

Abstract
Although Prader-Willi syndrome (PWS) is a well-described clinical dysmorphic syndrome, DNA testing is required for a definitive diagnosis. A definitive diagnosis can be made in approximately 99% of cases using DNA testing; there are a number of DNA tests that can be used for this purpose, although there is no set standard algorithm of testing. The dilemma arises because of the complex genetic mechanisms at the basis of PWS, which need to be elucidated. To establish the molecular mechanism with a complete work up, involves at least 2 tests. Here we discuss the commonly used tests currently available and suggest a cost—effective approach to diagnostic testing.
Keywords: Cytogenetics, chromosome microarray, methylation
Introduction
Prader-Willi syndrome (PWS) is a well described clinical dysmorphic syndrome but due to some overlap of features with both normal obese individuals and those with other developmental handicaps (1-5), a definitive diagnosis on clinical grounds alone is uncertain and requires DNA testing. There are various molecular genetic tests available and DNA testing is approximately (~)99% accurate for the diagnosis of PWS—this ranks as one of the best percentage accuracy of testing for any syndromic genetic condition. Thus, while the actual diagnosis is straightforward on the whole, the dilemma arises due to the complexity of establishing the molecular mechanism. It is necessary to establish the mechanism as this will provide information on the possible clinical features (4), the prognosis (6-8) and the recurrence risk (9,10). While there are various molecular tests available which can establish the mechanism, there is no set standard algorithm of testing; different laboratories use different approaches and often their own set pattern of testing, making the interpretation of test results difficult for clinicians. Here, the aim is to lay out the various possible tests currently in use and show the value of each one individually and in combination, to provide a useful guide for clinicians in understanding the genetic scenario of PWS diagnostic testing.
This discussion does not include a clinical appraisal of PWS, first described in 1956 by Prader et al. (11), which has been given in many previous publications since (1,4,12-15). The basic defect is in the malfunctioning of the hypothalamic nuclei controlling appetite. PWS occurs in approximately one in 20,000 live births (16) and occurs equally in males and females and in all ethnic groups. Angelman syndrome with a different phenotype, paternal imprinting, similar genetic mechanisms but extended to include a known gene (UBE3A) (17) is not considered in detail, but mentioned where it impinges on the interpretation of genetic testing for PWS.
The genetic abnormality in PWS
The genetic region for PWS on chromosome 15 was first recognised through chromosomal rearrangements involving the centromeric region of chromosome 15 (18). Since then, the critical region for PWS has been established to lie within chromosome 15(q11-13) [OMIM No. 176270] a region spanning 4–6 Mb (Figure 1). A number of genes have been mapped within this region, the critical one for PWS is SNURF/SNRPN gene, which contains 10 exons, many snoRNAs, the imprinting centre (IC) and the open reading frame (ORF) of the gene. Of these, exon 1 is critical for imprinting (10,19-20) and SNORD 116 RNA is important for many of the clinical features of PWS (21,22) (Figure 1). PWS is maternally imprinted and is caused by loss of paternal alleles within this region (9,23). However, as yet no abnormal gene product has been identified which could be the cause of PWS. SNRPN is a small nuclear ribonucleoprotein involved in alternate mRNA splicing. The critical region for Angelman Syndrome (AS) lies within this same 4–6 Mb region, but AS is caused by loss of maternal alleles (17).
Figure 1.

The critical region for PWS on chromosome 15, with the main genes indicated. MKRN3 (or ZNF127) is a zinc finger protein, expressed only from the paternal chromosome; MAGEL2 is expressed only from the paternal chromosome mainly in the brain; NECDIN encodes a DNA binding protein; C15ORF2 is the open reading frame of the SNURF/SNRPN gene; MAGEL2, NDN and MKRN3 are all small intronless genes. Black lines and light blue ovals between BP2 and BP3 indicate imprinted genes in PWS, Black lines between BP1 and BP2 indicate non-imprinted genes and the 2 yellow circles are the maternally imprinted genes in Angelman syndrome. Exons 1-10 are within SNRPN; snoRNAs are here depicted pictorially. IPW, an RNA transcript lies within the snoRNA region, does not encode a protein but is paternally expressed only; SNORD116 also lies within the snoRNA region and is paternally expressed only. BP, breakpoint; Cen, centromere; tel, telomere.
The genetic mechanisms leading to PWS are 3 fold—either there is a deletion of the critical region (~75% of patients), uniparental disomy (UPD) (~25%) or an imprinting centre (IC) defect (~1% of PWS) (9,19,20,24) (Figure 2). These mechanisms can be arrived at by occurring de novo or as a result of a structural abnormality, such as a translocation, ring formation, isochromosome, or inversion, involving chromosomes 15 alone or with some other chromosome. A rearrangement occurs in ~5% of individuals with PWS (25). These structural rearrangements may be balanced or unbalanced, the commonest being Robertsonian translocations (14;15) and (15;15) (25-30).
Figure 2.

The three molecular classes of PWS. Blue chromosome is paternal, pink is maternal. The yellow circle represents the 4–6 Mb critical region for PWS. The open circles indicate an active gene, the red cross an inactive (imprinted) gene. The black box indicates an IC defect. PWS, Prader-Willi syndrome; CI, imprinting centre.
Deletions
The PWS chromosome critical region spans 4–6 Mb and deletions within this region may be large, small or microdeletions. Large deletions of approximately 4–6 Mb have two major proximal breakpoints but a consistent single distal breakpoint (4,31). Class I deletions extend from D15S541 to D15S12 (BP1 to BP3) and class II from D15S543 to D15S12 (BP2 to BP3)—both take out the IC. (Figure 1). These deletions are considered collectively as the common deletion; there are subtle phenotypic difference between them (32); they are present in approximately equal proportions, and are the mechanism of PWS in ~75% of patients; there is no report of recurrence (4,19). Small deletions are those smaller than 4 Mb; their effect is similar to a large deletion and there is also no report of recurrence. Microdeletions are within the SNRPN gene and span from 6–250 kb DNA, with exon 1 deleted (20,33). These are sometimes referred to as IC mutations (or IC defects) (see below) and have a potentially high (50%) recurrence risk (10,34).
UPD
The chromosome count is normal, with normal appearing chromosomes 15—band for band—but both have come from the mother—there is no paternal input. UPD (mat) in PWS was first described in 1989 (24) and while initially thought to be rare, accounts for ~25% of individuals with PWS. In UPDmat, the commonest mechanism is frequently referred to as a trisomy rescue—there are two abnormal events, the first is meiotic, resulting in a zygote with trisomy 15 and the second is somatic after fertilisation with loss of the paternal chromosome 15. Heterodisomy occurs when the 2 chromosomes 15 in the trisomy 15 zygote are different; isodisomy is the term used when they are the same. Advanced maternal age is correlated with UPD as the first error is a maternal non disjunction event, leading to maternal meiotic trisomy 15 (35). There are other rarer chromosome breakage and segregation abnormalities, which can result in UPDmat (30).
Imprinting centre defect (ICD)
Essentially ICD (also referred to as IC mutations) are microdeletions within SNRPN, involving exon 1. The size of these deletions varies only a little, from exon 1–10. DNA testing is the only way to detect ICD, and this is usually performed in a specialised referral laboratory (20,33). Fortunately, ICD occur rarely (~1% of individuals with PWS), as this testing may take some time and may involve another sample from the patient and one from each parent. If the mechanism of the common 4–6 Mb deletion or UPD is established, it is unnecessary to look for an ICD; otherwise, it is necessary, due to the high recurrence risk (50%), if it is found to be inherited. Testing for an IC deletion can be done by sequence analysis at the smallest region of overlap (SRO) for the PWS IC region (~4.3 kb) (33,36).
Absence of the paternally derived SNORD116 gene cluster has been described as a rare cause of PWS with multiple clinical features as seen in typical PWS (37). Most (85%) of the imprinting centre defects are due to epimutations (i.e., two parental alleles but only maternal DNA methylation pattern), with small recurrence risk. The remaining ~15% of PWS with imprinting centre defects are found to have very small deletions in the IC region (7.5~>100 kb) with recurrence risk as high as 50% if the IC deletion is inherited from an unaffected father (10,23,24).
The dilemma for diagnostic genetic laboratories
There are 4 issues to consider:
To separate out the few patients with PWS from the great majority of laboratory referrals falling within the differential diagnosis, at birth and later (1,3,38,39);
Having diagnosed PWS, to separate out those few individuals with a high recurrence risk (an ICD) from the much larger number with a very low recurrence risk (deletions and UPDmat);
To make the testing as “user-friendly” as possible—it is notoriously difficult to obtain blood, or other tissue, from the patient, and family is not always available;
To keep the cost of testing to a minimum.
Tests commonly utilised at present
Cytogenetics
Cytogenetics after setting up tissue culture (usually from a blood sample) chromosomes are released from their cells (hypotonic and colchicine treatment), spread on a slide (cytogenetic harvest), and banding techniques applied which can identify each chromosome accurately band for band to a level of resolution of 5–10 Mb. This is a useful test to check for structural rearrangements (e.g., Robertsonian translocations or small supernumerary chromosome 15 which are associated with ~5% of UPD) (30,36,40); it is not useful for the detection of deletions or UPD (40).
Fluorescence in-situ hybridisation (FISH)
FISH utilises a fluorescence microscope to visualise with a bright signal whether a probe hybridises to an homologous sequence or not—i.e., deleted or not deleted (Figure 3). Several probes from within the critical region can be used for FISH studies (e.g., SNRPN & D15S10). FISH, with the appropriate probes, is a useful way to differentiate type 1 from type 2 deletions. These probes can be applied to chromosome spreads prepared after cell culture or directly to smears, which are whole cells on a slide without culture. FISH can be performed on cells in the suspension remaining from the cytogenetic harvest, retained in fixative at –20 °C (41). Thus, slides can be made later, using the same suspension, which can be useful for checking on some other result e.g., microarray. FISH on chromosome spreads effectively can characterise a balanced structural abnormality and is the test of choice for this purpose. FISH cannot be used to pick up UPD and will not distinguish between PWS and AS deletions.
Figure 3.
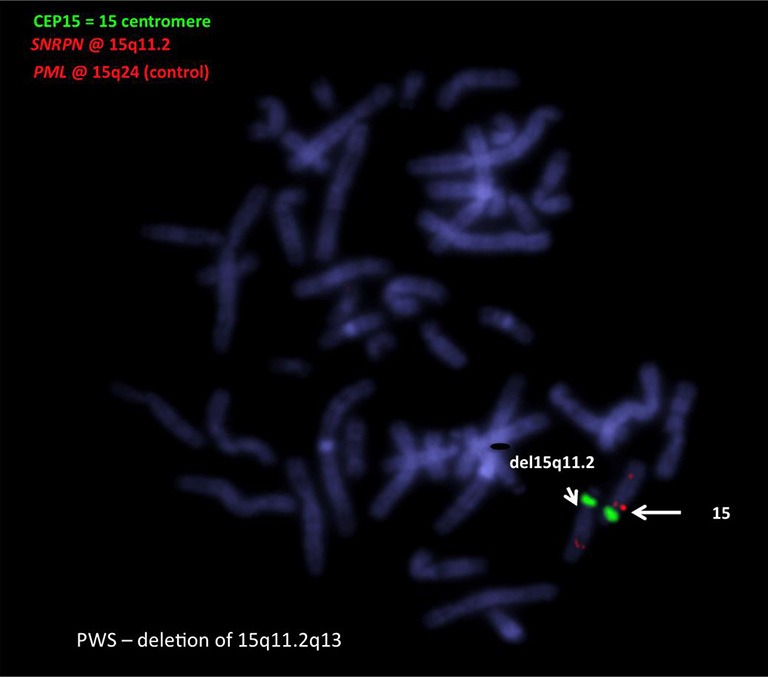
A FISH picture of deletion: the deleted chromosome has no red signal under the centromere (green). The PML red signal is an extra control for the terminal end of chromosome 15. FISH, fluorescence in-situ hybridisation; FISH, fluorescence in-situ hybridisation; PML, promyelocytic leukemia.
Methylation polymerase chain reaction (PCR)
Methylation PCR can diagnose PWS unequivocally when the abnormal pattern is present, and can also exclude AS (Figure 4). Methylation does not define the PWS molecular mechanism, as it is abnormal in all 3 scenarios—the common large and small deletions, UPD and IC defects (42,43). When an abnormal methylation result is shown, further testing is required to indicate the mechanism. This further testing is commonly done with FISH—a single probe will detect a deletion; if the FISH result is normal i.e., not deleted, DNA testing is required to show UPD. If UPD is not shown, an imprinting defect is suspected, and further special testing is necessary if the IC defect is to be characterised. At present >95% of IC defects are microdeletions within SNRPN (33). Rare patients have been reported who have PWS microdeletion by chromosome rearrangement (28,29).
Figure 4.

Methylation-PCR DNA was modified by bisulfite treatment and the SNRPN exon 1 region was amplified by PCR. Normal control has both the maternal (313 bp) and paternal (221 bp) bands. Only the maternal band is visible in a Prader-Willi patient and the paternal band is present in an Angelman patient. PCR, polymerase chain reaction.
Comparative genomic hybridisation (CGH) microarray
CGH microarray compares the DNA of a patient with a normal control. It can be either a CGH array (detects copy number change only) or CGH + single nucleotide polymorphism (SNP) array which can detect both copy number change as well as single nucleotide polymorphism change. Both can detect deletions of all sizes and unbalanced chromosome rearrangements (Figure 5). The whole genome is visible with microarrays, not just the PWS region, so that other abnormalities elsewhere in the genome can be detected. The precise deletion size and its corresponding genomic content may be important for genotype-phenotype correlations. CMA combined with SNPs (CGH + SNP array) can detect approximately 75% of UPDs by demonstrating long (>13.5 Mb) contiguous stretches of homozygosity limited to the chromosome 15 s (44). In these cases DNA polymorphism analysis is probably not necessary to diagnose UPD. One of the limitations of CGH + SNP is that it cannot detect all cases of uniparental heterodisomy UPD or a balanced chromosome rearrangement (5% of patients) (45). Furthermore, interpretation of the microarray result may be inaccurate if the patient has a homozygous mutation or mosaicism (46), both quite rare in PWS.
Figure 5.

CGH chromosome microarray with type 1 deletion [between BP1–BP3 (GRCh37 chr15:22,749,354-28,438,266)], the CGH data showing Log2ratio “−1” between chromosome 15q11. 2q13. 1 regions indicating a heterozygous deletion. CGH + SNP chromosome microarray with type 2 deletion [between BP2-BP3 (GRCh37 chr15:23,619,912-28,438,266)], the CGH data showing Log2 ratio “−1” between chromosome 15q11. 2q13. 1 region indicating a heterozygous deletion. The SNP data also showed loss of one allele [displaying only the 0-uncut (AA) & 1-uncut allele (AB) track]. (A) CGH microarray-Type1 deletion; (B) CGH + SNP Microarray-Type2 deletion.
Methylation sensitive multiplex ligation-dependent probe amplification (MS-MLPA)
Each MS-MLPA reaction generates two samples that require analysis by capillary electrophoresis:
One is an undigested sample to check for copy number change and the other is a digested sample (with HhaI endonuclease enzyme) for methylation detection. MS-MLPA can detect copy number changes as well as determining methylation status of the 15q11 region (47). It can also pick up small imprinting centre (IC) deletions and microdeletions within the SNORD116 cluster (Figure 6). MS-MLPA will detect UPD, but cannot definitively distinguish UPD from imprinting centre mutation (epigenetic mechanism). These results are depicted in MRC-Holland product description: https://www.mlpa.com/WebForms/WebFormProductDetails. aspx?Tag=_tz2fAPIAupKyMjaDF-E-t9bmuxqlhe_Lgqfk8Hkjuss. &ProductOID=_HukvlkjzRho.
Figure 6.

MS-MLPA showing normal control, PWS large common deletion, IC deletion and UPD.
RNA
The measurement of the expression of the SNRPN gene is another approach and considered by some to be more important than the actual presence of an abnormality (48). This is a complicated growing area as there are many RNA transcripts (with no apparent protein coding function) apart from the classical messenger RNA. There are 3 non coding RNA (ncRNA) of importance in the PWS region (HB13, 52 and 85) (Figure 1)—these may not be translated because the RNA binds to other molecules, or is methylated. Starting with an RNA test (not shown), lack of gene expression will diagnose PWS, but there is still the need to establish the mechanism and a cytogenetic analysis is also required to check for a balanced rearrangement. RNA extraction may be unreliable and extracted RNA unstable, so that this approach is not commonly used for diagnosis.
Testing algorithms
A comprehensive, definitive diagnosis requires a minimum of two tests, and depending on the choice of tests, these same two tests could exclude a balanced rearrangement. These two tests could be performed from one given specimen, or from 2 specimens taken at different points in time. If the specimen was sufficiently large, e.g., 10–15 mLs, then it could be split—part for a cytogenetic karyotype analysis or FISH, looking for a balanced chromosome rearrangement and the other part for DNA studies.
Starting with the DNA methylation test (from DNA extraction) a diagnosis will be made immediately, and AS excluded, but further testing is required for the mechanism. Frequently, FISH is the next test, for the common 4–6 Mb deletion as the mechanism, (from cells on a microscope slide, either from a smear or cell culture); this will cover 75% of patients. If the FISH test is normal, then proceed to DNA studies for UPD; this entails going back to the original specimen, or obtaining another specimen, or using the cell suspension remaining from the cytogenetic harvest and extracting DNA; then also obtaining blood from the parents (4,49).
Starting with a microarray, a deletion will be detected immediately but will not distinguish PWS from AS. A CGH + SNP microarray can detect UPD by loss of heterozygosity, but cannot determine the parent of origin for UPD unless a trio-analysis of the proband and both parents are performed simultaneously. However, if the UPD15 has resulted from a maternal uniparental heterodisomy (trisomy rescue) then, a trio-analysis would not be useful. Microsatellite analysis in the patient and parents are still required. However, a CGH array has an advantage in that it will detect abnormality across the whole genome.
Starting with MS-MLPA, a definitive diagnosis can be achieved in one test for ~75% of patients (Figure 6). The diagnosis plus deletion is achieved without the need for parental DNA and can distinguish AS from PWS, detecting all types of deletion (4,15). The limitation is that cases with UPD require another molecular test (either CGH + SNP array to detect long contiguous stretches of homozygosity, or DNA polymorphism studies by microsatellite analysis). The parents are still required to check for UPD. Thus, MS-MLPA cannot discriminate between UPD and IC epimutation. MS-MLPA assay does not detect copy number changes across the entire genome and it also cannot detect balanced rearrangements. Due to the rarity of ICD and balanced rearrangements in PWS, some centres consider it sufficient to diagnose PWS and establish deletion as the mechanism (50).
Cost
The cost of these various tests varies from one laboratory or genetic service to another. Some will charge for a complete diagnosis, others charge for each step of the process (e.g., DNA extraction, a cytogenetic harvest, slide making, cytogenetic analysis) so that a useful comparison cannot be made. The less tests required, the more cost effective the diagnosis becomes. Retesting after an incomplete diagnosis is costly (51). As a baseline for comparison (52), in our genetic service in Sydney, Australia, the cost of a cytogenetic karyotype is $A365.00; a CMA is $A365.00; FISH test is $A175.00; methylation test is $A300.00 and MS-MLPA is $A300.00. A charge can be made for one or the other of cytogenetic karyotype or CMA but not both, as the CMA is classified as a molecular karyotype. The patient may not need to pay for all this cost, as the Medicare scheme in Australia will cover a percentage of the cost (but not for the methylation or MS-MLPA) and some families have private health care cover. However, apart from the testing itself, there is considerable paperwork involved in tracking the case for the laboratory scientist, and a secretarial cost should also be added.
Thus, a comprehensive laboratory diagnosis remains a dilemma; in Figure 7, we present a pathway which will definitely diagnose and elicit the molecular mechanism of PWS in a cost-effective and timely manner, with MS-MLPA as the first test. This will be valid for the majority of patients (75% deletion). The compromise is that if UPD studies are to be proven and not just assumed when no deletion is found, and a balanced rearrangement is also to be excluded, further testing is necessary and may involve another specimen and the parents.
Figure 7.

Proposed testing strategy to diagnose and establish the molecular class in PWS using MS-MLPA as the first procedure.
Acknowledgements
The authors would like to thank the Nucleic Acid Unit for DNA extractions, the Molecular Genetics and the Cytogenetics Department within Sydney Genome diagnostics at The Children’s Hospital at Westmead for supporting the PWS/AS methylation training and chromosome microarray work performed by the author.
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Zellweger H. Differential Diagnosis in Prader-Willi Syndrome. In: Greenswag LR, Alexander RC, editors. Management of Prader-Willi Syndrome. Springer US;1988:15-22. [Google Scholar]
- 2.Schinzel A, Robinson WP, Bottani A, et al. Prader-Willi or Angelman syndrome in familial 15q11----q13 deletion of maternal origin? Hum Genet 1992;88:361-2. 10.1007/BF00197279 [DOI] [PubMed] [Google Scholar]
- 3.Smith A, Jauch A, Slater H, et al. Syndromal obesity due to paternal duplication 6(q24.3-q27). Am J Med Genet 1999;84:125-31. 10.1002/(SICI)1096-8628(19990521)84:2<125::AID-AJMG8>3.0.CO;2-W [DOI] [PubMed] [Google Scholar]
- 4.Cassidy SB, Schwartz S, Miller JL, et al. Prader-Willi syndrome. Genet Med 2012;14:10-26. 10.1038/gim.0b013e31822bead0 [DOI] [PubMed] [Google Scholar]
- 5.Hashemi B, Bassett A, Chitayat D, et al. Deletion of 15q11.2(BP1-BP2) region: further evidence for lack of phenotypic specificity in a pediatric population. Am J Med Genet A 2015;167A:2098-102. 10.1002/ajmg.a.37134 [DOI] [PubMed] [Google Scholar]
- 6.Boer H, Holland A, Whittington J, et al. Psychotic illness in people with Prader Willi syndrome due to chromosome 15 maternal uniparental disomy. Lancet 2002;359:135-6. 10.1016/S0140-6736(02)07340-3 [DOI] [PubMed] [Google Scholar]
- 7.Stauder JE, Boer H, Gerits RH, et al. Differences in behavioural phenotype between parental deletion and maternal uniparental disomy in Prader-Willi syndrome: an ERP study. Clin Neurophysiol 2005;116:1464-70. 10.1016/j.clinph.2005.02.019 [DOI] [PubMed] [Google Scholar]
- 8.Sinnema M, Schrander-Stumpel CT, Maaskant MA, et al. Aging in Prader-Willi syndrome: twelve persons over the age of 50 years. Am J Med Genet A 2012;158A:1326-36. 10.1002/ajmg.a.35333 [DOI] [PubMed] [Google Scholar]
- 9.Nicholls RD, Saitoh S, Horsthemke B. Imprinting in Prader-Willi and Angelman syndromes. Trends Genet 1998;14:194-200. 10.1016/S0168-9525(98)01432-2 [DOI] [PubMed] [Google Scholar]
- 10.Buiting K, Cassidy SB, Driscoll DJ, et al. Clinical utility gene card for: Prader-Willi Syndrome. Eur J Hum Genet 2014;22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Prader A, Labhart A, Willi H. Ein Syndrom von Adipositas, Kleinwuchs, Kryptorchismus und Oligophrenie nach myatonie-artigem Zustand im Neugeborenalter. Schweiz Med Wochenschr 1956;86:1260-1. [Google Scholar]
- 12.Gunay-Aygun M, Schwartz S, Heeger S, et al. The changing purpose of Prader-Willi syndrome clinical diagnostic criteria and proposed revised criteria. Pediatrics 2001;108:E92. 10.1542/peds.108.5.e92 [DOI] [PubMed] [Google Scholar]
- 13.Holm VA, Cassidy SB, Butler MG, et al. Prader-Willi syndrome: consensus diagnostic criteria. Pediatrics 1993;91:398-402. [PMC free article] [PubMed] [Google Scholar]
- 14.Smith A. The disability associated with Prader-Willi syndrome. Austr Disab Review 1996;4:29-35. [Google Scholar]
- 15.Goldstone AP, Holland AJ, Hauffa BP, et al. Recommendations for the diagnosis and management of Prader-Willi syndrome. J Clin Endocrinol Metab 2008;93:4183-97. 10.1210/jc.2008-0649 [DOI] [PubMed] [Google Scholar]
- 16.Smith A, Egan J, Ridley G, et al. Birth prevalence of Prader-Willi syndrome in Australia. Arch Dis Child 2003;88:263-4. 10.1136/adc.88.3.263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clayton-Smith J, Laan L. Angelman syndrome: a review of the clinical and genetic aspects. J Med Genet 2003;40:87-95. 10.1136/jmg.40.2.87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hawkey CJ, Smithies A. The Prader-Willi syndrome with a 15/15 translocation. Case report and review of the literature. J Med Genet 1976;13:152-7. 10.1136/jmg.13.2.152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buiting K, Saitoh S, Gross S, et al. Inherited microdeletions in the Angelman and Prader-Willi syndromes define an imprinting centre on human chromosome 15. Nat Genet 1995;9:395-400. 10.1038/ng0495-395 [DOI] [PubMed] [Google Scholar]
- 20.Buiting K, Barnicoat A, Lich C, et al. Disruption of the bipartite imprinting center in a family with Angelman syndrome. Am J Hum Genet 2001;68:1290-4. 10.1086/320120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sahoo T, del Gaudio D, German JR, et al. Prader-Willi phenotype caused by paternal deficiency for the HBII-85 C/D box small nucleolar RNA cluster. Nat Genet 2008;40:719-21. 10.1038/ng.158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bervini S, Herzog H. Mouse models of Prader-Willi Syndrome: a systematic review. Front Neuroendocrinol 2013;34:107-19. 10.1016/j.yfrne.2013.01.002 [DOI] [PubMed] [Google Scholar]
- 23.Nicholls RD. New insights reveal complex mechanisms involved in genomic imprinting. Am J Hum Genet 1994;54:733-40. [PMC free article] [PubMed] [Google Scholar]
- 24.Nicholls RD, Knoll JH, Glatt K, et al. Restriction fragment length polymorphisms within proximal 15q and their use in molecular cytogenetics and the Prader-Willi syndrome. Am J Med Genet 1989;33:66-77. 10.1002/ajmg.1320330109 [DOI] [PubMed] [Google Scholar]
- 25.Smith A, Lindeman R, Volpato F, et al. A de novo unbalanced reciprocal translocation identified as paternal in origin in the Prader-Willi syndrome. Hum Genet 1991;86:534-6. 10.1007/BF00194651 [DOI] [PubMed] [Google Scholar]
- 26.Robinson WP, Binkert F, Giné R, et al. Clinical and molecular analysis of five inv dup(15) patients. Eur J Hum Genet 1993;1:37-50. [DOI] [PubMed] [Google Scholar]
- 27.Reeve A, Norman A, Sinclair P, et al. True telomeric translocation in a baby with the Prader-Willi phenotype. Am J Med Genet 1993;47:1-6. 10.1002/ajmg.1320470102 [DOI] [PubMed] [Google Scholar]
- 28.Sun Y, Nicholls RD, Butler MG, et al. Breakage in the SNRPN locus in a balanced 46,XY,t(15;19) Prader-Willi syndrome patient. Hum Mol Genet 1996;5:517-24. 10.1093/hmg/5.4.517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuslich CD, Kobori JA, Mohapatra G, et al. Prader-Willi syndrome is caused by disruption of the SNRPN gene. Am J Hum Genet 1999;64:70-6. 10.1086/302177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yip MY. Uniparental disomy in Robertsonian translocations: strategies for uniparental disomy testing. Transl Pediatr 2014;3:98-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Amos-Landgraf JM, Ji Y, Gottlieb W, et al. Chromosome breakage in the Prader-Willi and Angelman syndromes involves recombination between large, transcribed repeats at proximal and distal breakpoints. Am J Hum Genet 1999;65:370-86. 10.1086/302510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Varela MC, Kok F, Setian N, et al. Impact of molecular mechanisms, including deletion size, on Prader-Willi syndrome phenotype: study of 75 patients. Clin Genet 2005;67:47-52. 10.1111/j.1399-0004.2005.00377.x [DOI] [PubMed] [Google Scholar]
- 33.Horsthemke B, Buiting K. Genomic imprinting and imprinting defects in humans. Adv Genet 2008;61:225-46. 10.1016/S0065-2660(07)00008-9 [DOI] [PubMed] [Google Scholar]
- 34.Ming JE, Blagowidow N, Knoll JH, et al. Submicroscopic deletion in cousins with Prader-Willi syndrome causes a grandmatrilineal inheritance pattern: effects of imprinting. Am J Med Genet 2000;92:19-24. 10.1002/(SICI)1096-8628(20000501)92:1<19::AID-AJMG4>3.0.CO;2-# [DOI] [PubMed] [Google Scholar]
- 35.Robinson WP, Langlois S, Schuffenhauer S, et al. Cytogenetic and age-dependent risk factors associated with uniparental disomy 15. Prenat Diagn 1996;16:837-44. 10.1002/(SICI)1097-0223(199609)16:9<837::AID-PD956>3.0.CO;2-7 [DOI] [PubMed] [Google Scholar]
- 36.Robinson WP, Wagstaff J, Bernasconi F, et al. Uniparental disomy explains the occurrence of the Angelman or Prader-Willi syndrome in patients with an additional small inv dup(15) chromosome. J Med Genet 1993;30:756-60. 10.1136/jmg.30.9.756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Duker AL, Ballif BC, Bawle EV, et al. Paternally inherited microdeletion at 15q11.2 confirms a significant role for the SNORD116 C/D box snoRNA cluster in Prader-Willi syndrome. Eur J Hum Genet 2010;18:1196-201. 10.1038/ejhg.2010.102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dulka S, Choudhary AK, Methratta S, et al. Blount disease in a case of Prader-Willi syndrome: why is it not as prevalent as in obesity without Prader-Willi syndrome? World J Pediatr 2013;9:182-4. 10.1007/s12519-011-0315-3 [DOI] [PubMed] [Google Scholar]
- 39.Forsythe E, Beales PL. Bardet-Biedl syndrome. Eur J Hum Genet 2013;21:8-13. 10.1038/ejhg.2012.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith A, Prasad M, Deng ZM, et al. Comparison of high resolution cytogenetics, fluorescence in situ hybridisation, and DNA studies to validate the diagnosis of Prader-Willi and Angelman's syndromes. Arch Dis Child 1995;72:397-402. 10.1136/adc.72.5.397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jauch A, Robson L, Smith A. Investigations with fluorescence in situ hybridization (FISH) demonstrate loss of the telomeres on the reciprocal chromosome in three unbalanced translocations involving chromosome 15 in the Prader-Willi and Angelman syndromes. Hum Genet 1995;96:345-9. 10.1007/BF00210421 [DOI] [PubMed] [Google Scholar]
- 42.Buchholz T, Jackson J, Smith A. Methylation analysis at three different loci within the imprinted region of chromosome 15q11-13. Am J Med Genet 1997;72:117-9. 10.1002/(SICI)1096-8628(19971003)72:1<117::AID-AJMG24>3.0.CO;2-T [DOI] [PubMed] [Google Scholar]
- 43.Runte M, Färber C, Lich C, et al. Comprehensive methylation analysis in typical and atypical PWS and AS patients with normal biparental chromosomes 15. Eur J Hum Genet 2001;9:519-26. 10.1038/sj.ejhg.5200661 [DOI] [PubMed] [Google Scholar]
- 44.Papenhausen P, Schwartz S, Risheg H, et al. UPD detection using homozygosity profiling with a SNP genotyping microarray. Am J Med Genet A 2011;155A:757-68. 10.1002/ajmg.a.33939 [DOI] [PubMed] [Google Scholar]
- 45.Kotzot D. Supernumerary marker chromosomes (SMC) and uniparental disomy (UPD): coincidence or consequence? J Med Genet 2002;39:775-8. 10.1136/jmg.39.10.775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kearney HM, Kearney JB, Conlin LK. Diagnostic implications of excessive homozygosity detected by SNP-based microarrays: consanguinity, uniparental disomy, and recessive single-gene mutations. Clin Lab Med 2011;31:595-613, ix. 10.1016/j.cll.2011.08.003 [DOI] [PubMed] [Google Scholar]
- 47.Bittel DC, Kibiryeva N, Butler MG. Methylation-specific multiplex ligation-dependent probe amplification analysis of subjects with chromosome 15 abnormalities. Genet Test 2007;11:467-75. 10.1089/gte.2007.0061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hüttenhofer A, Schattner P, Polacek N. Non-coding RNAs: hope or hype? Trends Genet 2005;21:289-97. 10.1016/j.tig.2005.03.007 [DOI] [PubMed] [Google Scholar]
- 49.Smith A, Robson L, Neumann A, et al. Fluorescence in-situ hybridisation and molecular studies used in the characterisation of a Robertsonian translocation (13q15q) in Prader-Willi syndrome. Clin Genet 1993;43:5-8. 10.1111/j.1399-0004.1993.tb04416.x [DOI] [PubMed] [Google Scholar]
- 50.Diagnostic testing for Prader-Willi and Angleman syndromes: Report of the ASHG/ACMG Test and Technology Transfer Committee. Am J Hum Genet 1996;58:1085-8. [PMC free article] [PubMed] [Google Scholar]
- 51.Saitoh S, Mutirangura A, Kuwano A, et al. Isochromosome 15q of maternal origin in two Prader-Willi syndrome patients previously diagnosed erroneously as cytogenetic deletions. Am J Med Genet 1994;50:64-7. 10.1002/ajmg.1320500114 [DOI] [PubMed] [Google Scholar]
- 52.Monaghan KG, Van Dyke DL, Feldman G, et al. Diagnostic testing: a cost analysis for Prader-Willi and Angelman syndromes. Am J Hum Genet 1997;60:244-7. [PMC free article] [PubMed] [Google Scholar]