are restricted from passing freely into CNS parenchyma by molecular functional barriers presented by parenchymal basement membrane and astrocyte endfeet. Bb| When astrocyte endfeet and parenchymal basement membrane are disrupted or rendered dysfunctional by experimental transgenic manipulations, cytotoxic inflammation spreads from the SAS into CNS parenchyma. Ca| At tissue lesions, leukocytes freely traffic and collect in non-neural lesion cores, but are largely excluded from adjacent viable CNS parenchyma when astrocytes scars and parenchymal basement membrane are intact and functional. Cb| When astrocyte scars and parenchymal basement membrane are disrupted or rendered dysfunctional by experimental transgenic manipulations, cytotoxic inflammation spreads from lesion cores into adjacent CNS parenchyma resulting in neural degeneration and increased lesion size.
Astrocyte recruitment and restriction of leukocytes
Entry of leukocytes into CNS parenchyma involves interactions among multiple cell types and requires active recruitment across several different cellular and molecular functional barriers (Fig. 3)3, 21, 24. As in other tissues, initial events involve rolling and adhesion of leukocytes to CNS endothelia. In contrast with other tissues, diapedesis in CNS requires passing through an endothelial blood-brain barrier whose permeability is regulated by numerous molecules that can be produced by diverse cell types, including astrocytes (Fig. 31)1, 3, 21, 24. Once leukocytes have extravasated, they do not pass directly into CNS parenchyma but collect in perivascular spaces formed by retraction of astrocyte endfeet and parenchymal basement membrane that surround blood vessels (Figs. 2Aa, 32)35, where they may receive molecular instructions from various cell types including astrocytes (Fig. 33). Passage beyond this barrier of basement membrane and astrocyte endfeet into CNS parenchyma normally requires active recruitment, which is regulated by various potential molecules that can be produced by different cell types21, 23, 24, including by astrocytes (Fig. 34). In this manner, astrocytes take part both in recruiting and restricting the migration of leukocytes in the CNS, and these functions are purposefully regulated in a context specific manner by specific signaling events (Fig. 34,5). Thus, in response to different signaling mechanisms, astrocytes can on the one hand exert pro-inflammatory effects by releasing molecules that disrupt the blood-brain barrier, promote leukocyte extravasation and help to recruit leukocytes into CNS parenchyma (Fig. 31–4, Tables 2,3); but on the other hand astrocytes can also exert anti-inflammatory effects by releasing molecules that promote blood-brain barrier repair, restrict or confine leukocyte migration and promote the resolution of inflammation over time (Fig. 35–8, Tables 1,2).
Figure 3. Pro- and anti-inflammatory molecular mechanisms of astrocyte borders and functional barriers.
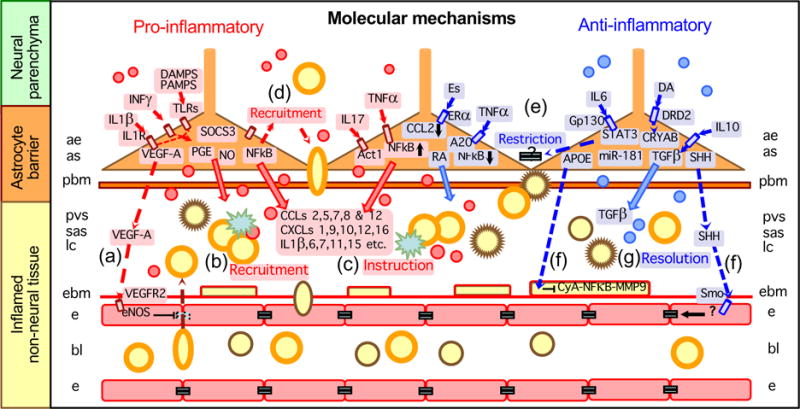
Entry of leukocytes into CNS parenchyma requires active recruitment across multiple cellular barriers, including an endothelial blood-brain barrier, endothelial (EBM) and parenchymal (PBM) basement membranes, and astrocytic functional barriers. Different molecules produced by astrocytes can take part in recruiting, restricting and instructing leukocytes in the CNS, as well as in promoting the resolution of inflammation over time. Thus, in response to different signaling mechanisms that are generated in different settings and at different times in relation to insult progression, astrocytes can in different contexts release molecules that (1) disrupt the blood-brain barrier by signaling to endothelia, (2) promote leukocyte extravasation into perivascular space (PVS), subarachnoid space (SAS) or lesion cores, (3) instruct leukocytes, (4) recruit leukocytes into CNS parenchyma, (5) restrict leukocyte entry into CNS parenchyma, or (6) repair the blood-brain barrier by signaling to pericytes, (7) promote resolution of inflammation or (8) repair the blood-brain barrier by signaling to endothelia. For molecular abbreviations see tables 1–3 and main text.
Table 3.
Pro-inflammatory astrocyte functions and molecular mechanisms demonstrated in vivo by transgenic loss-of function studies in mice
| Disease Model | Deleted or downregulated astrocyte molecule | Functions revealed | References |
|---|---|---|---|
| Spinal cord injury | SOCS3 | Recruits inflammation. | 41 |
| Spinal cord injury | NFκB | Recruits inflammation. | 40 |
| EAE | NFκB | Recruits inflammation. | 42 |
| EAE | Act1 | Recruits IL-17 induced inflammation. | 48 |
| EAE | CCL2 | Recruits inflammation. | 43, 44 |
| EAE | CXCL10 | Recruits inflammation. | 45 |
| EAE | VEGF | Opens blood-brain barrier; Recruits inflammation. |
46, 47 |
Astrocyte pro-inflammatory mechanisms
Several decades of investigation36 have shown that astrocytes can produce numerous pro-inflammatory molecules, including diverse cytokines, chemokines, growth factors and small molecules such as prostaglandins (PGE) and nitric oxide (NO) (Table 2). Recent technologies that allow cell type specific transcriptome analysis have begun to define specific contexts in which astrocytes produce a broad repertoire of pro-inflammatory molecules in vivo and to identify combinations of molecular triggers that regulate their production in vitro37–39. For example, analysis of astrocyte transcriptome profiles indicates that astrocyte exposure either in vivo or in vitro to pathogen associated molecular patterns (PAMPs) such as lipopolysaccharide (LPS) and associated cytokines markedly skew astrocyte transcriptome changes towards pro-inflammatory and potentially cytotoxic profiles37–39. In contrast, the ischemia caused by experimental stroke in vivo shifts the astrocyte transcriptome towards neuroprotective mechanisms37.
Transgenic approaches such as Cre-loxP mediated cell-type specific loss-of-function models are being used to dissect intra-cellular signaling cascades and identify inter-cellular effector molecules that mediate astrocyte pro-inflammatory functions (Table 3). For example, NFκB and SOCS3 are pro-inflammatory transcriptional regulators in astrocytes in CNS traumatic injury and CNS autoimmune inflammation (Table 3, Fig. 3)40–42. As inter-cellular effectors, CCL2 and CXCL10 released specifically by astrocytes are important recruiters of perivascular leukocytes in CNS autoimmune inflammation (Fig. 32)43–45. CCL2 and CCL7 production by astrocytes is heterogeneous38, which may contribute to selective direction of leukocyte migration in CNS parenchyma.
Multi-molecular signaling cascades are also being defined. For example, in response to stimulation by the pro-inflammatory cytokine IL-1β, astrocytes generate and release vasoactive endothelial growth factor (VEGF) that increases blood-brain barrier permeability and promotes leukocyte extravasation (Fig. 31)46, 47. Astrocyte Act1 signaling is critical for mediating IL-17 inflammatory gene induction in CNS autoimmune inflammation (Fig. 32,3)48. Certain types of increased neuronal activity can trigger inflammation in epilepsy, stress and neuropathic pain, and astrocytes contribute to mediating these effects49.
Astrocyte anti-inflammatory mechanisms
Essential anti-inflammatory roles of astrocytes have now been demonstrated by numerous transgenic loss-of-function experiments in diverse models of CNS injury and disease (Table 1), and specific molecular mechanisms are gradually being identified (Fig. 35–8; Table 2). An early observation using transgenic models indicated that blood-brain barrier repair after traumatic injury is critically dependent on the presence of newly proliferated scar forming astrocytes28, 30. Astrocyte impact on blood-brain barrier function has a long history of investigation50, and recent findings are beginning to define specific molecular mechanism through which astrocytes can influence blood-brain barrier integrity in different ways. For example, as discussed above, IL1β stimulated astrocytes release VEGF that disrupts the endothelial blood-brain barrier and increases leukocyte extravasation (Fig. 31)46, 47. Under different circumstances, astrocytes release molecules that promote endothelial blood-brain barrier repair, such as Sonic hedge hog (Shh) (Fig. 38)51, 52. In addition, apolipoprotein E (APOE) secretion by astrocytes critically suppresses a cyclophilin A-NFκB-matrix metaloproteinase-9 pathway in pericytes that increases blood-brain barrier permeability (Fig. 36)53. Thus, astrocytes are emerging as pivotal regulators of endothelial blood-brain barrier properties that can, via specific molecular mechanisms, either open or maintain barrier functions, and do so in a context dependent manner as regulated by specific signaling events.
As discussed above, astrocyte borders and scars form functional barriers that restrict leukocyte migration after diverse CNS insults including trauma, ischemia, autoimmune attack and neurodegeneration (Fig. 2C)28, 30–34. Transgenic loss-of-function approaches are beginning to define intra-cellular signaling cascades that mediate astrocyte anti-inflammatory functions (Table 1). Studies from multiple laboratories indicate that the gp130-JAK2-STAT3 signaling pathway is a critical regulator of astrocyte anti-inflammatory functions after a variety of CNS insults, and does so by mediating scar formation and barrier functions that restrict the spread of leukocytes and microbial pathogens after traumatic injury26, 41, 54, infection55 and autoimmune attack56. In this regard it is interesting that the astrocyte structural proteins, GFAP and vimentin57, are required for astrocyte scar formation8 and their absence markedly exacerbates the inflammation and tissue pathologies associated with autoimmune attack58, stroke31, 34 and neurodegeneration due either to lipid storage defects59 or amyloid-beta accumulation60.
Inter-cellular effector molecules that mediate astrocyte anti-inflammatory functions are also being identified. For example, in response to IL10, astrocytes release molecules such as TGFβ that promote resolution of inflammation61–63 (Fig. 37). Certain molecules released by astrocytes have anti-inflammatory effects on microglia and monocytes64, 65. Retinoic acid (RA) released from astrocytes is implicated in protecting blood-brain barrier function and attenuating inflammation (Fig. 3)66.
An indirect form of anti-inflammatory regulation also appears to be operational in the form of intra-cellular signaling mechanisms that suppress pro-inflammatory activators (Fig. 3). For example, TNFα signaling not only induces production of the pro-inflammatory transcriptional regulator NFκB, but also induces the ubiquitin modifying protein A20, which acts to suppress NFκB signaling67. A20 production by astrocytes is a critical anti-inflammatory regulator in autoimmune inflammation that suppresses astrocyte production of leukocyte-recruiting chemokines68. In an analogous fashion, dopamine (DA) signaling thorough the astrocytic dopamine D2 receptor (DRD2) acting through alphaβ-crystallin (CRYAB) attenuates inflammation associated MPTP-toxicity69. CRYAB produced by astrocytes is a potent negative regulator acting as a brake on various inflammatory pathways in autoimmune inflammation70. Similarly, galectin-9 (Gal-9) expression by astrocytes is induced by pro-inflammatory cytokines such as IL-1β, INFγ and TNFα, but Gal-9 released by astrocytes induces the apoptosis and downregulation of encephalitogenic T cells in autoimmune inflammation71. It is interesting to speculate that temporal regulation within astrocytes of production of molecules such as CRYAB, A20 and Gal-9 may contribute to mechanisms that bring about the resolution of CNS inflammation over time (Fig. 37). Small interfering RNAs (siRNAs) such as miR-181 are also implicated in attenuating astrocyte expression of certain pro-inflammatory molecules72 (Table 2).
Attenuation of astrocyte pro-inflammatory effects also appears to be an important mechanism underlying anti-inflammatory effects of sex steroids (Fig. 37). Both estrogen (Es) and testosterone have potent anti-inflammatory and neuroprotective effects in experimental and clinical CNS autoimmune inflammation73. Transgenic loss-of-function studies show that activation of estrogen receptor alpha (ERα), but not ERβ, specifically on astrocytes, potently attenuates autoimmune inflammation and reduces astrocyte production of the pro-inflammatory cytokines CCL2 and CCL774, 75.
Astrocyte roles in CNS leukocyte aggregation and instruction
The immune system consists of billions of motile leukocytes that roam throughout the body76. Their navigation, interactions and functions are orchestrated by chemokines and cytokines76, which are produced by leukocytes themselves and by resident cells in organs such as the CNS. Various aspects of leukocyte instruction are dependent on stochastic interactions, which are facilitated by formation of leukocyte aggregates in lymph nodes or in specialized structures within certain organs77, 78. Organized aggregates of leukocytes within the CNS are of interest with regard to their roles in infections and autoimmune inflammation3, 78–82, as well as their potential roles after CNS injury or during degenerative disease.
As discussed above, astrocytes form borders and functional barriers to non-neural tissues that exhibit increased immune cell trafficking. Because astrocytes have the combined capacity for recruitment and restriction of leukocytes (Fig. 3), they are well placed to facilitate both the formation and instruction of CNS of leukocyte aggregates in (1) perivascular spaces (also known as Virchow-Robin spaces), (2) perimeningeal locations, and (3) areas of tissue damage after trauma, ischemia, infection, degenerative disease or autoimmune inflammation (Figs. 2Aa–Ca).
Perivascular lymphocyte aggregates are characteristic of CNS viral infections. Recent studies show that during recovery from viral encephalomyelitis, multiple changes in the brain microenvironment not only facilitate B-cell entry, but also support the proliferation, differentiation and long-term survival of antiviral antibody-secreting cells in perivascular spaces that are inside the blood-brain barrier82. Perivascular lymphocyte aggregates are surrounded by barrier forming reactive astrocytes, which can produce many of the chemokines and cytokines implicated in B cell recruitment and retention in the CNS, including CCL2, CCL5, CXCL9, CXCL10, IL10 and BAFF (Fig. 2Aa, Table 2).
Meningeal and perivascular aggregates of inflammatory leukocytes are characteristic of autoimmune inflammation3, 78–81. Meningeal leukocyte aggregates are also separated from adjacent neural parenchyma by barrier forming reactive astrocytes, and these astrocytes are similarly well positioned to contribute to the formation and persistence of meningeal leukocyte aggregates through their ability to produce numerous chemokines and cytokines involved in recruitment and instruction of diverse leukocytes (Table 2) and their ability to restrict leukocyte migration during autoimmune inflammation (Fig. 2Ba, Table 1).
Although less well studied, aggregates of inflammatory leukocytes are also found in the non-neural lesion cores that form after CNS damage caused by trauma, ischemia, chronic epilepsy and chronic neurodegenerative insults (Fig. 2Ca)1. These leukocyte aggregates are also surrounded by scar-forming reactive astrocytes that separate them from adjacent neural parenchyma (Fig. 2Ca), and these border forming astrocytes not only contribute to the formation of leukocyte aggregates through their ability to produce numerous molecules that recruit and restrict leukocyte migration (Fig. 31,2,5), but these astrocytes have the potential to instruct leukocytes via the molecules they produce (Fig. 33, Table 2).
The formation of leukocyte aggregates during CNS inflammation could have various roles, such as facilitating the transfer of molecular instructions (i) among leukocytes, (ii) from locally resident CNS cells to leukocytes, and (iii) from leukocytes to local CNS cells including astrocytes. In addition, release of antibodies and other molecules produced by leukocyte aggregates within the blood-brain barrier could be important in clearing CNS infections or clearing debris and achieving repair after CNS damage. Formation and ongoing instruction of leukocyte aggregates could also have maladaptive roles that exacerbate and perpetuate CNS autoimmune inflammation. In this regard, certain evidence suggests that T cell-activation may occur in leukocyte aggregates within CNS lesions in neuromyelitis optica83, and astrocytes play causal roles in this disorder as discussed below.
Thus, multiple lines of evidence implicate important, but underappreciated roles for astrocytes in the formation, retention and instruction of leukocyte aggregates within specific CNS tissue compartments in different contexts. In microbial infections and after CNS damage due to trauma or ischemia, these astrocyte roles appear likely to serve adaptive functions involved in clearing debris, clearing pathogens and achieving tissue repair. In the light of these important, potent and widely exercised roles, it is perhaps not surprising that certain astrocyte effects, dysfunctions or loss of functions may contribute to diverse disease mechanisms.
Astrocyte impact on inflammation in CNS disorders
Given the many ways in which astrocytes can interact with immune and inflammatory cells, there is increasing interest in the impact of astrocytes on inflammation in diverse CNS disorders. It is important to emphasize that astrocyte roles in CNS inflammation are not uniform or stereotypic, but instead are heterogeneous and context dependent as determined by a multitude of specific signaling events. Thus, astrocyte effects on inflammation may vary not only among different CNS disorders, but may also vary within the same disorder as determined by specific circumstances, such as time course of the pathogenic mechanisms, location within the CNS or concomitant exposure to PAMPs and cytokines that derive from local or peripheral pathogens. In this regard it is also interesting to consider that astrocyte dysfunctions, and perhaps astrocyte genetic polymorphisms, have the potential to contribute to CNS disorders7, 15.
Trauma and stroke
CNS tissue damage caused by focal traumatic injury or ischemia gives rise to focal inflammation surrounded by astrocyte scars27. Under normal circumstances in otherwise healthy subjects, astrocytes contribute to regulating this inflammation in different ways, first by mounting pro-inflammatory responses that help to recruit leukocytes, and second by forming essential barriers that limit the spread of inflammatory cells, as discussed above (Figures 1–3). These astrocyte roles serve adaptive functions by helping to attract robust inflammation to sites of tissue damage where it is needed to clear debris, neutralize pathogens and promote tissue repair, while at the same time protecting immediately adjacent viable neural tissue. Experimental disruption of astrocyte scar formation after focal trauma or stroke is associated with spread of neurotoxic inflammation, increased lesion size, loss of neurons and decreased function (Fig. 2, Table 1). Such findings suggest that astrocyte dysfunctions could contribute to pathogenic mechanisms, and do so in a variety of ways. Astrocyte dysfunctions could be caused by genetic polymorphisms or by exposure to molecular signals deriving from infections, which might alter astrocyte inflammatory regulation during the response to trauma or stroke in ways that lead to detrimental effects. There is evidence for such possibilities. For example, astrocytes are major sources of APOE, and astrocyte derived APOE signaling to pericytes is important in maintaining a functioning blood-brain barrier (Fig. 36)53. In humans, polymorphisms of APOE are associated with various disease mechanisms, such that APOE4 is a risk factor associated with worse prognosis not only in Alzheimer’s disease, but also in stroke and traumatic injuries84, 85. In transgenic mouse experiments, astrocyte derived APOE4 was associated with poor blood-brain barrier maintenance53. Astrocyte signaling mechanisms may also be altered by infections. There is compelling evidence that inflammatory mediators produced both by local and peripheral infections can markedly skew astrocytes towards pro-inflammatory and cytotoxic responses37, 38, which could have detrimental effects in trauma or stroke, as discussed below. Lastly, it is noteworthy that CNS lesions after trauma and stroke are surrounded by large areas of perimeter tissue in which hypertrophic reactive astrocytes are intermingled with functioning neurons, and these astrocytes will also be responding to inflammatory stimuli emanating from the lesions27. Increasing evidence implicates important roles for astrocytes in maintaining and modulating neuronal synaptic functions86, 87. The extent to which inflammation acting on reactive astrocytes located among functioning neurons impacts on local circuit function is an important but understudied area88.
Astrocytes and CNS autoimmune disorders
Compelling experimental and clinical evidence indicates that astrocytes play critical and complex roles in CNS autoimmune inflammation. Numerous experimental loss-of-function studies using the EAE model demonstrate that astrocytes contribute both to attracting autoimmune inflammation and restricting its spread in vivo (Fig. 3, Tables 1, 3). These experimental observations are likely to help illuminate important pathophysiological mechanisms in clinical CNS autoimmune diseases. An illustrative example is neuromyelitis optica (NMO), a CNS inflammatory demyelinating and degenerative disease with pronounced tissue destruction and severe symptoms that can include vision loss and paralysis. NMO has been causally linked to autoantibodies that bind specifically to aquaporin-4 (AQP4) on astrocytes and are implicated in compliment-mediated astrocyte lysis89, 90. There is now persuasive experimental and clinical evidence that AQP4 is not simply a passive autoimmune CNS antigen, but that binding of AQP4 antibodies to astrocytes leads to disruption of astrocyte pro- and anti-inflammatory functions that play causal and disease-defining roles in NMO pathophysiology and disease progression. Experimental evidence for this notion includes observations that (i) combined exposure of astrocytes to AQP4/NMO-IgG and complement in vivo leads to lytic astrocyte destruction in experimental animals91; (ii) experimentally induced astrocyte destruction or dysfunction by diverse transgenically targeted mechanisms markedly exacerbates the spread and severity of inflammation during experimental CNS autoimmune inflammation32, 56 (see also Table 1); and (iii) binding of NMO-causing AQP4 antibodies (AQP4/NMO-IgG) to astrocytes in vitro triggers the release from astrocytes of a broad spectrum of pro-inflammatory molecules92. These experimental findings illuminate clinical evidence that both clinical progression and tissue lesions are markedly more severe in NMO patients that have autoimmune mediated CNS pathology associated with anti-AQP4 autoantibodies, when compared with patients that have anti-myelin autoantibodies but not anti-AQP4 in what is referred to by some as an NMO spectrum disorder93–95 but is likely to be a different mechanistic clinical entity96.
Thus, both experimental and clinical evidence support the notion of NMO as an astrocytopathy in which autoimmune pathophysiology is complicated by autoimmune disruption of astrocyte immune regulatory functions. Taken together, available evidence supports a working model of cellular and molecular pathophysiological mechanisms in which astrocytes play active and causal roles in different phases of NMO progression (Fig. 4). According to this model, focal pathology begins with binding of AQP4/NMO-IgG to astrocyte membranes (Fig. 4a). CNS entry of AQP4/NMO-IgG could occur through sporadically or disease-specific breaches of the blood-brain barrier. Low-level breaches of blood-brain barrier are well recognized to occur sporadically in otherwise healthy CNS through various mechanisms that can allow CNS entry of small but functional amounts of circulating IgG22, 97. Disease specific breaches could include B cell production of AQP4/NMO-IgG inside the CNS98. As AQP4/NMO-IgG bound to astrocyte membranes accumulates, it either directly or indirectly provokes astrocyte production of multiple molecules that help to recruit and activate inflammatory infiltrates (Fig. 4b). In this context it is noteworthy that there is evidence for activation and instruction of T-cells within the CNS in NMO lesions83. With time, AQP4/NMO-IgG leads to complement-mediated lytic astrocyte destruction89, 90, which in turn leads to loss of barrier function and the spread of neurotoxic inflammation (Fig. 4c). This spread of inflammation would be associated with the spread of additional blood-brain barrier breakdown, entry of more AQP4/NMO-IgG, and the subsequent cascade of astrocyte gain- and loss- of function with its associated inflammatory pathology, driving the local disease process in a positive feedback manner and leading to contiguous spread through adjacent CNS tissue with the generation of the large contiguous lesions that are characteristic of AQP4-associated NMO. It is also noteworthy that early binding of anti-AQP4 to astrocyte membranes (Fig. 4a) may alter astrocyte homeostatic functions such as regulation of extracellular fluid and ion balance. Disruption of astrocyte maintenance of extracellular ion homeostasis can impact on neuronal excitability and neural circuit function99. Such disruptions could explain early neurological symptoms associated with NMO prior to the onset of overt tissue degeneration. Recognition of NMO as an astrocytopathy with different stages and mechanisms of disease progression as they relate to different perturbations of astrocyte immune-regulatory functions (Fig. 4a–c) may also reveal new opportunities for treatment strategies in addition to those already under consideration100. From a broader perspective, recognition of the central role of astrocyte functions in CNS immune and inflammatory responses has fundamental implications for other CNS autoimmune disorders in which astrocyte roles are only beginning to emerge.
Figure 4. Model of NMO as an astrocytopathy with causal contributions of astrocyte gain- and loss- of functions to different stages of disease progression.
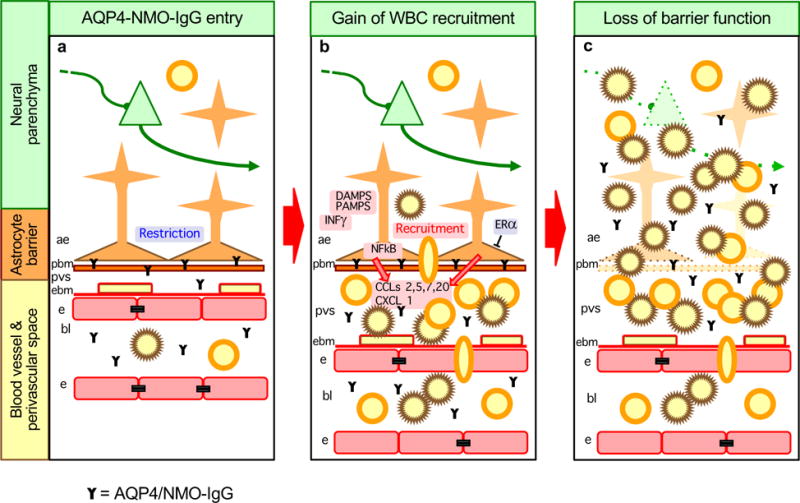
a| Stage 1. Sporadic or disease specific breaches of the blood-brain barrier lead to binding of AQP4/NMO-IgG to astrocyte membranes, which results in astrocyte functional changes. b| Stage 2. AQP4/NMO-IgG binding leads to astrocyte production of pro-inflammatory molecules and recruitment of leukocytes, initially into perivascular spaces (PVS). This process may be exacerbated by concomitant exposure to inflammatory mediators deriving from local or peripheral infections, such as DAMPs, PAMPs or INFγ. This process may be attenuated by estrogen related signaling mechanisms. c| Stage 3. Compliment-mediated astrocyte destruction leads to loss of astrocyte barrier functions and subsequent spread of neurotoxic inflammation into adjacent neural parenchyma.
Multiple sclerosis is a major CNS autoimmune disorder. For much of the long history of investigating multiple sclerosis, astrocytes were regarded as non-participatory bystanders that responded to, but played little or no role in disease mechanisms35. This view is now changing and there is abundant and steadily growing evidence indicating that astrocytes actively participate in both lesion development and repair, as reviewed recently elsewhere35. Neuropathological specimens of active multiple sclerosis lesions exhibit unambiguous early damage to perivascular astrocyte endfeet35. Although causation cannot be inferred in such specimens, the astrocyte appearance and concomitant local inflammation in such clinical specimens is comparable with the experimentally induced loss of astrocyte barrier functions described above (Fig. 2, Table 1). There is also compelling evidence that astrocytes take part in both pro- and anti-inflammatory mechanisms in multiple sclerosis and its experimental models35. These findings fit well with the experimental findings and models discussed here (Tables 1–3; Figures 1–3). Given the many ways in which astrocytes can participate in pro- and anti-inflammatory mechanisms associated with multiple sclerosis and its experimental models, it is interesting to consider that astrocyte dysfunctions, or differences in astrocyte functions secondary to genetic polymorphisms or induced by concomitant local or peripheral infections, may influence the susceptibility of different individuals to multiple sclerosis, or may contribute to the waxing and waning of symptoms. From a therapeutic perspective reducing astrocyte pro-inflammatory effects may be useful targets. The roles of auto-antibodies are less well defined in multiple sclerosis than in neuromyelitis optica. The most conspicuous autoantibodies in multiple sclerosis are against myelin peptides, which could at present be regarded as passive antigens. Recently, auto-antibodies against the astrocyte membrane potassium channel Kir4.1 have been reported in nearly half of a cohort of multiple sclerosis patients101, although another report did not find this in a different cohort of patients102. The functional consequences of anti-Kir4.1 antibodies with respect to astrocyte homeostatic or immune regulatory functions are not yet known. Work is needed to identify potential effects of these and other auto-antibodies on astrocytes in multiple sclerosis and other autoimmune disorders with CNS involvement, such as lupus. Identification of diverse antibodies among different patient subsets and recognition that certain antibodies may alter immune regulatory functions of CNS intrinsic cells such as astrocytes may help to shed light on the diversity of clinical syndromes associated with CNS autoimmunity.
Astrocytes and sex differences in CNS inflammatory disorders
A striking feature of CNS autoimmune diseases is their markedly increased prevalence among women103, 104. Conservative estimates are that female to male ratios are at least 7:1 in neuromyelitis optic and 2:1 in multiple sclerosis. The cellular and molecular mechanisms that underlie these differences are incompletely understood and likely to be multifactorial. Intriguing evidence suggests that astrocytes may contribute to these differences. As discussed throughout this article, astrocytes are well positioned to exert powerful effects on CNS inflammation. Signalling through estrogen receptor alpha on astrocytes mediates potent anti-inflammatory and neuroprotective effects during autoimmune inflammation74, 75. As discussed above, in neuromyelitis optica, which exhibits a remarkable prevalence for females, there is strong evidence that astrocytes play active causal roles via both gain and loss of functions mediated by astrocytes that facilitate the spread of neurotoxic inflammation (Fig. 4). These findings strongly suggest that further investigation of potential astrocyte contributions to sex differences in mechanisms of CNS inflammation is warranted.
Astrocytes and inflammation associated with neurodegenerative diseases
Astrogliosis is a feature of all neurodegenerative pathologies, but astrocyte roles in most neurodegenerative disorders are only beginning to be investigated. Inflammation occurs during the course of most neurodegenerative pathologies and its impact is of increasing interest. Various lines of evidence are emerging and potentially converging as molecular dissection of various processes illuminates cellular and molecular mechanisms. For example, APOE secretion by astrocytes suppresses a cyclophilin A-NFκB-matrix metaloproteinase-9 pathway in pericytes that increases blood-brain barrier permeability and is pro-inflammatory (Fig. 36)53. The APOE4 allele is associated with increased risk of Alzheimer’s disease, and experimental modulation of astrocytes towards APOE4 leads to blood-brain barrier disruption, predisposing to inflammation53. Synapse loss is a key feature of Alzheimer’s disease, and astrocytes take part in induction and pruning of synapses using molecular cues that in part overlap with immune system cues105, 106. Astrocyte expression of some of these cues is modified by inflammatory stimuli. Attenuation of reactive astrogliosis exacerbates amyloid-beta accumulation and inflammation in mouse models of Alzheimer’s disease60. In a mouse model of a neurodegenerative lipid storage defect, experimental attenuation of reactive astrogliosis markedly exacerbates inflammation and degeneration59. In Huntington’s disease and its models, astrocytes as well as neurons accumulate nuclear inclusions of mutant huntingtin protein (mHtt). In transgenic mouse models, astrocytes with mHtt downregulate the potassium channel Kir4.1, leading to increased extracellular potassium and increased neuronal excitability, which is likely to contribute to and exacerbate direct effects of mHtt on neurons87, 99. It is interesting that these changes occur before the onset of detectable astrogliosis, leading to the important recognition that astrogliosis and astrocyte dysfunction are independent and separable events99. In Parkinson’s disease and models, astrocytes are also implicated as regulating inflammation and having both neuroprotective and neurodegenerative potential107. It is beyond the scope of this article to examine the many ways in which astrocytes may impact on the broad spectrum of neurodegenerative diseases. Nevertheless, a rapidly growing literature points towards critical roles for astrocytes in modulating inflammation and disease progression in diverse neurodegenerative disorders, and that both loss or gain of astrocyte functions have the potential to attenuate or exacerbate these conditions in context specific manners. The over-simplified assumption that astrogliosis is uniformly harmful and should be broadly attenuated in neurodegenerative conditions is incorrect and no longer tenable. Specific mechanisms need to be identified and addressed.
Astrocytes and CNS intrinsic infections
Astrocytes contribute to the control and clearance of CNS intrinsic infections in multiple ways. Astrocyte barrier functions along blood vessels and meninges are critical for limiting the spread of infections through CNS parenchyma (Table 1, Fig 2)55. As discussed above, in addition to barrier functions, astrocytes take part in the recruitment and instruction of CNS intrinsic leukocyte aggregates (Fig. 3b,c), and such aggregates are thought to contribute to immune responses that control and clear microbial infections, particularly as regards the production inside the CNS endothelial blood-brain barrier of antibodies that are critical for viral clearance82. Astrocyte functions that help control and clear infections and related inflammation are particularly important along meningeal borders where leukocyte trafficking for immune surveillance and inflammatory activity are high for detection and rapid response to pathogens that gain entry into the CNS protective meningeal shield3, 23. Restricting these processes to the meninges is critical so as to protect immediately adjacent functioning CNS parenchyma, for example along the vast expanses of cerebral cortex. The transition from meningitis to cerebritis is a well-known devastating progression in CNS clinical infections.
Impact of peripheral infections on astrocytes and CNS inflammation
Limiting microbial infections is likely to have powerfully shaped the evolution of CNS injury responses. As a consequence, cells responding to CNS insults are exquisitely sensitive to PAMPs and molecular cues associated with infections27. There is growing awareness that such molecular cues released not only by local, but also by distant peripheral infections can markedly alter the character of inflammation associated with diverse CNS insults both in experimental and clinical settings27. Astrocytes are exquisitely sensitive to infection-associated PAMPs and cytokines, which can drive astrocyte transcriptome profiles towards highly pro-inflammatory and cytotoxic phenotypes37–39. While such responses may be essential and beneficial if triggered by local microbial infections, they could be detrimental if triggered via circulating PAMPs generated by peripheral infections. Thus, astrocytes exposed to circulating PAMPs could inappropriately contribute to recruiting cytotoxic leukocytes (neutrophils, eosinophils and macrophages) to CNS insults that what would otherwise be sterile (uninfected) tissue responses. Notably, there is clinical epidemiological evidence that peripheral infections have a negative impact on neurological outcome after spinal cord injury108.
It is also interesting to consider that astrocytes stimulated by PAMPs and inflammatory cytokines might contribute to autoimmune potential by producing local levels of molecular cues that recruit and instruct leukocytes within the CNS. The signaling events that initiate CNS autoimmune disorders or trigger their relapses are not well understood. It is noteworthy that induction of experimental autoimmune encephalomyelitis with myelin antigens is markedly accelerated by, and in some cases is dependent upon, concomitant inoculation of PAMP-related immune stimulatory molecules109. Although these immune stimulatory molecules are generally thought to act entirely through the peripheral immune system, they will also, as just described, push astrocyte transcription towards a markedly pro-inflammatory profile, which may contribute to the induction or persistence of autoimmune pathology. In support of this notion, exposure to lipopolysaccharide (LPS), the prototypical PAMP, drives astrocytes towards highly pro-inflammatory molecular phenotypes37, 38, and deletion of CCL2 production selectively from astrocytes attenuates autoimmune clinical symptoms and inflammation43, 44. Such astrocyte-related mechanisms are only beginning to be explored but hold much potential for adding to the understanding of CNS autoimmune pathophysiology. In general, the impact of concomitant local or peripheral microbial infections and inflammation on astrocyte and other glial cell responses to diverse CNS insults and their potential contribution to neurodegenerative and autoimmune events is an important emerging topic that warrants further investigation.
Astrocytes, Inflammatory cytokines and behavioral disturbances
It deserves brief mention that astrocytes may contribute to behavioral effects associated with inflammatory stimuli. Astrocytes can modulate neuronal function and impact on complex behaviors including sleep86, pain110, mood and depression86, 111–113, and certain childhood behavioral syndromes that involve disorders of synapse development105, 106, 114. Cytokines and inflammatory stimuli are increasingly implicated in sickness behaviors, pain, sleep disturbances, mood disorders and certain behavioral developmental disorders115–121. Cytokines and inflammatory stimuli exert powerful effects on astrocyte gene expression and physiology in ways that can impact markedly on synaptic and neuronal functions38, 88. The broad mixture of astrocyte signaling capabilities provides astrocytes with the potential to mediate crosstalk between immune/inflammatory and neural systems88. Although much work is still needed, these types of interactions represent an exciting emerging area of research that has the potential to deepen our understanding of how signaling events associated with inflammation can impact on behavior and its disturbances.
Astrocyte dysfunctions, CNS inflammation and the concept of astrocytopathies
There is steadily accumulating evidence that astrocyte dysfunction can provoke or contribute to diverse clinical neurological disorders6, 7, 15. Potential causes of astrocyte dysfunction include genetic mutations, genetic polymorphisms, metabolic disturbances, comorbid infections or autoimmune attack. For example, Alexander disease, a dominant mutation of the GFAP gene is associated with inflammatory degenerative changes, particularly of white matter, and severe neurological dysfunction122, 123. In familial amyotrophic lateral sclerosis (ALS), a dominant mutation of the superoxide dismutase (SOD) gene leads to production of neurotoxic molecules. Targeting mutant SOD selectively to astrocytes in transgenic mice is sufficient to cause neuronal dysfunction and degeneration124–126, but in the full disease, mutant SOD in neurons also contributes to neuronal degeneration. Other examples that have been discussed in more detail above include Huntington’s disease, where astrocytes accumulate mHtt nuclear inclusions, leading to Kir4.1 downregulation in genetic mouse models, with increased extracellular potassium and increased neuronal excitability87, 99, which is likely to contribute to and exacerbate direct effects of mHtt on neurons. In addition, genetic polymorphisms of the astrocyte protein APOE in the form of allele 4, is associated with blood-brain barrier disruption and increased risk and severity in Alzheimer’s disease and traumatic brain injury53. Lastly, in NMO, autoimmune targeting of astrocytes leads to both gain- and loss- of astrocyte immune regulatory functions that lead to spread of neurotoxic inflammation (Fig. 4). Observations such as these provide compelling support for the emerging concept of astrocytopathies, in which gain or loss of astrocyte functions is either the primary cause or makes a significant contribution to the pathophysiology and neurological symptoms of certain CNS disorders.
Concluding remarks
Inflammation is an essential response to CNS insults that is critical for clearance of potentially toxic cellular debris and pathogens. CNS parenchyma is particularly vulnerable to rapid spread of inflammatory destruction. Astrocytes form borders and functional barriers that help restrict the trafficking of immune and inflammatory cells to non-neural cellular compartments in both healthy CNS and during the response to CNS insults, thereby limiting the spread of potentially neurotoxic inflammation. Astrocytes can also contribute to the recruitment, retention and instruction of leukocytes in CNS tissue. Although astrogliosis and astrocyte scars are sometimes thought of as detrimental to CNS recovery, both experimental and clinical observations now indicate that loss or dysfunction of scar forming astrocytes can severely exacerbate CNS inflammation and tissue damage. Compelling evidence implicates astrocytes as critical regulators of multiple aspects of CNS inflammation in ways that have the potential to impact significantly on outcome after traumatic or ischemic damage, infection, neurodegenerative disease and autoimmune disorders. There is a growing awareness that understanding and safely modulating CNS inflammation in diverse CNS disorders will benefit from a better understanding of astrocyte immune regulatory mechanisms and how they can dysfunction.
Acknowledgments
Work in the author’s laboratory is supported by the National Institutes of Health (Grants NS057624 and NS084030), the Dr. Miriam and Sheldon G. Adelson Medical Foundation, the Hilton Foundation and Wings for Life.
Footnotes
Competing interests statement
The author declares no competing financial interests.
References
- 1.Burda JE, Sofroniew MV. Reactive gliosis and the multicellular response to CNS damage and disease. Neuron. 2014;81:229–48. doi: 10.1016/j.neuron.2013.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goverman J. Autoimmune T cell responses in the central nervous system. Nat Rev Immunol. 2009;9:393–407. doi: 10.1038/nri2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ransohoff RM, Engelhardt B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat Rev Immunol. 2012;12:623–35. doi: 10.1038/nri3265. [DOI] [PubMed] [Google Scholar]
- 4.Prinz M, Priller J. Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat Rev Neurosci. 2014;15:300–12. doi: 10.1038/nrn3722. [DOI] [PubMed] [Google Scholar]
- 5.Barres BA. The mystery and magic of glia: a perspective on their roles in health and disease. Neuron. 2008;60:430–440. doi: 10.1016/j.neuron.2008.10.013. [DOI] [PubMed] [Google Scholar]
- 6.Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119:7–35. doi: 10.1007/s00401-009-0619-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sofroniew MV. Astrogliosis. Cold Spring Harb Perspect Biol. 2014;7 doi: 10.1101/cshperspect.a020420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pekny M, Nilsson M. Astrocyte activation and reactive gliosis. Glia. 2005;50:427–434. doi: 10.1002/glia.20207. [DOI] [PubMed] [Google Scholar]
- 9.Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009;32:638–647. doi: 10.1016/j.tins.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kang W, Hebert JM. Signaling pathways in reactive astrocytes, a genetic perspective. Mol Neurobiol. 2011;43:147–54. doi: 10.1007/s12035-011-8163-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anderson MA, Ao Y, Sofroniew MV. Heterogeneity of reactive astrocytes. Neurosci Lett. 2014;565C:23–29. doi: 10.1016/j.neulet.2013.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sofroniew MV. Reactive astrocytes in neural repair and protection. Neuroscientist. 2005;5:400–407. doi: 10.1177/1073858405278321. [DOI] [PubMed] [Google Scholar]
- 13.Silver J, Miller JH. Regeneration beyond the glial scar. Nature Rev Neurosci. 2004;5:146–156. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]
- 14.Hamby ME, Sofroniew MV. Reactive astrocytes as therapeutic targets for CNS disorders. Neurotherapeutics. 2010;7:494–506. doi: 10.1016/j.nurt.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Verkhratsky A, et al. Neurological diseases as primary gliopathies: a reassessment of neurocentrism. ASN Neuro. 2012;4 doi: 10.1042/AN20120010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bianchi MG, Bardelli D, Chiu M, Bussolati O. Changes in the expression of the glutamate transporter EAAT3/EAAC1 in health and disease. Cell Mol Life Sci. 2014;71:2001–15. doi: 10.1007/s00018-013-1484-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kono H, Rock KL. How dying cells alert the immune system to danger. Nat Rev Immunol. 2008;8:279–89. doi: 10.1038/nri2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Janeway CA, Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 19.Iwasaki A, Medzhitov R. Regulation of adaptive immunity by the innate immune system. Science. 2010;327:291–5. doi: 10.1126/science.1183021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Galea I, Bechmann I, Perry VH. What is immune privilege (not)? Trends Immunol. 2007;28:12–18. doi: 10.1016/j.it.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 21.Owens T, Bechmann I, Engelhardt B. Perivascular spaces and the two steps to neuroinflammation. J Neuropathol Exp Neurol. 2008;67:1113–21. doi: 10.1097/NEN.0b013e31818f9ca8. [DOI] [PubMed] [Google Scholar]
- 22.Broadwell RD, Sofroniew MV. Serum proteins by-pass the blood brain barrier for extracellular entry to the CNS. Exp Neurol. 1993;120:246–263. doi: 10.1006/exnr.1993.1059. [DOI] [PubMed] [Google Scholar]
- 23.Engelhardt B, Coisne C. Fluids and barriers of the CNS establish immune privilege by confining immune surveillance to a two-walled castle moat surrounding the CNS castle. Fluids Barriers CNS. 2011;8:4. doi: 10.1186/2045-8118-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wilson EH, Weninger W, Hunter CA. Trafficking of immune cells in the central nervous system. J Clin Invest. 2010;120:1368–79. doi: 10.1172/JCI41911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kawano H, et al. Role of the lesion scar in the response to damage and repair of the central nervous system. Cell Tissue Res. 2012;349:169–80. doi: 10.1007/s00441-012-1336-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wanner IB, et al. Glial scar borders are formed by newly proliferated, elongated astrocytes that interact to corral inflammatory and fibrotic cells via STAT3-dependent mechanisms after spinal cord injury. J Neurosci. 2013;33:12870–86. doi: 10.1523/JNEUROSCI.2121-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burda JE, Radulovic M, Yoon H, Scarisbrick IA. Critical role for PAR1 in kallikrein 6-mediated oligodendrogliopathy. Glia. 2013;61:1456–70. doi: 10.1002/glia.22534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bush TG, et al. Leukocyte infiltration, neuronal degeneration and neurite outgrowth after ablation of scar-forming, reactive astrocytes in adult transgenic mice. Neuron. 1999;23:297–308. doi: 10.1016/s0896-6273(00)80781-3. [DOI] [PubMed] [Google Scholar]
- 29.Bush TG, et al. Fulminant jejuno-ileitis following ablation of enteric glia in adult transgenic mice. Cell. 1998;93:189–201. doi: 10.1016/s0092-8674(00)81571-8. [DOI] [PubMed] [Google Scholar]
- 30.Faulkner JR, et al. Reactive astrocytes protect tissue and preserve function after spinal cord injury. J Neurosci. 2004;24:2143–2155. doi: 10.1523/JNEUROSCI.3547-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li L, et al. Protective role of reactive astrocytes in brain ischemia. J Cereb Blood Flow Metab. 2008;28:468–481. doi: 10.1038/sj.jcbfm.9600546. [DOI] [PubMed] [Google Scholar]
- 32.Voskuhl RR, et al. Reactive astrocytes form scar-like perivascular barriers to leukocytes during adaptive immune inflammation of the CNS. J Neurosci. 2009;29:11511–11522. doi: 10.1523/JNEUROSCI.1514-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Toft-Hansen H, Fuchtbauer L, Owens T. Inhibition of reactive astrocytosis in established experimental autoimmune encephalomyelitis favors infiltration by myeloid cells over T cells and enhances severity of disease. Glia. 2011;59:166–76. doi: 10.1002/glia.21088. [DOI] [PubMed] [Google Scholar]
- 34.Liu Z, et al. Beneficial effects of gfap/vimentin reactive astrocytes for axonal remodeling and motor behavioral recovery in mice after stroke. Glia. 2014;62:2022–33. doi: 10.1002/glia.22723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brosnan CF, Raine CS. The astrocyte in multiple sclerosis revisited. Glia. 2013;61:453–65. doi: 10.1002/glia.22443. [DOI] [PubMed] [Google Scholar]
- 36.Eddleston M, Mucke L. Molecular profile of reactive astrocytes – implications for their role in neurological disease. Neuroscience. 1993;54:15–36. doi: 10.1016/0306-4522(93)90380-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zamanian JL, et al. Genomic analysis of reactive astrogliosis. J Neurosci. 2012;32:6391–410. doi: 10.1523/JNEUROSCI.6221-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hamby ME, et al. Inflammatory mediators alter the astrocyte transcriptome and calcium signaling elicited by multiple g-protein-coupled receptors. J Neurosci. 2012;32:14489–510. doi: 10.1523/JNEUROSCI.1256-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.John GR, Lee SC, Song X, Rivieccio M, Brosnan CF. IL-1-regulated responses in astrocytes: relevance to injury and recovery. Glia. 2005;49:161–176. doi: 10.1002/glia.20109. [DOI] [PubMed] [Google Scholar]
- 40.Brambilla R, et al. Inhibition of astroglial nuclear factor kappaB reduces inflammation and improves functional recovery after spinal cord injury. J Exp Med. 2005;202:145–156. doi: 10.1084/jem.20041918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Okada S, et al. Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nature Med. 2006;12:829–834. doi: 10.1038/nm1425. [DOI] [PubMed] [Google Scholar]
- 42.Brambilla R, et al. Transgenic inhibition of astroglial NF-kappaB improves functional outcome in experimental autoimmune encephalomyelitis by suppressing chronic central nervous system inflammation. J Immunol. 2009;182:2628–2640. doi: 10.4049/jimmunol.0802954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim RY, et al. Astrocyte CCL2 sustains immune cell infiltration in chronic experimental autoimmune encephalomyelitis. J Neuroimmunol. 2014;274:53–61. doi: 10.1016/j.jneuroim.2014.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moreno M, et al. Conditional ablation of astroglial CCL2 suppresses CNS accumulation of M1 macrophages and preserves axons in mice with MOG peptide EAE. J Neurosci. 2014;34:8175–85. doi: 10.1523/JNEUROSCI.1137-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ko EM, et al. Deletion of astroglial CXCL10 delays clinical onset but does not affect progressive axon loss in a murine autoimmune multiple sclerosis model. J Neuroinflammation. 2014;11:105. doi: 10.1186/1742-2094-11-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Argaw AT, Gurfein BT, Zhang Y, Zameer A, John GR. VEGF-mediated disruption of endothelial CLN-5 promotes blood-brain barrier breakdown. Proc Natl Acad Sci USA. 2009;106:1977–1982. doi: 10.1073/pnas.0808698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Argaw AT, et al. Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. J Clin Invest. 2012;122:2454–68. doi: 10.1172/JCI60842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kang Z, et al. Astrocyte-restricted ablation of interleukin-17-induced Act1-mediated signaling ameliorates autoimmune encephalomyelitis. Immunity. 2010;32:414–25. doi: 10.1016/j.immuni.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xanthos DN, Sandkuhler J. Neurogenic neuroinflammation: inflammatory CNS reactions in response to neuronal activity. Nat Rev Neurosci. 2014;15:43–53. doi: 10.1038/nrn3617. [DOI] [PubMed] [Google Scholar]
- 50.Abbott NJ, Ronnback L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7:41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- 51.Alvarez JI, et al. The Hedgehog pathway promotes blood-brain barrier integrity and CNS immune quiescence. Science. 2011;334:1727–31. doi: 10.1126/science.1206936. [DOI] [PubMed] [Google Scholar]
- 52.Alvarez JI, Katayama T, Prat A. Glial influence on the blood brain barrier. Glia. 2013;61:1939–58. doi: 10.1002/glia.22575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bell RD, et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. 2012;485:512–6. doi: 10.1038/nature11087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Herrmann JE, et al. STAT3 is a critical regulator of astrogliosis and scar formation after spinal cord injury. J Neurosci. 2008;28:7231–7243. doi: 10.1523/JNEUROSCI.1709-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Drogemuller K, et al. Astrocyte gp130 expression is critical for the control of Toxoplasma encephalitis. J Immunol. 2008;181:2683–2693. doi: 10.4049/jimmunol.181.4.2683. [DOI] [PubMed] [Google Scholar]
- 56.Haroon F, et al. Gp130-dependent astrocytic survival is critical for the control of autoimmune central nervous system inflammation. J Immunol. 2011;186:6521–31. doi: 10.4049/jimmunol.1001135. [DOI] [PubMed] [Google Scholar]
- 57.Middeldorp J, Hol EM. GFAP in health and disease. Prog Neurobiol. 2011;93:421–43. doi: 10.1016/j.pneurobio.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 58.Liedtke W, Edelmann W, Chiu FC, Kucherlapati R, Raine CS. Experimental autoimmune encephalomyelitis in mice lacking glial fibrillary acidic protein is characterized by a more severe clinical course and an infiltrative central nervous system lesion. Am J Pathol. 1998;152:251–259. [PMC free article] [PubMed] [Google Scholar]
- 59.Macauley SL, Pekny M, Sands MS. The role of attenuated astrocyte activation in infantile neuronal ceroid lipofuscinosis. J Neurosci. 2011;31:15575–85. doi: 10.1523/JNEUROSCI.3579-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kraft AW, et al. Attenuating astrocyte activation accelerates plaque pathogenesis in APP/PS1 mice. FASEB J. 2013;27:187–98. doi: 10.1096/fj.12-208660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Norden DM, Fenn AM, Dugan A, Godbout JP. TGFbeta produced by IL-10 redirected astrocytes attenuates microglial activation. Glia. 2014;62:881–95. doi: 10.1002/glia.22647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cekanaviciute E, et al. Astrocytic transforming growth factor-beta signaling reduces subacute neuroinflammation after stroke in mice. Glia. 2014;62:1227–40. doi: 10.1002/glia.22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cekanaviciute E, et al. Astrocytic TGF-beta signaling limits inflammation and reduces neuronal damage during central nervous system Toxoplasma infection. J Immunol. 2014;193:139–49. doi: 10.4049/jimmunol.1303284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Min KJ, Yang MS, Kim SU, Jou I, Joe EH. Astrocytes induce hemeoxygenase-1 expression in microglia: a feasible mechanism for preventing excessive brain inflammation. J Neurosci. 2006;26:1880–7. doi: 10.1523/JNEUROSCI.3696-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kostianovsky AM, Maier LM, Anderson RC, Bruce JN, Anderson DE. Astrocytic regulation of human monocytic/microglial activation. J Immunol. 2008;181:5425–5432. doi: 10.4049/jimmunol.181.8.5425. [DOI] [PubMed] [Google Scholar]
- 66.Mizee MR, et al. Astrocyte-derived retinoic acid: a novel regulator of blood-brain barrier function in multiple sclerosis. Acta Neuropathol. 2014;128:691–703. doi: 10.1007/s00401-014-1335-6. [DOI] [PubMed] [Google Scholar]
- 67.Catrysse L, Vereecke L, Beyaert R, van Loo G. A20 in inflammation and autoimmunity. Trends Immunol. 2014;35:22–31. doi: 10.1016/j.it.2013.10.005. [DOI] [PubMed] [Google Scholar]
- 68.Wang X, et al. Astrocytic A20 ameliorates experimental autoimmune encephalomyelitis by inhibiting NF-kappaB- and STAT1-dependent chemokine production in astrocytes. Acta Neuropathol. 2013 doi: 10.1007/s00401-013-1183-9. [DOI] [PubMed] [Google Scholar]
- 69.Shao W, et al. Suppression of neuroinflammation by astrocytic dopamine D2 receptors via alphaB-crystallin. Nature. 2013;494:90–4. doi: 10.1038/nature11748. [DOI] [PubMed] [Google Scholar]
- 70.Ousman SS, et al. Protective and therapeutic role for alphaB-crystallin in autoimmune demyelination. Nature. 2007;448:474–9. doi: 10.1038/nature05935. [DOI] [PubMed] [Google Scholar]
- 71.Steelman AJ, Smith R, 3rd, Welsh CJ, Li J. Galectin-9 protein is upregulated in astrocytes by tumor necrosis factor and promotes encephalitogenic T-cell apoptosis. J Biol Chem. 2013;288:23776–87. doi: 10.1074/jbc.M113.451658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hutchison ER, et al. Evidence for miR-181 involvement in neuroinflammatory responses of astrocytes. Glia. 2013;61:1018–28. doi: 10.1002/glia.22483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Spence RD, Voskuhl RR. Neuroprotective effects of estrogens and androgens in CNS inflammation and neurodegeneration. Front Neuroendocrinol. 2012;33:105–15. doi: 10.1016/j.yfrne.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Spence RD, et al. Neuroprotection mediated through estrogen receptor-{alpha} in astrocytes. Proc Natl Acad Sci U S A. 2011;108:8867–72. doi: 10.1073/pnas.1103833108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Spence RD, et al. Estrogen mediates neuroprotection and anti-inflammatory effects during EAE through ERalpha signaling on astrocytes but not through ERbeta signaling on astrocytes or neurons. J Neurosci. 2013;33:10924–33. doi: 10.1523/JNEUROSCI.0886-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rot A, von Andrian UH. Chemokines in innate and adaptive host defense: basic chemokinese grammar for immune cells. Annu Rev Immunol. 2004;22:891–928. doi: 10.1146/annurev.immunol.22.012703.104543. [DOI] [PubMed] [Google Scholar]
- 77.Allen CD, Okada T, Cyster JG. Germinal-center organization and cellular dynamics. Immunity. 2007;27:190–202. doi: 10.1016/j.immuni.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Aloisi F, Pujol-Borrell R. Lymphoid neogenesis in chronic inflammatory diseases. Nat Rev Immunol. 2006;6:205–17. doi: 10.1038/nri1786. [DOI] [PubMed] [Google Scholar]
- 79.Franciotta D, Salvetti M, Lolli F, Serafini B, Aloisi F. B cells and multiple sclerosis. Lancet Neurol. 2008;7:852–8. doi: 10.1016/S1474-4422(08)70192-3. [DOI] [PubMed] [Google Scholar]
- 80.Peters A, et al. Th17 cells induce ectopic lymphoid follicles in central nervous system tissue inflammation. Immunity. 2011;35:986–96. doi: 10.1016/j.immuni.2011.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lucchinetti CF, et al. Inflammatory cortical demyelination in early multiple sclerosis. N Engl J Med. 2011;365:2188–97. doi: 10.1056/NEJMoa1100648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Metcalf TU, Baxter VK, Nilaratanakul V, Griffin DE. Recruitment and retention of B cells in the central nervous system in response to alphavirus encephalomyelitis. J Virol. 2013;87:2420–9. doi: 10.1128/JVI.01769-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pohl M, et al. T cell-activation in neuromyelitis optica lesions plays a role in their formation. Acta Neuropathol Commun. 2013;1:85. doi: 10.1186/2051-5960-1-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yamagata K, Nakayama C, Suzuki K. Dietary beta-carotene regulates interleukin-1beta-induced expression of apolipoprotein E in astrocytes isolated from stroke-prone spontaneously hypertensive rats. Neurochem Int. 2013;62:43–9. doi: 10.1016/j.neuint.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 85.Mahley RW, Huang Y. Apolipoprotein e sets the stage: response to injury triggers neuropathology. Neuron. 2012;76:871–85. doi: 10.1016/j.neuron.2012.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Halassa MM, Haydon PG. Integrated brain circuits: astrocytic networks modulate neuronal activity and behavior. Annu Rev Physiol. 2010;72:335–355. doi: 10.1146/annurev-physiol-021909-135843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tong X, et al. Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington’s disease model mice. Nat Neurosci. 2014;17:694–703. doi: 10.1038/nn.3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sofroniew MV. Multiple roles for astrocytes as effectors of cytokines and inflammatory mediators. Neuroscientist. 2014;20:160–72. doi: 10.1177/1073858413504466. [DOI] [PubMed] [Google Scholar]
- 89.Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med. 2005;202:473–477. doi: 10.1084/jem.20050304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Roemer SF, et al. Pattern-specific loss of aquaporin-4 immunoreactivity distinguishes neuromyelitis optica from multiple sclerosis. Brain. 2007;130:1194–205. doi: 10.1093/brain/awl371. [DOI] [PubMed] [Google Scholar]
- 91.Saadoun S, et al. Intra-cerebral injection of neuromyelitis optica immunoglobulin G and human complement produces neuromyelitis optica lesions in mice. Brain. 2010;133:349–61. doi: 10.1093/brain/awp309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Howe CL, et al. Neuromyelitis optica IgG stimulates an immunological response in rat astrocyte cultures. Glia. 2014;62:692–708. doi: 10.1002/glia.22635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kitley J, et al. Neuromyelitis Optica Spectrum Disorders With Aquaporin-4 and Myelin-Oligodendrocyte Glycoprotein Antibodies: A Comparative Study. JAMA Neurol. 2014 doi: 10.1001/jamaneurol.2013.5857. [DOI] [PubMed] [Google Scholar]
- 94.Sato DK, et al. Distinction between MOG antibody-positive and AQP4 antibody-positive NMO spectrum disorders. Neurology. 2014 doi: 10.1212/WNL.0000000000000101. online. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hoftberger R, et al. Antibodies to MOG and AQP4 in adults with neuromyelitis optica and suspected limited forms of the disease. Mult Scler. 2014 doi: 10.1177/1352458514555785. pii:1352458514555785. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zamvil SS, Slavin AJ. Does MOG Ig-positive AQP4-seronegative opticospinal inflammatory disease justify a diagnosis of NMO spectrum disorder? Neurology: Neuroimmunology & Neuroinflammation. 2015 doi: 10.1212/NXI.0000000000000062. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Diamond B, Huerta PT, Mina-Osorio P, Kowal C, Volpe BT. Losing your nerves? Maybe it’s the antibodies. Nat Rev Immunol. 2009;9:449–56. doi: 10.1038/nri2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kowarik MC, et al. The cerebrospinal fluid immunoglobulin transcriptome and proteome in neuromyelitis optica reveals central nervous system-specific B cell populations. J Neuroinflammation. 2015;12:19. doi: 10.1186/s12974-015-0240-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Khakh BS, Sofroniew MV. Astrocytes and Huntington’s Disease. ACS Chem Neurosci. 2014;5:494–6. doi: 10.1021/cn500100r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Papadopoulos MC, Bennett JL, Verkman AS. Treatment of neuromyelitis optica: state-of-the-art and emerging therapies. Nat Rev Neurol. 2014;10:493–506. doi: 10.1038/nrneurol.2014.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Srivastava R, et al. Potassium channel KIR4.1 as an immune target in multiple sclerosis. N Engl J Med. 2012;367:115–23. doi: 10.1056/NEJMoa1110740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Brickshawana A, et al. Investigation of the KIR4.1 potassium channel as a putative antigen in patients with multiple sclerosis: a comparative study. Lancet Neurol. 2014;13:795–806. doi: 10.1016/S1474-4422(14)70141-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ngo ST, Steyn FJ, McCombe PA. Gender differences in autoimmune disease. Front Neuroendocrinol. 2014;35:347–69. doi: 10.1016/j.yfrne.2014.04.004. [DOI] [PubMed] [Google Scholar]
- 104.Voskuhl R. Sex differences in autoimmune diseases. Biol Sex Differ. 2011;2:1. doi: 10.1186/2042-6410-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Stephan AH, Barres BA, Stevens B. The complement system: an unexpected role in synaptic pruning during development and disease. Annu Rev Neurosci. 2012;35:369–89. doi: 10.1146/annurev-neuro-061010-113810. [DOI] [PubMed] [Google Scholar]
- 106.Clarke LE, Barres BA. Emerging roles of astrocytes in neural circuit development. Nat Rev Neurosci. 2013;14:311–21. doi: 10.1038/nrn3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Rappold PM, Tieu K. Astrocytes and therapeutics for Parkinson’s disease. Neurotherapeutics. 2010;7:413–23. doi: 10.1016/j.nurt.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Failli V, et al. Functional neurological recovery after spinal cord injury is impaired in patients with infections. Brain. 2012;135:3238–50. doi: 10.1093/brain/aws267. [DOI] [PubMed] [Google Scholar]
- 109.Gold R, Linington C, Lassmann H. Understanding pathogenesis and therapy of multiple sclerosis via animal models: 70 years of merits and culprits in experimental autoimmune encephalomyelitis research. Brain. 2006;129:1953–71. doi: 10.1093/brain/awl075. [DOI] [PubMed] [Google Scholar]
- 110.Hansen RR, Malcangio M. Astrocytes-Multitaskers in chronic pain. Eur J Pharmacol. 2013;716:120–128. doi: 10.1016/j.ejphar.2013.03.023. [DOI] [PubMed] [Google Scholar]
- 111.Paradise MB, Naismith SL, Norrie LM, Graeber MB, Hickie IB. The role of glia in late-life depression. Int Psychogeriatr. 2012;24:1878–90. doi: 10.1017/S1041610212000828. [DOI] [PubMed] [Google Scholar]
- 112.Martin JL, Magistretti PJ, Allaman I. Regulation of Neurotrophic Factors and Energy Metabolism by Antidepressants in Astrocytes. Curr Drug Targets. 2013 doi: 10.2174/1389450111314110009. [DOI] [PubMed] [Google Scholar]
- 113.Czeh B, Di Benedetto B. Antidepressants act directly on astrocytes: evidences and functional consequences. Eur Neuropsychopharmacol. 2013;23:171–85. doi: 10.1016/j.euroneuro.2012.04.017. [DOI] [PubMed] [Google Scholar]
- 114.Chung WS, et al. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature. 2013;504:394–400. doi: 10.1038/nature12776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Miller DB, O’Callaghan JP. Depression, cytokines, and glial function. Metabolism. 2005;54:33–8. doi: 10.1016/j.metabol.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 116.Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9:46–56. doi: 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Dowlati Y, et al. A meta-analysis of cytokines in major depression. Biol Psychiatry. 2010;67:446–57. doi: 10.1016/j.biopsych.2009.09.033. [DOI] [PubMed] [Google Scholar]
- 118.Irwin MR, Cole SW. Reciprocal regulation of the neural and innate immune systems. Nature Reviews Immunology. 2011;11:625–632. doi: 10.1038/nri3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bauer S, Kerr BJ, Patterson PH. The neuropoietic cytokine family in development, plasticity, disease and injury. Nat Rev Neurosci. 2007;8:221–32. doi: 10.1038/nrn2054. [DOI] [PubMed] [Google Scholar]
- 120.Deverman BE, Patterson PH. Cytokines and CNS development. Neuron. 2009;64:61–78. doi: 10.1016/j.neuron.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 121.Patterson PH. Maternal infection and autism. Brain Behav Immun. 2012;26:393. doi: 10.1016/j.bbi.2011.09.008. [DOI] [PubMed] [Google Scholar]
- 122.Brenner M, et al. Mutations in GFAP, encoding glial fibrillary acidic protein, are associated with Alexander disease. Nat Genet. 2001;27:117–120. doi: 10.1038/83679. [DOI] [PubMed] [Google Scholar]
- 123.Messing A, Brenner M, Feany MB, Nedergaard M, Goldman JE. Alexander disease. J Neurosci. 2012;32:5017–23. doi: 10.1523/JNEUROSCI.5384-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lobsiger CS, Cleveland DW. Glial cells as intrinsic components of non-cell-autonomous neurodegenerative disease. Nat Neurosci. 2007;10:1355–1360. doi: 10.1038/nn1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Nagai M, et al. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci. 2007;10:615–622. doi: 10.1038/nn1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Yamanaka K, et al. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci. 2008;11:251–253. doi: 10.1038/nn2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Myer DJ, Gurkoff GG, Lee SM, Hovda DA, Sofroniew MV. Essential protective roles of reactive astrocytes in traumatic brain injury. Brain. 2006;129:2761–2772. doi: 10.1093/brain/awl165. [DOI] [PubMed] [Google Scholar]
- 128.Sahni V, et al. BMPR1a and BMPR1b signaling exert opposing effects on gliosis after spinal cord injury. J Neurosci. 2010;30:1839–55. doi: 10.1523/JNEUROSCI.4459-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Shimada IS, Borders A, Aronshtam A, Spees JL. Proliferating reactive astrocytes are regulated by Notch-1 in the peri-infarct area after stroke. Stroke. 2011;42:3231–7. doi: 10.1161/STROKEAHA.111.623280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Shimada IS, Lecomte MD, Granger JC, Quinlan NJ, Spees JL. Self-renewal and differentiation of reactive astrocyte-derived neural stem/progenitor cells isolated from the cortical peri-infarct area after stroke. J Neurosci. 2012;32:7926–40. doi: 10.1523/JNEUROSCI.4303-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Meeuwsen S, Persoon-Deen C, Bsibsi M, Ravid R, van Noort JM. Cytokine, chemokine and growth factor gene profiling of cultured human astrocytes after exposure to proinflammatory stimuli. Glia. 2003;43:243–53. doi: 10.1002/glia.10259. [DOI] [PubMed] [Google Scholar]
- 132.Jensen CJ, Massie A, De Keyser J. Immune players in the CNS: the astrocyte. J Neuroimmune Pharmacol. 2013;8:824–39. doi: 10.1007/s11481-013-9480-6. [DOI] [PubMed] [Google Scholar]
- 133.Cooley ID, Chauhan VS, Donneyz MA, Marriott I. Astrocytes produce IL-19 in response to bacterial challenge and are sensitive to the immunosuppressive effects of this IL-10 family member. Glia. 2014;62:818–28. doi: 10.1002/glia.22644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Pitter KL, et al. The SHH/Gli pathway is reactivated in reactive glia and drives proliferation in response to neurodegeneration-induced lesions. Glia. 2014;62:1595–607. doi: 10.1002/glia.22702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Sarafian TA, et al. Disruption of astrocyte STAT3 signaling decreases mitochondrial function and increases oxidative stress in vitro. PLoS One. 2010;5:e9532. doi: 10.1371/journal.pone.0009532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hong P, Jiang M, Li H. Functional requirement of dicer1 and miR-17-5p in reactive astrocyte proliferation after spinal cord injury in the mouse. Glia. 2014;62:2044–60. doi: 10.1002/glia.22725. [DOI] [PubMed] [Google Scholar]
- 137.Krumbholz M, et al. BAFF is produced by astrocytes and up-regulated in multiple sclerosis lesions and primary central nervous system lymphoma. J Exp Med. 2005;201:195–200. doi: 10.1084/jem.20041674. [DOI] [PMC free article] [PubMed] [Google Scholar]