

Dermatomyositis (DM) is a multisystem disease with symptoms spanning diverse body functions including the skin, the joints, the heart and the lungs. The skin manifestations involve a sun-sensitive rash on the face, neck and upper trunk, and erythema of knees, elbows and knuckles. In classical cases, where these typical skin manifestations combine with muscle weakness, diagnosis can be set on clinical grounds. In other cases, diagnosis is confirmed by assessing the level of serum muscle enzymes, electromyographic findings, and a muscle biopsy (1). In the skeletal muscle tissue of DM, the primary target of the immune response is the vascular endothelium of perimysial and perifascicular blood vessels, and inflammatory infiltrates often occur at perivascular sites. Complement deposition of the terminal C5b-9 membrane attack complex (MAC) leads to blood vessel necrosis (2). The trigger that initiates complement activation remains however unclear.
In a recently published article in Brain, Lahoria and colleagues (3) touch upon two of the main research questions remaining in inflammatory muscle disease. Firstly, why does DM display this unique feature of endothelial damage, which is conspicuously in contrast with other subtypes of inflammatory myopathy? In two other main subgroups, polymyositis and sporadic inclusion body myositis, the primary target of the immune response are the muscle fibers themselves, as illustrated by active invasion of nonnecrotic muscle fibers by auto aggressive cytotoxic T-cells and macrophages (4). Secondly, does this endotheliopathy precede the perifascicular muscle atrophy (Figure 1) that characterizes the disease, or are these myopathological features simultaneous or more or less unrelated events?
Figure 1.
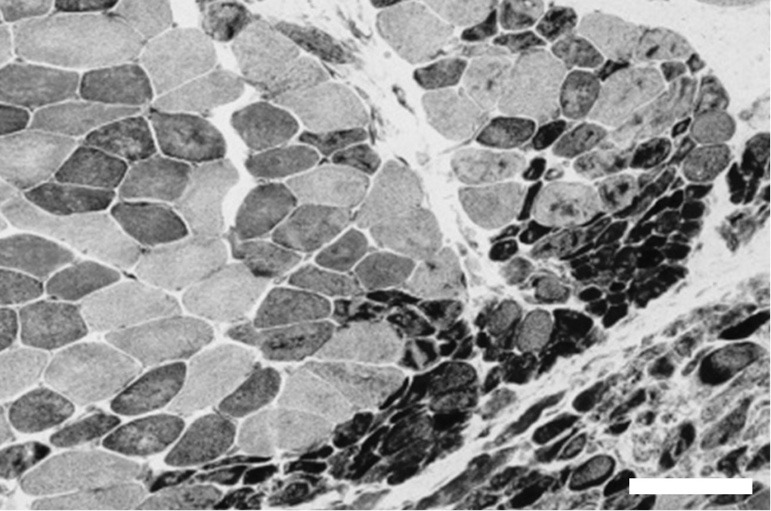
Histochemical fiber-type-staining detecting the reduced form of nicotinamide adenine dinucleotide shows the perifascicular muscle fiber atrophy characteristic of muscle biopsies from dermatomyositis patients. Scale bar =100 µM.
The authors investigated in detail the microvascular changes in 50 patients diagnosed with DM, both of the juvenile (n=15) and the adult form (n=35). Capillary density was found to be reduced 2-fold and transverse vessel density reduced 3-fold in regions with perifascicular atrophy. Material from patients at different disease stages was evaluated, as were patients that had received treatment prior to the biopsy. In a patient in acute phase of the disease, severe capillary depletion was present in the biopsy even in fascicles without perifascicular atrophy. In contrast, a patient treated for 5 months with 20 mg pro day of prednisone displayed a capillary density that was least decreased, and the subject’s muscle weakness was mild. The authors found perifascicular inflammatory cell infiltration to be a rare event, and much less frequent than MAC positive perifascicular capillaries. The latter were encountered in the majority of DM patients and could be regarded as one of the most important diagnostic features. The article presents several important observations in favor of DM being an antibody-dependent, complement-mediated disease characterized by capillary injury that results in perifascicular muscle fiber atrophy. An early study in favor of a sequence of events starting with blood vessel dropout was published way back in 1990 (5), and DM biopsies show evidence of focal ischemia by way of strong expression of markers of oxidative stress and lipid peroxidation (6).
The pro-inflammatory environment in DM is starting to become more and more clear, and many indications point to dysregulated innate immunity as an important pathological factor. Perifascicular pathology is associated with interferon (IFN) type 1-induced alterations, though it needs to be said that inflammatory myopathies are generally associated with increased IFN type 1 levels (7), as are indeed also other rheumatic diseases. However, it appears that the trigger that initiates this IFN signature seems specific for the individual disease entities, leading to the characteristic inflammatory patterns in the different diseases. Sera from DM patients contains higher levels of IFNα and expression is inversely correlated with duration of disease, suggesting that IFNα plays a role in disease initiation (8). The plasmacytoid dendritic cells inside the muscle tissue are thought to be the main local source of IFN type 1 (9), but research results suggest important additional systemic production also exists in patients. Several IFN type 1 signature genes are overexpressed specifically in the perifascicular areas of DM tissues, as is the associated factor retinoic acid-inducible gene-1 (RIG-1). Interestingly, RIG-1 was found absent from the affected muscle fibers of polymyositis and sporadic inclusion body myositis tissues (10). In a subgroup of DM patients, usually presenting an amyopathic phenotype with interstitial lung disease, autoantibodies can be detected that recognize IFN-induced with helicase C domain protein 1 (IFIH1), one of the RIG-I-like receptors (11,12). RIG-I and IFIH1 can interact with viral RNA, activating IFN type 1-inducible cytokine gene expression (13). Based upon these observations, it has been put forward that upregulated IFIH1 might liberate otherwise cryptic epitopes, causing breakage of self-tolerance and consequently autoimmune tissue damage. We are conscious that the affected muscle fibers need to be regarded as active contributors to the inflammatory process, initiating and/or perpetuating a persistent local immune response, and contributing to the associated dysregulation of innate immunity. The vast potential of muscle fibers to produce immune stimulators and the differential expression of cytokines between the subtypes of inflammatory myopathies has been described extensively (14). Of interest in this respect is that Lahoria et al. (3) describe the expression of MxA, another type 1 IFN-inducible antiviral protein, on both perifascicular capillaries and muscle fibers. This raises the possibility that a common mechanism could produce these abnormalities rather than one leading to the other, adding nuance to the story of muscle fiber atrophy as merely the result of blood vessel dropout.
Non-immune mechanisms equally are contributors to the muscle tissue damage in DM. Viperin, another IFN-induced anti-viral protein and a modulator of mitochondrial and endoplasmic reticulum (ER) stress, is upregulated in DM (10). Viperin thus represents a link between inflammatory and ER stress response pathways, the latter being significantly elevated in muscle tissue of patients with polymyositis and DM alike (15). Chaperoning heat shock proteins (HSP) are also known to regulate skeletal muscle physiology and adaptation to stress, and are involved in clearing the tissues of damaged, misfolded and aggregated proteins. Expression of HSP70 and 90 families was found strongly increased in perifascicular atrophic fibers and regenerating fibers in DM, a feature shared with the affected fibers in polymyositis, sporadic inclusion body myositis and muscular dystrophy biopsies (16). Most of these non-immune mechanisms could therefore be regarded as universal (failing) tissue protection programs in muscle disease.
The jury is still out on the precise relationship between vascular and fiber damage in DM, but the article by Lahoria et al. (3) offers valuable data and insight, and is yet another illustration of how descriptive myopathological analysis is able to aid our understanding of disease mechanisms. Though this particular type of research delivers static data, it offers indirect evidence into the sequence of events concerned.
Acknowledgements
The author is recipient of a research grant from the Association belge contre les maladies neuromusculaires (ABMM)—Aide à la recherche ASBL 2016.
Provenance: This is a guest commentary commissioned by section editor Dr. Mingzhu Gao (Department of Laboratory Medicine, Wuxi Second Hospital, Nanjing Medical University, Wuxi, Jiangsu, China).
Conflicts of Interest: The author has no conflicts of interest to declare.
References
- 1.De Bleecker JL, De Paepe B. The idiopathic inflammatory myopathies. In: Adv Clin Neurosci 2003;13:353-78. [Google Scholar]
- 2.Kissel JT, Mendell JR, Rammohan KW. Microvascular deposition of complement membrane attack complex in dermatomyositis. N Engl J Med 1986;314:329-34. 10.1056/NEJM198602063140601 [DOI] [PubMed] [Google Scholar]
- 3.Lahoria R, Selcen D, Engel AG. Microvascular alterations and the role of complement in dermatomyositis. Brain 2016;139:1891-903. 10.1093/brain/aww122 [DOI] [PubMed] [Google Scholar]
- 4.De Paepe B, De Bleecker JL. The nonnecrotic invaded muscle fibers of polymyositis and sporadic inclusion body myositis: on the interplay of chemokines and stress proteins. Neurosci Lett 2013;535:18-23. 10.1016/j.neulet.2012.11.064 [DOI] [PubMed] [Google Scholar]
- 5.Emslie-Smith AM, Engel AG. Microvascular changes in early and advanced dermatomyositis: a quantitative study. Ann Neurol 1990;27:343-56. 10.1002/ana.410270402 [DOI] [PubMed] [Google Scholar]
- 6.Gitiaux C, Kostallari E, Lafuste P, et al. Whole microvascular unit deletions in dermatomyositis. Ann Rheum Dis 2013;72:445-52. 10.1136/annrheumdis-2012-201822 [DOI] [PubMed] [Google Scholar]
- 7.Haq SA, Tournadre A. Idiopathic inflammatory myopathies: from immunopathogenesis to new therapeutic targets. Int J Rheum Dis 2015;18:818-25. 10.1111/1756-185X.12736 [DOI] [PubMed] [Google Scholar]
- 8.Greenberg SA, Pinkus JL, Pinkus GS, et al. Interferon-alpha/beta-mediated innate immune mechanisms in dermatomyositis. Ann Neurol 2005;57:664-78. 10.1002/ana.20464 [DOI] [PubMed] [Google Scholar]
- 9.Siegal FP, Kadowaki N, Shodell M, et al. The nature of the principal type 1 interferon-producing cells in human blood. Science 1999;284:1835-7. 10.1126/science.284.5421.1835 [DOI] [PubMed] [Google Scholar]
- 10.Suárez-Calvet X, Gallardo E, Nogales-Gadea G, et al. Altered RIG-I/DDX58-mediated innate immunity in dermatomyositis. J Pathol 2014;233:258-68. 10.1002/path.4346 [DOI] [PubMed] [Google Scholar]
- 11.Sato S, Hoshino K, Satoh T, et al. RNA helicase encoded by melanoma differentiation-associated gene 5 is a major autoantigen in patients with clinically amyopathic dermatomyositis: Association with rapidly progressive interstitial lung disease. Arthritis Rheum 2009;60:2193-200. 10.1002/art.24621 [DOI] [PubMed] [Google Scholar]
- 12.Nakashima R, Imura Y, Kobayashi S, et al. The RIG-I-like receptor IFIH1/MDA5 is a dermatomyositis-specific autoantigen identified by the anti-CADM-140 antibody. Rheumatology (Oxford) 2010;49:433-40. 10.1093/rheumatology/kep375 [DOI] [PubMed] [Google Scholar]
- 13.Yoneyama M, Fujita T. Function of RIG-I-like receptors in antiviral innate immunity. J Biol Chem 2007;282:15315-8. 10.1074/jbc.R700007200 [DOI] [PubMed] [Google Scholar]
- 14.De Paepe B, Creus KK, De Bleecker JL. Role of cytokines and chemokines in idiopathic inflammatory myopathies. Curr Opin Rheumatol 2009;21:610-6. 10.1097/BOR.0b013e3283317b31 [DOI] [PubMed] [Google Scholar]
- 15.Nagaraju K, Casciola-Rosen L, Lundberg I, et al. Activation of the endoplasmic reticulum stress response in autoimmune myositis: potential role in muscle fiber damage and dysfunction. Arthritis Rheum 2005;52:1824-35. 10.1002/art.21103 [DOI] [PubMed] [Google Scholar]
- 16.Paepe BD, Creus KK, Weis J, et al. Heat shock protein families 70 and 90 in Duchenne muscular dystrophy and inflammatory myopathy: balancing muscle protection and destruction. Neuromuscul Disord 2012;22:26-33. 10.1016/j.nmd.2011.07.007 [DOI] [PubMed] [Google Scholar]