

Abstract
Objective
Proteins can exist as multiple proteoforms in vivo that can have important roles in physiological and pathological states.
Methods
We present the development and characterization of mass spectrometric immunoassay (MSIA) for quantitative determination of serum amyloid A (SAA) proteoforms.
Results
Intra- and inter-day precision revealed CVs<10%. Against existing SAA ELISA, the developed MSIA showed good correlation according to the Altman-Bland plot. Individual concentrations of the SAA proteoforms across a cohort of 170 samples, revealed 7 diverse SAA polymorphic types and 12 different proteoforms.
Conclusion
The new SAA MSIA enables parallel analysis of SAA polymorphisms and quantification of all expressed SAA proteoforms, in a high-throughput and time-efficient manner.
Keywords: inflammation biomarker, mass spectrometry, polymorphism, posttranslational modifications, proteoforms, baseline concentration
Introduction
Serum amyloid A (SAA, apo-SAA) represents a group of proteins recognized as inflammatory factors and biomarkers for multiple clinical conditions (Malle et al., 1993). Structurally, SAA proteins are products of four genes (SAA1, SAA2, SAA3 and SAA4) on the 11th chromosome, three of which are expressed in humans (SAA1, SAA2 and SAA4) (Sellar et al., 1994b). SAA1 and SAA2 genes express five (1.1, 1.2, 1.3, 1.4 and 1.5) and two (2.1 and 2.2) allelic variants, resulting in synthesis of multiple SAA protein products, while SAA4 gene product (SAA 4.1) is only constitutively expressed (Sellar et al., 1994a, Farwig et al., 2005) (SAA proteins are named according to the revised SAA nomenclature (Sipe, 1999)). When analyzed at the molecular level, SAA1 and SAA2 proteins differ only slightly in their amino acid sequences. Therefore, they present with molecular masses in a narrow range between 11.6 – 11.7 kDa.
Prior studies demonstrate that not all of the polymorphic SAA variants are expressed in individuals (Uhlar and Whitehead, 1999, Yamada et al., 1999, Bakkaloglu et al., 2004, MacGregor et al., 2004). Some of the SAA proteoforms expression is race dependent and can represent an individual SAA fingerprint (Baba et al., 1995). Therefore, regulatory mechanisms dictate the SAA expression in circulation in such a way that individuals possess unique SAA profiles represented by one, two or multiple SAA proteins, coded by SAA1 and/or SAA2 genes (Uhlar and Whitehead, 1999, Kluve-Beckerman et al., 1988).
SAA proteins have a variety of roles in vivo. After being synthetized in the liver, SAA proteins act as apolipoproteins (hence the acronym apo-SAA) and are an important component of high-density lipoprotein (HDL) particles (Artl et al., 2000, Liang and Sipe, 1995, Banka et al., 1995). In an onset of acute inflammatory conditions, cytokine signaling system can induce SAA release from the liver in the circulation at a high rate (increasing in concentration up to 1000-fold) (Malle et al., 1993). Excess SAA has the ability to incorporate into HDL particles, replacing apolipoprotein A-I (apoA-I), hence changing the lipid particles morphology (Marsche et al., 2007). In turn, this causes changes in the lipid metabolism, causing decreases in apoA-I, paraoxonase 1 (PON1), cholesteryl ester transfer protein (CETP) and lecithin—cholesterol acyltransferase (LCAT), and increases in free fatty acids, triglycerides (TGs) and free cholesterol in circulation (Andersson, 1997, Kisilevsky and Subrahmanyan, 1992, Manley et al., 2006, Steinmetz et al., 1989). SAA association with lipid metabolism has been confirmed in multiple studies that investigate the role of SAA in coronary artery disease (CAD) and production of atherosclerotic plaques in blood vessels (Xie et al., 2010, Fyfe et al., 1997, Johnson et al., 2004). The systematic effects of the increased SAA concentration in circulation are also induced by several chronic inflammatory conditions, such as rheumatoid arthritis, multiple sclerosis, type-2 diabetes and some types of cancer (Cunnane, 2001, Artl et al., 2000, Zhao et al., 2010, Ristori et al., 1998). In addition, immune-related and anti-inflammatory roles of SAA have been reported (Badolato et al., 1994, Aldo-Benson and Benson, 1982, Eklund et al., 2012).
SAA protein structure has been highly conserved through evolution, however, in humans, the terminal amino-acids are subjected to posttranslational processing (Uhlar and Whitehead, 1999). Detecting posttranslationally modified SAA proteins has significant implications, as the terminally cleaved SAA 1.1 protein (missing 28 amino acids from the C-terminus) is known to form amyloid fibrils and cause AA amyloidosis (Husby et al., 1994). In addition, accumulation of SAA fibrils can occur as a result of benign catabolic processes in cells (associated with serine proteases), as well as due to chronic inflammation (Bakkaloglu et al., 2004, Skogen and Natvig, 1981). Published evidence support the strong structure – function relation between SAA polymorphism, SAA proteins and susceptibility to amyloidosis (Takase et al., 2014, Booth et al., 1998).
Current methodologies for analysis of SAA proteoforms provide information about the total concentration, and do not distinguish between the polymorphic SAA profile and the posttranslational modifications. Commercially available, as well as in-house enzyme-linked immunoassays (ELISAs) and immunonephelometry methods are widely used to assess the SAA concentrations in clinical settings (Wu et al., 2007, Yamada et al., 1993). Genetic analyses, primarily genotyping and allele frequency analyses are used to investigate the SAA polymorphism and amyloidosis (Betts et al., 1991). These methodologies, however, lack the capability to distinguish between SAA proteins and detect posttranslational modifications. Mass-spectrometry-based assays are efficient in overcoming this issue. In the past decade several methods that use mass spectrometry detection for SAA proteins analyses, such as 2D-gel electrophoresis coupled with liquid chromatography/ESI mass spectrometry (Ducret et al., 1996), protein chip immunoaffinity capture with SELDI mass spectrometry (SELDI-MS) (Tolson et al., 2004), as well as in-gel trypsin digestion, followed by ESI-MS of SAA peptides (Loo et al., 2011) have been proposed.
Mass spectrometric immunoassay (MSIA) offers the advantage of combining the selectivity of immunoassays with the specificity of mass spectrometry detection (Nelson et al., 1995, Nelson and Borges, 2011). MSIA has been used for both qualitative and quantitative analyses of multiple protein entities (Kiernan et al., 2006, Trenchevska et al., 2014). In previous work, we reported several truncated SAA proteoforms originating from SAA 1.1 and SAA 2.1, lacking one or more amino acids from the N- and/or C-terminus by qualitative MSIA (Kiernan et al., 2003). In addition, in recent work, we addressed the importance of the SAA posttranslational modifications, by indicating an association between the relative ratio of SAA 1.1R (truncated SAA 1.1 lacking one N-terminal arginine (R) residue) to the non-truncated SAA 1.1, and plasma glucose levels (Yassine et al., 2015).
In this work we go a step further and present the development, validation and application of a fully quantitative MSIA for analysis of SAA proteins in human plasma samples. The developed assay enables determination of SAA polymorphic profiles, and quantification of all present SAA proteoforms in a single run.
Methods
Reagents
Monoclonal mouse anti-human antibody to serum amyloid A (SAA; Cat. No. MO-C40028A) was obtained from ANOGEN (Mississauga, ON, Canada,). Polyclonal affinity purified rabbit anti-bovine beta lactoglobulin antibody (Cat. No. GTX77272) was obtained from GeneTex, Inc (Irvine, CA). Recombinant human APO-serum amyloid A (Cat. No. CRS300B) was purchased from Cell Sciences (Canton, MA). Protein calibration standard I (Cat. No. 206355) was purchased from Bruker (Billerica, MA). Phosphate buffered saline (PBS) buffer (Cat. No. 28372), MES buffered saline (28390), 1,1′ Carbonyldiimidazole (97%) (CDI; 530-62-1), acetonitrile (ACN; A955-4), hydrochloric acid (HCl; A144-212), N-methylpyrrolidinone (NMP; BP1172-4) and affinity pipettes fitted with porous microcolumns (991CUS01) were obtained from Thermo Scientific (Waltham, MA). β-Lactoglobulin from bovine milk (Cat. No. L8005), tween20 (P7949), trifluoracetic acid (TFA; 299537-100G), sinapic acid (85429), sodium chloride (S7653), HEPES (H3375), ethanolamine (ETA; 398136), albumin from bovine serum (BSA; A7906), ammonium acetate (A7330) and octyl β-D glucopyranoside (NOG; 08001), polyethylene glycol (P3015), calcium chloride (C1016), and glycine buffer (G8898) were obtained from Sigma Aldrich (St. Louis, MO). Acetone (Cat. No. 0000017150) was obtained from Avantor Performance Materials (Center Valley, PA).
Study subjects
For the method development and validation, normal human EDTA plasma samples were used, obtained from ProMed DX (Norton, MA). For the method comparison and application total of 170 human EDTA plasma samples were obtained from University of Arizona (Phoenix, AZ, USA). The study was approved by the University of Arizona IRB and all participants provided written informed consent. To ensure proper privacy protection, the samples were labeled only with a specific code and supplied with accompanying information about the age, gender, ethnicity and existing medical condition (if any). Prior to analysis, samples were centrifuged (5 min at 14.5×1000 rpm), aliquoted in low-profile 96-well trays and stored at −80°C.
Standards and Samples preparation
Recombinant human apo-SAA standard was used to generate an eight-point standard curve. Serial dilutions were done using PBS, 3 g/L BSA buffer, preparing standards with concentrations of 2 mg/L (standard #1), 1 mg/L (standard #2), 0.5 mg/L (standard #3), 0.25mg/L (standard 4), 0.125 mg/L (standard #5), 0.0625 mg/L (standard #6), 0.0313 mg/L (standard #7) and 0.0156 mg/L (standard #8). For the standard curve, 50 μL of each standard were added to the wells of a microplate containing 50 μL assay buffer (PBS, 0.1%Tween) and 50 μL 10 mg/L beta-lactoglobulin (BL was used as an IRS). For the analytical samples, the standard solutions were substituted with human plasma (diluted 5-fold into PBS,0.1%Tween buffer). A control standard sample with known total SAA concentration was analyzed in triplicate with each run.
Affinity pipettes derivatization
Using a Multimek 96 pipettor (Beckman Coulter, Brea, CA ) affinity pipettes were initially activated as previously described (Trenchevska et al., 2010). Following activation, the affinity pipettes were immersed into the well of a microplate containing SAA and BL antibodies solution (anti-SAA: anti-BL = 3:1 (w/w; 100 μL, 5 μg anti-SAA/well). The derivatization process consisted of 750 cycles of aspirations and dispenses of 100 μL volumes in order to bind the antibodies to the activated surfaces. One rinse with ETA followed (50 cycles each, 100 μL), ending with two final rinses with PBS w/0.1% TWEEN (50 cycles each, 100 μL). Antibody-derivatized pipettes were stored at +4°C until used.
Mass spectrometric immunoassay
The quantitative SAA MSIA was applied to all plasma samples. Initially, the derivatized affinity pipettes were pre-rinsed with PBS, 0.1%Tween (50 aspiration/dispense cycles, 100 μL each), followed by two 60 mM HCL rinses and another buffer rinse. Next, the pipettes were immersed into a microplate containing the samples and 250 aspirations and dispense cycles were performed (100 μL volumes each) allowing for affinity capture of SAA and BL from the samples. The pipettes were then rinsed with assay buffer (100 cycles), and twice with water (10 cycles and 20 cycles respectively, 100 μL aspiration/dispense each). SAA-loaded tips were then exposed to six-microliter aliquots of MALDI matrix solution (25 g/L sinapic acid in aqueous solution containing 33% (v/v) acetonitrile, and 0.4 % (v/v) trifluoroacetic acid). After a 10 second delay (to allow for the dissociation of the proteins from the capturing antibodies), the eluates were dispensed directly onto a 96-well formatted MALDI target.
Following drying, linear-mode mass spectra were acquired from each sample spot, each consisting of five thousand laser shots using an Autoflex III MALDI-TOF mass spectrometer (Bruker, Billerica, MA), operating in positive-ion, delayed extraction linear mode, with ion source 1 at 20.00 kV, ion source 2 at 18.45 kV, lens at 6.00 kV, 90 ns delay, and signal suppression up to 3000 Da. Prior to acquisition of the mass spectra, the target mass range (m/z = 5,000 – 30,000) was externally calibrated using a mixture of calibrants (protein calibration standard I, Cat. No. 8206355) obtained from Bruker Daltonics (Billerica, MA). The calibrant mix was diluted 15-fold in sinapic acid matrix and spotted onto the MALDI target. Additionally, before processing, all mass spectra were subjected to internal calibration using the BL+1 and BL+2 signals, with mass accuracy up to 0.001 Da.
Data analysis
All obtained mass spectra were imported into FlexAnalysis software (Bruker Daltonics, MA), smoothed (Savitzky-Golay algorithm) and baseline subtracted (Tophat algorithm). All peaks representing SAA proteoforms and BL were identified and peak intensities were used to analyze the data. A standard curve was constructed using the intensity ratio between SAA standard peak and BL peak, against SAA standard concentration. In all samples, first, SAA – to – BL+1 signal intensity ratios were calculated for each proteoform. Next, ratios of all proteoforms were summed. Generated standard curve equation was applied to calculate the total SAA protein concentration. Finally, the concentration of each individual proteoform was calculated based on its percentage of the total SAA.
To assess SAA polymorphism among the human plasma samples, obtained MW of the proteoforms were compared to the theoretical MW of the SAA gene products using SAA protein amino acid sequences and the program PAWS (Genomic Solutions, Waltham, MA, USA).
Results and Discussion
Development and validation of a quantitative MSIA for serum amyloid A (SAA)
Quantification of SAA proteoforms was achieved through generation of standard curves, utilizing recombinant SAA protein standard and BL as an internal reference standard (IRS) for quantification. SAA standard curves consisted of eight standard concentrations, ranging from 0.0156 mg/L to 2 mg/L prepared as described in the previous section. Signals from each standard were normalized towards the BL signal. The choice of IRS was critical step in the quantitative SAA MSIA method development, because it had to satisfy all the requirements in accordance to the recently established “fit-for-purpose” approach (Carr et al., 2014). Beta lactoglobulin is a non-human protein, therefore, does not cross-react with the anti-human SAA antibody or the analyzed protein. Also, its signal in the mass spectrum (at m/z = 18,281 Da) is in close proximity to the signals from the SAA (in range between 11,500 – 11,750 Da), thus ensuring similar MS instrument settings use. The IRS was added in constant concentration in all analytical samples (standards and plasma), optimized to achieve saturation of the anti-BL antibody and produce a constant signal in the mass spectra throughout the analyses.
For the standard curve generation, first peak intensities for the SAA standards and the BL internal standard were tabulated. Separate standard curve was generated in each analysis from the peak intensity ratio between the SAA protein standard and the BL (SAA std/BL [peak intensity ratio], y-axis), versus the SAA standard concentration (c(SAA std) [mg/L], x-axis). Standard curve equation was generated using a linear fit curve in Sigma Plot v.11.0 (Systat Software, Inc., San Jose, CA), and was further used to calculate SAA proteoforms concentration in the plasma samples.
The physiological concentrations of SAA vary depending on the gene polymorphic variants that are expressed, as well as the presence of inflammatory condition. According to published data, physiological concentrations are in the low mg/L range, but can significantly increase in the presence of acute or chronic inflammation (Pizzini et al., 2000). To account for SAA concentration variations, the generated standard curve was prepared in a wide concentration range that was sufficient for quantification of all the proteoforms from the analytical samples. An example of a SAA/BL standard curve, along with the corresponding mass spectra are presented in Fig. 1. The response is linear across the entire range (r2 = 0.9902), with a standard error of estimate, SEE = 0.8019, and an average percent of relative standard deviation (%RSD) of 16.4.
Fig. 1.
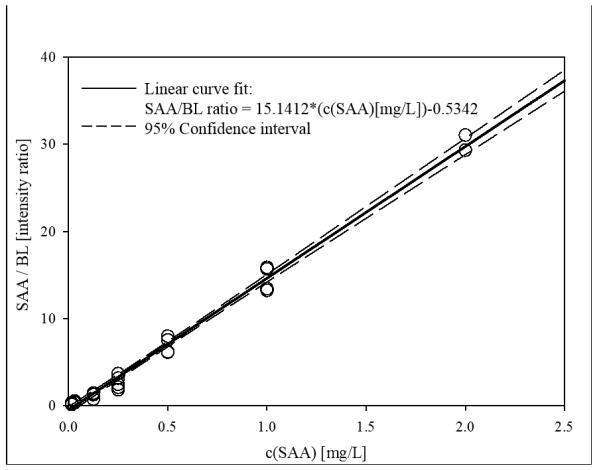
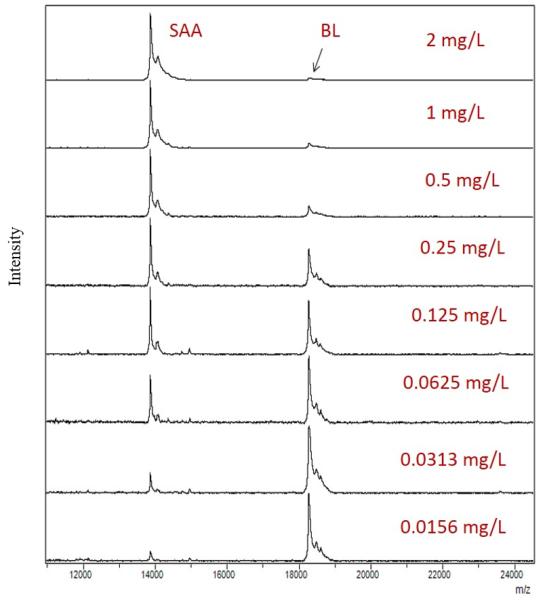
a) Example SAA standard curve with BL as an IRS; b) Representative mass spectra from generated standard curve, showing peaks for SAA and BL in different SAA concentrations.
The lower limit of detection (LOD) was calculated from the limit of blank (LoB) as a sum from the mean of blank and 1.645 standard deviations from blank samples, and was found to be 0.007 mg/L. The lower limit of quantification (LLOQ), as determined from the LOD (Armbruster and Pry, 2008) and the standard curve was 0.016 mg/L.
To validate the reproducibility of the developed MSIA, a single sample was analyzed on five different days with five standard curves. Inter- and intra-day variability was assessed by analyzing the standard control sample with known SAA concentration (0.4 mg/L) in triplicate on the five different days, each time with a different standard curve. SAA concentration of the control sample was calculated from the corresponding standard curve equations and used to assess within and between-run CVs. For the intra- day precision, CVs between 1.6 % and 8.6 % were observed. Inter-day variability was calculated to be 4.7 % (observed average SAA concentration = 0.387 mg/L) (Table I).
Table I.
Inter and intra-day variability CVs
| Intra-assay CVs | Inter-assay CV | ||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Sample | 1 | 2 | 3 | 4 | 5 | ||
| STDEVS: | 0.011 | 0.036 | 0.006 | 0.013 | 0.024 | STDEVS: | 0.018 |
|
MEAN
[mg/L]: |
0.372 | 0.420 | 0.383 | 0.407 | 0.355 |
MEAN
[mg/L]: |
0.3874 |
| CV [%]: | 2.96 | 8.57 | 1.57 | 3.19 | 6.76 | CV [%]: | 4.65 |
There are different approaches which can be used to evaluate the accuracy of an assay (Carr et al., 2014, Chau et al., 2008, FDA, 2001). In this work we have adapted the protocol used in commercially available ELISAs for SAA that we have also used for our method comparison (http://www.hoelzelbiotech.com/media/import/pdf_manual/Anogen//EL10015.Manual.pdf ). In short, SAA standard was spiked in control EDTA human plasma sample in three different concentrations (low = 0.02 mg/L; medium = 0.2 mg/L and high = 1.5 mg/L) throughout the range of the assay, and retrieving it from the plasma sample. All spiked controls were run in triplicate, on a single day with a standard curve. Retrieved (observed) concentrations from the samples were compared to the expected values.
Results from the accuracy experiment are presented in Table II and show SAA standard percent recovery in the MSIA between 89 – 111 % for all concentrations.
Table II.
MSIA accuracy experiment – obtained percent recovery
| Expected | Observed | Recovery | |
|---|---|---|---|
|
| |||
| mg/L | mg/L | E/O [%] | |
| HC 1 | 1.50 | 1.67 | 90.1 |
| HC 2 | 1.50 | 1.62 | 92.7 |
| HC 3 | 1.50 | 1.58 | 95.2 |
|
| |||
| MC 1 | 0.200 | 0.215 | 93.1 |
| MC 2 | 0.200 | 0.224 | 89.3 |
| MC 3 | 0.200 | 0.184 | 108 |
|
| |||
| LC 1 | 0.020 | 0.022 | 92.5 |
| LC 2 | 0.020 | 0.018 | 111 |
| LC 3 | 0.020 | 0.021 | 93.1 |
HC – High concentration spike; MC – medium concentration spike; LC – low concentration spike
In a final experiment, SAA MSIA was benchmarked against existing ELISA for SAA analysis. To do a direct method comparison study, 30 human plasma samples were analyzed both with the developed MSIA and with a commercially available ELISA (Human SAA ELISA Kit, Cat. No. EL10015, Anogen, Mississauga, Ontario, Canada). Serum amyloid A ELISA was performed according to the instructions provided by the manufacturer. The human plasma samples were analyzed by quantitative SAA MSIA in parallel with an accompanying standard curve, as described in the previous section.
A good correlation was observed between ELISA SAA and MSIA total SAA concentrations. Passing Bablok (Passing and Bablok, 1983) correlation was obtained with regression equation of y=2.48+0.953x, and a Cusum linearity p value of 1.00 indicating no significant deviation from linearity. In addition, a slight positive bias, was revealed by the Altman-Bland (Bland and Altman, 1999) scatter plot (Fig. 2a), and the difference plot (Fig. 2b). The appearance of bias in method comparison analyses is not unusual (Niederkofler et al., 2013, Dossus et al., 2009), and it can be linked to several causes. One may be the possibility that the secondary (labeled) antibody used in the ELISA does not recognize the truncated SAA proteoforms (there was no information available for the secondary antibody in the ELISA). Such discrepancy will result in lower total SAA concentrations in ELISA (such as in presented results).
Fig. 2.
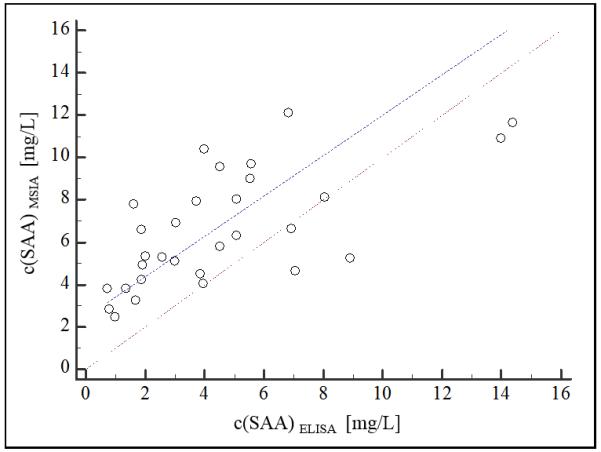
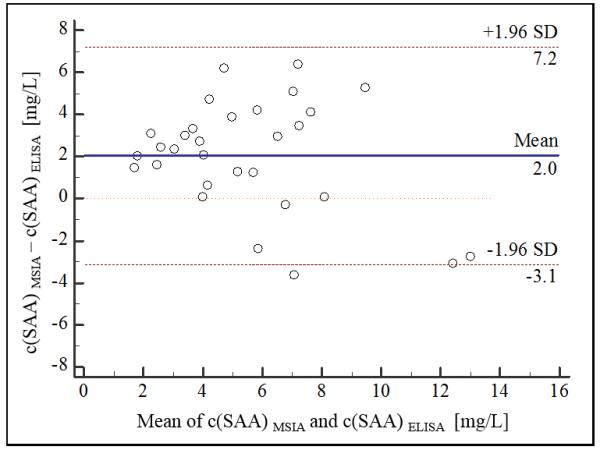
SAA method validation MSIA vs ELISA a) scatter plot (dashed line represents the obtained regression; dotted line represents identity (x=y); b) Bland-Altman difference plot (solid line represents the mean of concentration difference, 2 mg/L; dashed lines represent limits of agreement ±1.96 SD).
SAA profiling in human plasma samples
The combination of the selectivity of the immunoaffinity separation, with the specificity of the mass spectrometric detection and accurately-determined m/z values, were sufficient for accurate identification of the different SAA proteoforms. The proteoforms were, thus identified due to the following: 1) knowledge of the amino acid sequence of the SAA and 2) knowledge/estimation of the PTMs for SAA. We have done such assignments in numerous previously published studies (Trenchevska et al., 2011, Trenchevska et al., 2015a, Trenchevska et al., 2015b). Using this approach we determined the SAA protein profile and concentrations of several SAA proteoforms in 170 human plasma samples. Presented in Fig. 3 is an example of mass spectra obtained from a plasma sample using the developed SAA mass spectrometric immunoassay. Several signals originating from SAA proteoforms were identified in the m/z range between 11,000 – 11,800, as well as signal from the IRS – the beta lactoglobulin at m/z = 18,281. The inlets in Fig.3 represent examples of SAA profiles from three different human plasma samples. Presented mass spectra show qualitative differences regarding the expressed SAA proteoforms. This is a result of the SAA polymorphic types that is associated with each individual. It can be noted that the first sample (top inset) expresses only SAA 1.1 protein, the second expresses SAA 1.1 and SAA 2.1, whereas the third sample expresses three SAA proteins (SAA 1.1, SAA 1.3 and SAA 2.1) in parallel. Note that, in addition to the native SAA protein signals, additional peaks are presented in the mass spectra. The importance and assignment of these peaks is further explained.
Fig. 3.

Example mass spectra of serum amyloid A and beta lactoglobulin extracted from human plasma samples using MSIA. *The inlets in the mass spectra represent close-ups of three different SAA protein profiles from three human plasma samples. **Signals labeled with an asterisks (*) represent MALDI matrix adducts.
Depending on their molecular weight, as well as the mass-to-charge ratio, among the analyzed human plasma samples we were able to differentiate and analyze four of the full length SAA proteins – SAA 1.1 (MW = 11,682.7 Da), SAA 1.3 (MW = 11,654.6 Da), SAA 2.1 (MW = 11,628.0 Da) and SAA 2.2 (MW = 11,647.7 Da). In all samples, the resolution of the SAA signals (even though not baselined) was sufficient to determine each individual m/z peak maximum – i.e., the intensity which was used for the quantification. In addition to the native SAA proteoforms (that account for the full-length protein), we identified posttranslationally modified proteoforms representing N-terminal truncations, missing one, two or more amino acids in the SAA sequences. A total of eleven additional truncated SAA proteoforms were identified originating from SAA 1.1, SAA 1.3, SAA 2.1 and SAA 2.2 (Table III).
Table III.
SAA proteoforms identified using targeted mass spectrometric immunoassay
| SAA proteoform | Theoretical [M+H]+ |
Observed [M+H]+ |
Frequency [%] |
c(SAA) [mg/L] |
|
|---|---|---|---|---|---|
|
Native SAA proteoforms
| |||||
| SAA 1.1 | native SAA 1.1 | 11,682.7 | 11,683.7 | 99.4 | 3.15 (0.398 – 22.3) |
| SAA 1.3 | native SAA 1.3 | 11,654.6 | 11,655.2 | 11.2 | 1.60 (0.673 – 4.25) |
| SAA 2.1 | native SAA 2.1 | 11,628.7 | 11,629.1 | 57.6 | 0.969 (0.259 – 5.01) |
| SAA 2.2 | native SAA 2.2 | 11,647.7 | 11,648.6 | 11.8 | 1.69 (0.382 – 7.38) |
|
| |||||
|
Truncated SAA proteoforms
| |||||
| SAA 1.1 R | 1.1 lacking 1 N-terminal (R-) amino acid |
11,526.5 | 11,526.8 | 99.4 | 2.61 (0.396 – 13.4) |
| SAA 1.1 RS | 1.1 lacking N-terminal dipeptide (RS-) |
11,439.4 | 11,439.4 | 99.4 | 0.907 (0.344 – 6.67) |
| SAA 1.1 RSFF | 1.1 lacking N-terminal tetra peptide (RSFF-) |
11,147.1 | 11,146.4 | 11.8 | 0.269 (0.0851 – 0.812) |
| SAA 1.1 RSFFS | 1.1 lacking N-terminal penta peptide (RSFFS-) |
11,058.9 | 11,058.7 | 7.06 | 0.157 (0.101 – 0.395) |
| SAA 1.3 R | 1.3 lacking 1 N-terminal (R-) amino acid |
11,498.4 | 11,498.5 | 11.2 | 1.53 (0.711 – 3.79) |
| SAA 1.3 RS | 1.3 lacking N-terminal dipeptide (RS-) |
11,411.4 | 11,411.2 | 10.6 | 0.696 (0.253 – 1.69) |
| SAA 1.3 RSFF | 1.3 lacking N-terminal tetra peptide (RSFF-) |
11,117.0 | 11,116.9 | 1.18 | 0.231 (0.219 – 0.244) |
| SAA 2.1 R | 2.1 lacking 1 N-terminal (R-) amino acid |
11,472.5 | 11,472.6 | 57.6 | 0.894 (0.219 – 3.16) |
| SAA 2.1 RS | 2.1 lacking N-terminal dipeptide (RS-) |
11,385.4 | 11,384.9 | 42.9 | 0.456 (0.219 – 1.35) |
| SAA 2.1 RSFF | 2.1 lacking N-terminal tetra peptide (RSFF-) |
11,091.1 | 11,092.8 | 2.35 | 0.139 (0.103 – 0.160) |
| SAA 2.2 R | 2.2 lacking 1 terminal (R-) amino acid |
11,491.5 | 11,492.0 | 12.4 | 1.23 (0.344 – 4.44) |
| SAA 2.2 RS | 2.2 lacking N-terminal dipeptide (RS-) |
11,404.5 | 11,403.2 | 8.23 | 0.481 (0.219 – 1.35) |
The concentration of SAA proteins was determined by calculating the protein/BL peak intensity ratios for each proteoform, summing up the ratios of all proteoforms, determining each SAA protein concentration using the standard curve, and then calculating the concentration of individual proteoforms based on their percentage of the originating SAA. This approach worked especially well for those proteoforms whose proteoform/BL peak ratios were below those of the standard curve. The total SAA concentration was calculated as a sum of the individual SAA proteins and was in the range between 1.56 – 56.3 mg/L (average c(SAA) = 8.71 mg/L).
Significant differences between the SAA proteoforms depending on the polymorphic variants expressed were noticed between the samples.
Table III shows detailed description of the identified SAA proteoforms as well as their observed and theoretical MW and concentration ranges for the individual SAA proteoforms identified in the 170 human plasma samples.
From the native SAA proteins, SAA 1.1 was the most abundant, present in 169 out of the 170 samples (99.4 %), followed by SAA 2.1 (present in 98 samples). SAA 1.3 and SAA 2.2 proteins were observed less frequently and identified in 19 and 20 samples respectively. There were two most-abundant SAA proteoforms in the majority of samples - the full-length native SAA protein, and SAA lacking N-terminal arginine (des-R proteoform). SAA lacking arginine-serine dipeptide (des-RS) was identified in all samples as well, but was significantly lower in abundance. The truncated SAA proteoforms which represent N-terminal truncations lacking four (des-RSFF) and five (des-RSFFS) residues from the amino acid sequence of the native SAA protein were present in less than 11% of samples and in low concentration.
We further analyzed SAA concentration distribution depending on the SAA gene that coded for the proteoforms synthesized. After calculating SAA concentrations for SAA1 coded proteins and SAA2 coded proteins, we analyzed the average total concentration of SAA 1.1 and SAA 1.3 proteoforms vs SAA 2.1 and SAA 2.2 proteoforms. Results show an average total concentration of 5.23 mg/L (1.14 – 35.9 mg/L) for SAA1 products (SAA1.1 and SAA1.3 proteoforms) and 2.67 mg/L (0.505 – 12.0 mg/L) for SAA2 (SAA2.1 and SAA2.2 proteoforms). These findings are in accordance with literature data that point out a relationship between SAA polymorphism and SAA basal concentration (Yamada et al., 1999, MacGregor et al., 2004). Among the native SAA proteins, SAA 1.1 has the highest physiological concentration at baseline (6.66 mg/L), followed by SAA 1.3 (3.79 mg/L). SAA2 protein products exhibit slightly lower average SAA concentrations. These findings are important for accurate assignment of SAA reference concentration range – and have a potential to distinguish between normal concentration and onset of inflammatory condition on individual basis.
Analyzing serum amyloid A profile – an insight into SAA polymorphism and posttranslational modifications
MSIA enables for unambiguous detection of SAA proteoforms in a single run. SAA profile differs among individuals in association with genetic predisposition (Booth et al., 1998). In the analyzed sample cohort we were able to distinguish seven different SAA profiles (Fig. 4). Only SAA1 gene products (SAA 1.1 and SAA 1.3) were present as a single SAA proteoform, whereas SAA2 gene products were present as accompaniments to SAA1 proteins. The most abundant protein profile was SAA 1.1/2.1, representing expression of two SAA proteins (SAA 1.1 and SAA 2.1, coded by SAA1 and SAA2 genes respectably), followed by single SAA 1.1 protein. It is notable that 16.5 % of the analyzed samples (28 out of 170 samples) express three SAA proteins simultaneously and exhibit higher average total SAA concentration. Lowest total SAA concentration was observed in the single sample that expressed SAA 1.3 protein (Fig.4).
Fig. 4.
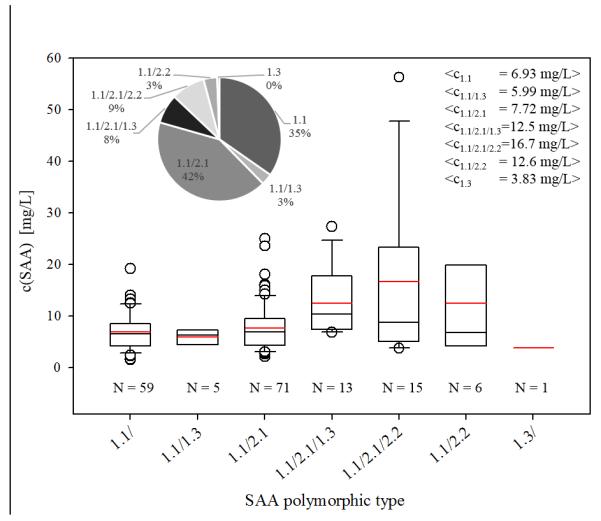
Total SAA concentration distribution among different SAA polymorphic profiles identified in the sample cohort; * The inlets present the percentage distribution of the SAA polymorphic profiles (left) and the total average SAA concentration in each sample subset (right).
Beside the total concentration and polymorphism, SAA MSIA enables for detection of posttranslational modifications, primarily N-truncated proteoforms. Posttranslationally modified proteoforms are significant due to their potential to be utilized as biomarkers for certain conditions (Trenchevska et al., 2010). Due to the confines of current methodologies, only limited number of SAA PTMs (primarily associated with C-truncations in amyloidosis, and not detected in this study) have been addressed, leaving out the additional N-truncated proteoforms. As a result, their role in inflammation and other clinical conditions is not well studied. Using SAA MSIA we were able to quantify 8 SAA PTMs originating from the 4 native SAA proteins (Table III). SAA lacking N-terminal arginine and N-terminal arginine-serine dipeptide residues were identified in all samples, while less abundant proteoforms lacking N-terminal tetra peptide (des-RSFF) was identified for SAA 1.1, SAA 1.3 and SAA 2.1 proteins. An interesting distribution was observed for the abundance of the des-R truncated SAA proteoforms (Fig. 5/a-d): SAA des-R proteoforms were present at high concentrations, regardless of the concentration of native SAA.
Fig. 5.

Concentration distribution of posttranslationally modified SAA proteoforms: a) average total concentrations of SAA 1.1 proteins; b) average total concentrations of SAA 1.3 proteins; c) average total concentrations of SAA 2.1 proteins; d) average total concentrations of SAA 2.2 proteins.
This high percentage of des-R – to – native SAA is important because it suggests existence of mechanisms that favor this N-terminal truncation in vivo. Although this PTM accounts for more than 50 % of the total SAA, a little is known regarding its role in inflammation and disease. We have previously addressed the importance of des-R SAA 1.1 and reported that the distribution of SAA 1.1R proteoform is gender related and inversely associated with plasma glucose levels in type 2 diabetes patients (Yassine et al., 2015). In this work, we show that a similar distribution ratio between native SAA and des-R SAA occurs for the other SAA proteins (SAA 1.3, SAA 2.1 and SAA 2.2). In addition, using the developed MSIA, we provide fully quantitative data and confirm that des-R truncated SAA proteoforms are highly abundant proteoforms in all SAA entities.
To the best of our knowledge, this is the first assay that provides in-depth analysis of both SAA profile and concentration of all SAA proteoforms in human plasma samples. Further analyses in clinical samples are necessary in order to investigate the potential relation between truncated SAA proteoforms in inflammation and specific clinical conditions.
Conclusion
Serum amyloid A is a highly heterogeneous protein that is subjected to posttranslational processing in vivo. Its role as an inflammation marker is related to the total concentration in plasma and/or serum. However, limited numbers of studies have exploited the importance of the polymorphism and posttranslationally modified SAA proteoforms in assessing the structure-function relation.
The new SAA MSIA enables parallel analysis of SAA polymorphisms and simultaneously identifies and quantifies all expressed SAA proteoforms. The assay was validated and demonstrated good correlation with a commercially available ELISA, but it provided a unique information about the concentration of each of the expressed SAA proteins and their PTMs, all in a single analysis. The developed MSIA is high-throughput – a total of 85 samples can be analyzed in a single run on an automated platform, and is time-efficient – results for SAA profile and concentration can be obtained in less than 2 hours.
Acknowledgements
This work was supported in part by Award Numbers R01DK082542, R24DK090958 to O.T., C.R.B., R.W.N and D.N., and K23HL107389 to H.N.Y from the National Institute of Diabetes and Digestive and Kidney Diseases. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Diabetes and Digestive and Kidney Diseases or the National Institutes of Health.
Footnotes
Declaration of interest
All authors report no declaration of interest.
References
- ALDO-BENSON MA, BENSON MD. SAA suppression of immune response in vitro: evidence for an effect on T cell-macrophage interaction. J Immunol. 1982;128:2390–2. [PubMed] [Google Scholar]
- ANDERSSON LO. Pharmacology of apolipoprotein A-I. Curr Opin Lipidol. 1997;8:225–8. doi: 10.1097/00041433-199708000-00006. [DOI] [PubMed] [Google Scholar]
- ARMBRUSTER DA, PRY T. Limit of blank, limit of detection and limit of quantitation. Clin Biochem Rev. 2008;29(Suppl 1):S49–52. [PMC free article] [PubMed] [Google Scholar]
- ARTL A, MARSCHE G, LESTAVEL S, SATTLER W, MALLE E. Role of serum amyloid A during metabolism of acute-phase HDL by macrophages. Arterioscler Thromb Vasc Biol. 2000;20:763–72. doi: 10.1161/01.atv.20.3.763. [DOI] [PubMed] [Google Scholar]
- BABA S, MASAGO SA, TAKAHASHI T, KASAMA T, SUGIMURA H, TSUGANE S, TSUTSUI Y, SHIRASAWA H. A novel allelic variant of serum amyloid A, SAA1 gamma: genomic evidence, evolution, frequency, and implication as a risk factor for reactive systemic AA-amyloidosis. Hum Mol Genet. 1995;4:1083–7. doi: 10.1093/hmg/4.6.1083. [DOI] [PubMed] [Google Scholar]
- BADOLATO R, WANG JM, MURPHY WJ, LLOYD AR, MICHIEL DF, BAUSSERMAN LL, KELVIN DJ, OPPENHEIM JJ. Serum amyloid A is a chemoattractant: induction of migration, adhesion, and tissue infiltration of monocytes and polymorphonuclear leukocytes. J Exp Med. 1994;180:203–9. doi: 10.1084/jem.180.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAKKALOGLU A, DUZOVA A, OZEN S, BALCI B, BESBAS N, TOPALOGLU R, OZALTIN F, YILMAZ E. Influence of Serum Amyloid A (SAA1) and SAA2 gene polymorphisms on renal amyloidosis, and on SAA/C-reactive protein values in patients with familial mediterranean fever in the Turkish population. J Rheumatol. 2004;31:1139–42. [PubMed] [Google Scholar]
- BANKA CL, YUAN T, DE BEER MC, KINDY M, CURTISS LK, DE BEER FC. Serum amyloid A (SAA): influence on HDL-mediated cellular cholesterol efflux. J Lipid Res. 1995;36:1058–65. [PubMed] [Google Scholar]
- BETTS JC, EDBROOKE MR, THAKKER RV, WOO P. The human acute-phase serum amyloid A gene family: structure, evolution and expression in hepatoma cells. Scand J Immunol. 1991;34:471–82. doi: 10.1111/j.1365-3083.1991.tb01570.x. [DOI] [PubMed] [Google Scholar]
- BLAND JM, ALTMAN DG. Measuring agreement in method comparison studies. Stat Methods Med Res. 1999;8:135–60. doi: 10.1177/096228029900800204. [DOI] [PubMed] [Google Scholar]
- BOOTH DR, BOOTH SE, GILLMORE JD, HAWKINS PN, PEPYS MB. SAA1 alleles as risk factors in reactive systemic AA amyloidosis. Amyloid. 1998;5:262–5. doi: 10.3109/13506129809007299. [DOI] [PubMed] [Google Scholar]
- CARR SA, ABBATIELLO SE, ACKERMANN BL, BORCHERS C, DOMON B, DEUTSCH EW, GRANT RP, HOOFNAGLE AN, HÜTTENHAIN R, KOOMEN JM, LIEBLER DC, LIU T, MACLEAN B, MANI DR, MANSFIELD E, NEUBERT H, PAULOVICH AG, REITER L, VITEK O, AEBERSOLD R, ANDERSON L, BETHEM R, BLONDER J, BOJA E, BOTELHO J, BOYNE M, BRADSHAW RA, BURLINGAME AL, CHAN D, KESHISHIAN H, KUHN E, KINSINGER C, LEE JS, LEE SW, MORITZ R, OSES-PRIETO J, RIFAI N, RITCHIE J, RODRIGUEZ H, SRINIVAS PR, TOWNSEND RR, VAN EYK J, WHITELEY G, WIITA A, WEINTRAUB S. Targeted peptide measurements in biology and medicine: best practices for mass spectrometry-based assay development using a fit-for-purpose approach. Mol Cell Proteomics. 2014;13:907–17. doi: 10.1074/mcp.M113.036095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHAU CH, RIXE O, MCLEOD H, FIGG WD. Validation of analytic methods for biomarkers used in drug development. Clin Cancer Res. 2008;14:5967–76. doi: 10.1158/1078-0432.CCR-07-4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CUNNANE G. Amyloid precursors and amyloidosis in inflammatory arthritis. Curr Opin Rheumatol. 2001;13:67–73. doi: 10.1097/00002281-200101000-00011. [DOI] [PubMed] [Google Scholar]
- DOSSUS L, BECKER S, ACHAINTRE D, KAAKS R, RINALDI S. Validity of multiplex-based assays for cytokine measurements in serum and plasma from "non-diseased" subjects: comparison with ELISA. J Immunol Methods. 2009;350:125–32. doi: 10.1016/j.jim.2009.09.001. [DOI] [PubMed] [Google Scholar]
- DUCRET A, BRUUN CF, BURES EJ, MARHAUG G, HUSBY G, AEBERSOLD R. Characterization of human serum amyloid A protein isoforms separated by two-dimensional electrophoresis by liquid chromatography/electrospray ionization tandem mass spectrometry. Electrophoresis. 1996;17:866–76. doi: 10.1002/elps.1150170508. [DOI] [PubMed] [Google Scholar]
- EKLUND KK, NIEMI K, KOVANEN PT. Immune functions of serum amyloid A. Crit Rev Immunol. 2012;32:335–48. doi: 10.1615/critrevimmunol.v32.i4.40. [DOI] [PubMed] [Google Scholar]
- FARWIG ZN, MCNEAL CJ, LITTLE D, BAISDEN CE, MACFARLANE RD. Novel truncated isoforms of constitutive serum amyloid A detected by MALDI mass spectrometry. Biochem Biophys Res Commun. 2005;332:352–6. doi: 10.1016/j.bbrc.2005.04.129. [DOI] [PubMed] [Google Scholar]
- FDA A Guidance for Industry - Bioanalytical method validation. 2001.
- FYFE AI, ROTHENBERG LS, DEBEER FC, CANTOR RM, ROTTER JI, LUSIS AJ. Association between serum amyloid A proteins and coronary artery disease: evidence from two distinct arteriosclerotic processes. Circulation. 1997;96:2914–9. doi: 10.1161/01.cir.96.9.2914. [DOI] [PubMed] [Google Scholar]
- HUSBY G, MARHAUG G, DOWTOR B, SLETTEN K, SIPE J,D. Serum amyloid A (SAA): Biochemistry, genetics and the pathogenesis of AA amyloidosis. Amyloid. 1994 [Google Scholar]
- JOHNSON BD, KIP KE, MARROQUIN OC, RIDKER PM, KELSEY SF, SHAW LJ, PEPINE CJ, SHARAF B, BAIREY MERZ CN, SOPKO G, OLSON MB, REIS SE, NATIONAL HEART, L. N. BLOOD INSTITUTE Serum amyloid A as a predictor of coronary artery disease and cardiovascular outcome in women: the National Heart, Lung, and Blood Institute-Sponsored Women's Ischemia Syndrome Evaluation (WISE) Circulation. 2004;109:726–32. doi: 10.1161/01.CIR.0000115516.54550.B1. [DOI] [PubMed] [Google Scholar]
- KIERNAN UA, NEDELKOV D, NELSON RW. Multiplexed mass spectrometric immunoassay in biomarker research: a novel approach to the determination of a myocardial infarct. J Proteome Res. 2006;5:2928–34. doi: 10.1021/pr060062+. [DOI] [PubMed] [Google Scholar]
- KIERNAN UA, TUBBS KA, NEDELKOV D, NIEDERKOFLER EE, NELSON RW. Detection of novel truncated forms of human serum amyloid A protein in human plasma. FEBS Lett. 2003;537:166–70. doi: 10.1016/s0014-5793(03)00097-8. [DOI] [PubMed] [Google Scholar]
- KISILEVSKY R, SUBRAHMANYAN L. Serum amyloid A changes high density lipoprotein's cellular affinity. A clue to serum amyloid A's principal function. Lab Invest. 1992;66:778–85. [PubMed] [Google Scholar]
- KLUVE-BECKERMAN B, DWULET FE, BENSON MD. Human serum amyloid A. Three hepatic mRNAs and the corresponding proteins in one person. J Clin Invest. 1988;82:1670–5. doi: 10.1172/JCI113779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIANG JS, SIPE JD. Recombinant human serum amyloid A (apoSAAp) binds cholesterol and modulates cholesterol flux. J Lipid Res. 1995;36:37–46. [PubMed] [Google Scholar]
- LOO D, MOLLEE PN, RENAUT P, HILL MM. Proteomics in molecular diagnosis: typing of amyloidosis. J Biomed Biotechnol. 2011;2011:754109. doi: 10.1155/2011/754109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MACGREGOR AJ, GALLIMORE JR, SPECTOR TD, PEPYS MB. Genetic effects on baseline values of C-reactive protein and serum amyloid a protein: a comparison of monozygotic and dizygotic twins. Clin Chem. 2004;50:130–4. doi: 10.1373/clinchem.2003.028258. [DOI] [PubMed] [Google Scholar]
- MALLE E, STEINMETZ A, RAYNES JG. Serum amyloid A (SAA): an acute phase protein and apolipoprotein. Atherosclerosis. 1993;102:131–46. doi: 10.1016/0021-9150(93)90155-n. [DOI] [PubMed] [Google Scholar]
- MANLEY PN, ANCSIN JB, KISILEVSKY R. Rapid recycling of cholesterol: the joint biologic role of C-reactive protein and serum amyloid A. Med Hypotheses. 2006;66:784–92. doi: 10.1016/j.mehy.2005.10.018. [DOI] [PubMed] [Google Scholar]
- MARSCHE G, FRANK S, RAYNES JG, KOZARSKY KF, SATTLER W, MALLE E. The lipidation status of acute-phase protein serum amyloid A determines cholesterol mobilization via scavenger receptor class B, type I. Biochem J. 2007;402:117–24. doi: 10.1042/BJ20061406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NELSON RW, BORGES CR. Mass spectrometric immunoassay revisited. J Am Soc Mass Spectrom. 2011;22:960–8. doi: 10.1007/s13361-011-0094-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NELSON RW, KRONE JR, BIEBER AL, WILLIAMS P. Mass spectrometric immunoassay. Anal Chem. 1995;67:1153–8. doi: 10.1021/ac00103a003. [DOI] [PubMed] [Google Scholar]
- NIEDERKOFLER EE, PHILLIPS DA, KRASTINS B, KULASINGAM V, KIERNAN UA, TUBBS KA, PETERMAN SM, PRAKASH A, DIAMANDIS EP, LOPEZ MF, NEDELKOV D. Targeted selected reaction monitoring mass spectrometric immunoassay for insulin-like growth factor 1. PLoS One. 2013;8:e81125. doi: 10.1371/journal.pone.0081125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PASSING H, BABLOK A new biometrical procedure for testing the equality of measurements from two different analytical methods. Application of linear regression procedures for method comparison studies in clinical chemistry, Part I. J Clin Chem Clin Biochem. 1983;21:709–20. doi: 10.1515/cclm.1983.21.11.709. [DOI] [PubMed] [Google Scholar]
- PIZZINI C, MUSSAP M, PLEBANI M, FANOS V. C-reactive protein and serum amyloid A protein in neonatal infections. Scand J Infect Dis. 2000;32:229–35. doi: 10.1080/00365540050165848. [DOI] [PubMed] [Google Scholar]
- RISTORI G, LAURENTI F, STACCHINI P, GASPERINI C, BUTTINELLI C, POZZILLI C, SALVETTI M. Serum amyloid A protein is elevated in relapsing-remitting multiple sclerosis. J Neuroimmunol. 1998;88:9–12. doi: 10.1016/s0165-5728(98)00037-x. [DOI] [PubMed] [Google Scholar]
- SELLAR GC, JORDAN SA, BICKMORE WA, FANTES JA, VAN HEYNINGEN V, WHITEHEAD AS. The human serum amyloid A protein (SAA) superfamily gene cluster: mapping to chromosome 11p15.1 by physical and genetic linkage analysis. Genomics. 1994a;19:221–7. doi: 10.1006/geno.1994.1051. [DOI] [PubMed] [Google Scholar]
- SELLAR GC, OGHENE K, BOYLE S, BICKMORE WA, WHITEHEAD AS. Organization of the region encompassing the human serum amyloid A (SAA) gene family on chromosome 11p15.1. Genomics. 1994b;23:492–5. doi: 10.1006/geno.1994.1530. [DOI] [PubMed] [Google Scholar]
- SIPE J. Revised nomenclature for serum amyloid A (SAA). Nomenclature Committee of the International Society of Amyloidosis. Part 2. Amyloid. 1999;6:67–70. doi: 10.3109/13506129908993291. [DOI] [PubMed] [Google Scholar]
- SKOGEN B, NATVIG JB. Degradation of amyloid proteins by different serine proteases. Scand J Immunol. 1981;14:389–96. doi: 10.1111/j.1365-3083.1981.tb00579.x. [DOI] [PubMed] [Google Scholar]
- STEINMETZ A, HOCKE G, SAÏLE R, PUCHOIS P, FRUCHART JC. Influence of serum amyloid A on cholesterol esterification in human plasma. Biochim Biophys Acta. 1989;1006:173–8. doi: 10.1016/0005-2760(89)90192-6. [DOI] [PubMed] [Google Scholar]
- TAKASE H, TANAKA M, MIYAGAWA S, YAMADA T, MUKAI T. Effect of amino acid variations in the central region of human serum amyloid A on the amyloidogenic properties. Biochem Biophys Res Commun. 2014;444:92–7. doi: 10.1016/j.bbrc.2014.01.029. [DOI] [PubMed] [Google Scholar]
- TOLSON J, BOGUMIL R, BRUNST E, BECK H, ELSNER R, HUMENY A, KRATZIN H, DEEG M, KUCZYK M, MUELLER GA, MUELLER CA, FLAD T. Serum protein profiling by SELDI mass spectrometry: detection of multiple variants of serum amyloid alpha in renal cancer patients. Lab Invest. 2004;84:845–56. doi: 10.1038/labinvest.3700097. [DOI] [PubMed] [Google Scholar]
- TRENCHEVSKA O, KAMCHEVA E, NEDELKOV D. Mass spectrometric immunoassay for quantitative determination of protein biomarker isoforms. J Proteome Res. 2010;9:5969–73. doi: 10.1021/pr1007587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TRENCHEVSKA O, KAMCHEVA E, NEDELKOV D. Mass spectrometric immunoassay for quantitative determination of transthyretin and its variants. Proteomics. 2011;11:3633–41. doi: 10.1002/pmic.201100023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TRENCHEVSKA O, PHILLIPS DA, NELSON RW, NEDELKOV D. Delineation of concentration ranges and longitudinal changes of human plasma protein variants. PLoS One. 2014;9:e100713. doi: 10.1371/journal.pone.0100713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TRENCHEVSKA O, SCHAAB MR, NELSON RW, NEDELKOV D. Development of multiplex mass spectrometric immunoassay for detection and quantification of apolipoproteins C-I, C-II, C-III and their proteoforms. Methods. 2015a doi: 10.1016/j.ymeth.2015.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TRENCHEVSKA O, SHERMA ND, ORAN PE, REAVEN PD, NELSON RW, NEDELKOV D. Quantitative mass spectrometric immunoassay for the chemokine RANTES and its variants. J Proteomics. 2015b;116:15–23. doi: 10.1016/j.jprot.2014.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UHLAR CM, WHITEHEAD AS. Serum amyloid A, the major vertebrate acute-phase reactant. Eur J Biochem. 1999;265:501–23. doi: 10.1046/j.1432-1327.1999.00657.x. [DOI] [PubMed] [Google Scholar]
- WU TL, I CHEN TSAI, CHANG PY, TSAO KC, SUN CF, WU LL, WU JT. Establishment of an in-house ELISA and the reference range for serum amyloid A (SAA): complementarity between SAA and C-reactive protein as markers of inflammation. Clin Chim Acta. 2007;376:72–6. doi: 10.1016/j.cca.2006.07.012. [DOI] [PubMed] [Google Scholar]
- XIE X, MA YT, YANG YN, FU ZY, LI XM, HUANG D, MA X, CHEN BD, LIU F. Polymorphisms in the SAA1/2 gene are associated with carotid intima media thickness in healthy Han Chinese subjects: the Cardiovascular Risk Survey. PLoS One. 2010;5:e13997. doi: 10.1371/journal.pone.0013997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAMADA T, NOMATA Y, SUGITA O, OKADA M. A rapid method for measuring serum amyloid A protein by latex agglutination nephelometric immunoassay. Ann Clin Biochem. 1993;30(Pt 1):72–6. doi: 10.1177/000456329303000112. [DOI] [PubMed] [Google Scholar]
- YAMADA T, WADA A, ITOH Y, ITOH K. Serum amyloid A1 alleles and plasma concentrations of serum amyloid A. Amyloid. 1999;6:199–204. doi: 10.3109/13506129909007327. [DOI] [PubMed] [Google Scholar]
- YASSINE HN, TRENCHEVSKA O, HE H, BORGES CR, NEDELKOV D, MACK W, KONO N, KOSKA J, REAVEN PD, NELSON RW. Serum amyloid a truncations in type 2 diabetes mellitus. PLoS One. 2015;10:e0115320. doi: 10.1371/journal.pone.0115320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHAO Y, HE X, SHI X, HUANG C, LIU J, ZHOU S, HENG CK. Association between serum amyloid A and obesity: a meta-analysis and systematic review. Inflamm Res. 2010;59:323–34. doi: 10.1007/s00011-010-0163-y. [DOI] [PubMed] [Google Scholar]