

Abstract
DNA repair defects create cancer predisposition in humans by fostering a higher rate of mutations. While DNA repair is quite well characterized, recent studies have identified previously unrecognized relationships between DNA repair and R-loop-mediated genome instability. R-loops are three stranded nucleic acid structures in which RNA binds to genomic DNA to displace a loop of single-stranded DNA. Mutations in homologous recombination, nucleotide excision repair, crosslink repair and DNA damage checkpoints have all now been linked to formation and function of transcription-coupled R-loops. This perspective will summarize recent literature linking DNA repair to R-loop mediated genomic instability and discuss how R-loops may contribute to mutagenesis in DNA-repair deficient cancers.
Graphical Abstract
Background
Faithful replication and segregation of DNA during cell division is essential for survival. While a low level of mutagenesis is tolerated across generations, higher mutational loads are deleterious1. In order to suppress the accumulation of mutations and maintain the genome, organisms have evolved a robust replication and mitotic apparatus and a semi-redundant network of DNA repair machinery and cell cycle control mechanisms. Defects in these pathways can be associated with severe developmental disorders that are often characterized in part by a dramatic increase in cancer incidence2. Indeed, heterozygous deficiencies of genome maintenance genes are well established contributors to an increased risk of cancer formation3. Finally, somatic mutations or silencing of genome maintenance genes is also frequently found in cancer genomes4 and likely contribute to the ongoing genome instability phenotypes that characterize most cancers.
Some of the best characterized groups of genes associated with genome maintenance and tumour suppression are those involved in the cellular response to DNA damage. Detection of DNA base damage or strand breaks engages one of several partially redundant DNA repair pathways, depending primarily on the type of lesion and the cell cycle stage. Homologous recombination (HR) or non-homologous end joining (NHEJ) corrects double-strand breaks; base excision repair (BER) or nucleotide excision repair (NER) corrects base damage; mismatch repair (MMR) corrects DNA replication errors; translesion synthesis (TLS) promotes damage tolerance during replication; and the Fanconi Anemia (FA) pathway corrects DNA interstrand crosslinks. These core pathways, along with some variations such as alternative-NHEJ5, ribonucleotide-excision repair6, or post-replication repair7, ensure a robust system in eukaryotic cells to tolerate or reverse a diverse array of potentially damaging DNA lesions and suppress their mutagenic effects.
One endogenously generated threat to genome maintenance that has gained prominence over the past decade is R-loops8; 9. R-loops refer to the formation of a stable DNA:RNA hybrid on genomic DNA such that single-stranded DNA (ssDNA) on the non-template strand is exposed (Figure 1A). R-loops cause genome instability by at least two mechanisms: first exposed ssDNA is more chemically labile and can also be inappropriately recognized by mutagenic cellular enzymes such as cytidine deaminases; second R-loops can block replication fork progression leading to replication stress and the possibility of triggering error prone modes of replication and repair8. To mitigate these risks eukaryotes express RNaseH1 and RNaseH2, two enzymes that specifically degrade the RNA moiety of an DNA:RNA hybrid10. Moreover, it has been established that many transcription, RNA 3′-end processing, termination, packaging and export associated factors work to prevent R-loop formation11; 12; 13; 14; 15. Remaining ‘constitutive’ R-loops do not cause genome instability and instead appear to play important regulatory roles in gene expression, telomere maintenance and other pathways 8; 16; 17.
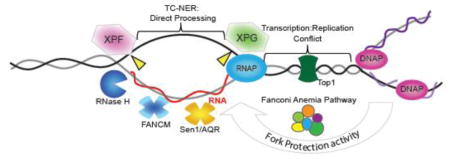
Figure 1.

Potential modes of action for DNA damage proteins in R-loop mitigation. (A) R-loop structure is shown (RNA = red; DNA = grey/black) occurring co-transcriptionally with RNA polymerase (RNAP). The RNA exits RNAPII at a separate position from the template DNA so the RNA must re-invade to form an R-loop. Also shown are core anti-R-loop enzymatic activities: RNase H which degrades the RNA moiety; Helicases such as Senataxin (Sen1)39 or Aquarius (AQR)25 which may unwind R-loops; and Topoisomerase which reduces topological strain favoring RNA invasion. (B) DNA damage induced R-loops. Transcription into a damaged DNA region would experience altered chromatin environment (i.e. remodelers actively suppress transcription next to breaks20), collision with repair proteins or be subject to orchestrated disassembly by DNA repair signaling (e.g. spliceosome displacement favoring R-loops21). (C) Direct processing of R-loops into DNA breaks by the transcription-coupled nucleotide excision repair (TC-NER) machinery. Putative cut sites are shown in yellow although the cut strand is not known25. (D) Interplay between replication fork protection activities and R-loop stability. At sites of transcription-replication conflict the Fanconi Anemia pathway, which includes BRCA1, BRCA2 and XPF, act to suppress R-loop formation36; 37. The FANCM helicase is shown as a mediator that could directly remove R-loops through its strand migration activity36.
Until 2013, R-loops and DNA repair proteins occupied related but parallel lines of inquiry in the genome maintenance field; DNA repair dealt with DNA damage while R-loops were ‘upstream’ of DNA damage and were controlled by the actions of RNA processing factors and RNaseHs. Since this time there has been growing evidence that DNA repair factors may actively be recruited to R-loops, or that R-loops collaborate with DNA repair proteins to send signals for repair. Here we describe the direct links between DNA repair and R-loops and discuss the potential implications of these links for the role of R-loops in cancer.
DNA damage causes R-loops
While the role of R-loops in causing DNA damage has been carefully established over the past decade, more recent evidence suggests that the reverse is also true – DNA damage favors the formation of R-loops. The role of DNA damage in initiating R-loops was illustrated first in vitro by the definition of R-loop Initiation Zones on plasmids18. The authors found that a single-stranded DNA nick downstream of the promoter served as a strong, transcription-dependant initiator of transient R-loop formation, most likely because the non-template strand can be more easily displaced to allow R-loop initiation. Triggering of R-loop formation by DNA damage was later observed in cells exposed to laser-induced DNA damage19. To measure R-loops in live cells, Britton et al., employed an mCherry-tagged catalytic mutant of E. coli RNaseHI that bound, but could not degrade, DNA:RNA hybrids. The authors found that R-loops accumulated transiently in live cells at tracks of laser-induced damage and coincided with canonical DNA damage marks such as γ-H2AX19. It seemed plausible that transcriptional machinery, present proximal to a lesion on DNA, could stall and thereby increase the chance occurrence of R-loops; indeed, it is known that lesion proximal transcription is actively suppressed by chromatin remodellers20 (Figure 1B). Tresini et al., recently painted a more complex and regulated picture of damage-induced R-loops21. These authors began by measuring the effects of ultraviolet irradiation on chromatin-binding proteins in fibroblasts and found that the U2 and U4 spliceosomes became more dynamic after irradiation. They showed that mature spliceosomes are evicted in the presence of bulky transcription-blocking DNA lesions and, using a mutant form of GFP-RNaseH1 to bind R-loops in cells, they showed that R-loops accumulate at sites of UV-induced damage. Finally, they found that R-loop formation led to ATM activation which propagated a local signal to the global transcriptional machinery to regulate alternative splicing. Thus, the initial spliceosome displacement leads to R-loop formation locally at the site of damage, and this is signaled via ATM to induce global changes in spliceosome dynamics and thus regulate gene expression21. These studies provide further evidence that certain types of DNA damage favour the formation of R-loops and highlight the potentially important signaling roles of R-loops in genome maintenance. Indeed, there are now several fascinating examples, outside the scope of this review, which show that RNAs act in cis at sites of DNA damage to promote repair by various mechanisms22; 23
DNA repair proteins suppress R-loop formation
Recently, the identification of roles for DNA repair proteins in suppressing R-loop formation has accelerated. In the past 5 years, defects in homologous recombination (HR), nucleotide excision repair (NER), and the Fanconi Anemia (FA) pathway have been implicated in R-loop suppression. In addition, the DNA damage response kinase ATM has been shown to regulate R-loop-dependent signaling in response to DNA damage.
NER – R-loops are known sources of DNA damage but the mechanisms by which the damage arises are not clear. One well known source of instability at R-loops is Activation Induced cytidine Deaminase (AID), which targets the R-loops at the immunoglobulin locus for damage24. However, AID is not expressed in most cells and there must be other mechanisms at play. Sollier et al., began by identifying a putative helicase called Aquarius (AQR) with homology to Senataxin whose depletion led to increased R-loops and DNA damage25. Previous work had shown that nucleases associated with NER called XPF and XPG have the capability of processing R-loops in vitro26. Therefore, the authors tested whether XPF or XPG were responsible for the DNA damage observed in AQR-deficient cells. Through a series of experiments it was shown that the nuclease activity of XPF or XPG could directly engage and cut DNA at R-loops. The authors went on to show that it is specifically the transcription coupled (TC) NER, rather than the global genome (GG) NER, pathway which is important for breaks at R-loops (Figure 1C). Depletion of the TC-NER specific factor CSB suppressed damage in AQR-depleted cells, while depletion of the GG-NER regulator XPC had no effect. The R-loop cleavage induced by the TC-NER pathway may play a normal role in R-loop clearance because co-depletion of XPG and AQR led to an even greater increase in R-loop formation. Interestingly, TC-NER factors appear to lead to double-strand breaks (DSBs) at R-loops rather than the expected single-strand breaks (SSBs). The authors suggest that a non-canonical activity of XPF/G could cut both strands, that another flap endonuclease is involved, or simply that progress through SSBs during DNA replication leads to the DSBs25. Currently, the detailed mechanisms are under exploration but it is important to note that, while NER proteins seem to promote R-loop clearance, they do so by causing DNA breaks and genome instability. Thus, normal R-loop clearance by NER factors differs from other anti-R-loop mechanisms by involving potentially genotoxic strand breakage events.
BRCA1 and BRCA2
Connections between R-loops and the breast cancer susceptibility genes BRCA1 and BRCA2 first emerged for BRCA2 in 201427. Both BRCA1 and BRCA2 play roles at early stages of homologous recombination, where BRCA2 regulates Rad51 loading onto ssDNA, and in the stability of stalled DNA replication forks28. Bhatia et al., began by characterizing the physical interaction between BRCA2 and PCID2, a member of an RNA packaging complex called TREX-2. TREX-2 was previously identified as a suppressor of R-loop formation and the physical interactions prompted the authors to analyze R-loops in BRCA2 depleted cells. R-loops accumulated in cells depleted for BRCA2 and, to a lesser extent, BRCA1. Importantly, to detect increased R-loops, the authors used both the S9.6 monoclonal antibody, which specifically recognizes DNA:RNA hybrids, and a hybrid-binding (HB) domain of RNaseH fused to GFP. Cells depleted for BRCA2, or bearing BRCA2 mutations, showed increased γ-H2Ax staining, a marker of DNA damage, that was dependent on RNaseH activity, suggesting that R-loops were driving at least some of the DNA damage seen. Interestingly, R-loops seemed to form regardless of whether the BRCA2 depleted cells were in S-phase, although the phenotype was stronger in S-phase cells. Finally, the authors found that BRCA2 binding to R-loop prone genes was partially RNaseH-dependent, suggesting that active recruitment to R-loop forming sites occurred27.
BRCA1 was separately linked to transcription coupled R-loop suppression when it was recognized that it binds the termination factor Senataxin, a helicase that specifically unwinds DNA:RNA hybrids14; 29. BRCA1 is recruited to transcriptional pause-site associated R-loops, where its interaction with Senataxin helps to prevent ssDNA damage. This DNA damage is R-loop dependent in BRCA1 depleted cells, directly linking R-loop driven DNA breaks to loss of BRCA1. Remarkably, these authors went one step further and showed that, in BRCA1-deficient human tumours, there is a significant enrichment of mutations at precisely the locations where BRCA1 is predicted to play a role in suppressing mutations during transcription termination29. These data strongly suggested that R-loops are responsible for at least some of the mutations observed in cancer genomes and is a strong step toward understanding the role of R-loops in cancer formation.
Fanconi Anemia Pathway
Emerging from the studies of BRCA1/2 was the question of which roles of the BRCA proteins were engaged to deal with R-loops. Hatchi et al. suggested that BRCA1 or 2 could have independent moonlighting functions in transcription that relate to R-loop suppression. Alternatively, R-loop suppressive functions could relate to coordinated functions of BRCA1 and 2 in homologous recombination30, or in replication fork protection as part of the Fanconi Anemia pathway31, where BRCA1 is called FANCS32 and BRCA2 is FANCD133. Two recent papers have implicated the Fanconi Anemia pathway as an active suppressor of R-loop formation. The Fanconi Anemia pathway involves at least 18 FANC genes (named A-T), promotes faithful DNA replication, and is essential for accurate repair of DNA interstrand crosslinks (ICLs)34; 35. Focussing on FANCD2, but also testing FANCA, FANCL and FANCM, Schwab et al. showed that the role of the FA pathway in replication fork stability is partly transcription dependent and that FA proteins localize at transcribed regions36. They went on to show that cells depleted of FA proteins accumulate excess R-loops and that replication impairment is RNaseH-sensitive in these cells, implicating R-loops directly. Finally, tests with purified protein and in DT-40 cells showed that the translocase activity of the helicase FANCM is required to suppress R-loops36. Garcia-Rubio et al., also showed that FA protein depletion led to R-loop accumulation in various cell models and that these R-loops directly contributed to genome instability37. They additionally showed that mitomycin C induced FANCD2 foci assemble in a manner that is dependent on R-loops. Helping replication forks navigate transcription-replication collisions is a function ascribed to many factors including topoisomerase I38, Senataxin39, and INO80/PAF140. Studies on the FA proteins suggest an important role for DNA repair proteins at the replication fork in suppressing R-loops and their deleterious consequences at these sites of collision (Figure 1D). Replication forks are comprised of dozens of proteins and a host of fork protection factors that respond to replication stress; going forward, it will be important to define which factors directly or indirectly act to destabilize and resolve R-loops.
R-loop promoting roles of DNA repair
Despite contradictory roles for other HR proteins in humans, in the yeast system, HR proteins Rad51 and Rad52 have actually been ascribed a pro-R-loop function. Wahba et al., described a role for Rad51/52 in promoting R-loops possibly by facilitating strand invasion of free RNA to its complementary DNA in the genome 41. This study was the first to describe a role for R-loops formed in trans, in other words not co-transcriptionally, in promoting genome instability. The authors found that some mutant strains increased R-loops in a manner that is dependent on Rad51, where others did not. The role of Rad51 in loading R-loops onto genomic DNA has not been established in any other systems and the regulation of this process is not clear. Nevertheless, it highlights how the balance of normal DNA repair activities is critical to prevent instability. Indeed, the same study highlights the anti-recombinase Srs2 as a novel anti-R-loop mechanism via its opposition to Rad51-mediated strand invasion 41.
Perspective
R-loops are known sources of genome instability and it is now clear that DNA repair and DNA replication fork progression factors, actively work to suppress R-loop-mediated DNA damage. Indeed, RNA processing factors and R-loops appear to have important roles in the DNA damage repair pathway42. Meanwhile genome-wide analyses of R-loop prone regions highlight the normal regulatory functions of R-loops across species17; 43; 44. The question of how cells distinguish normal R-loops from deleterious ones remains unanswered. Moreover, presumably many other canonical genome maintenance and fork protection proteins will emerge as direct or indirect R-loop regulators if these pathways inherently have properties that suppress R-loops.
Prior work on R-loops has focused on RNA processing defects as a source of R-loop formation. Here again there is a growing convergence of observations to suggest that some RNA processing factors play a role in the DNA damage response. For example, BRCA1 binds to several core splicing factors after DNA damage to regulate the transcriptional response. In addition, a host of RNA processing factors have been implicated directly at break sites where they regulate DNA repair (Reviewed in 42). Thus, not only are DNA repair proteins directly impacting R-loops, but RNA processing factors are regulating DNA repair. There is clearly much to learn at the interface of these pathways in the cellular response to genotoxic stress.
The identification of R-loop-mediated genome instability in so many different types of DNA repair deficient cells raises the possibility that these R-loops are driving mutational processes that lead to cancer formation or progression. Indeed, R-loops could be mutagenic or their accumulation could lead to dysregulation of gene expression in cancer, for example, through their epigenetic action at CpG island promoters17; 44. Additional analysis of R-loop biology in in vivo cancer models will shed light on their prospective roles in tumours. If excess R-loops do accumulate in cancer genomes and cause a phenotype of genetic or epigenetic instability, then they might also be actionable as anti-cancer therapeutic targets. Approaches that target anti-R-loop mechanisms such as RNaseH, Senataxin or Xrn145 could provide a therapeutic benefit in such cases. It is worth noting that a well-characterized anti-R-loop player, Topoisomerase I38; 46, has been inhibited clinically in various cancers for many years. Whether the clinical efficacy of Top1 inhibition involves R-loops is unknown and only time will tell if inhibiting other anti-R-loop proteins will prove to be effective in cancer treatment.
Research Highlights.
R-loops are an important source of DNA damage and genome instability
DNA repair proteins implicated in tumour suppression limit R-loop induced DNA damage
DNA replication fork protection may exert an anti-R-loop effect
Acknowledgments
We thank members of the Hieter and Stirling labs for helpful comments on this manuscript. This research is funded by the Canadian Cancer Society (grant 703263 to P.C.S.). P.C.S. and P.H. are funded by the Canadian Institutes of Health Research (P.C.S. MOP 136982; P.H. MOP 38096) and P.H. is funded by the National Institute of Health (RO1CA158162). P.C.S. is a CIHR New Investigator and a Michael Smith Foundation for Health Research Scholar.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Herr AJ, Kennedy SR, Knowels GM, Schultz EM, Preston BD. DNA replication error-induced extinction of diploid yeast. Genetics. 2014;196:677–91. doi: 10.1534/genetics.113.160960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cleaver JE. Cancer in xeroderma pigmentosum and related disorders of DNA repair. Nat Rev Cancer. 2005;5:564–73. doi: 10.1038/nrc1652. [DOI] [PubMed] [Google Scholar]
- 3.Reliene R, Bishop AJ, Schiestl RH. Involvement of homologous recombination in carcinogenesis. Advances in Genetics. 2007;58:67–87. doi: 10.1016/S0065-2660(06)58003-4. [DOI] [PubMed] [Google Scholar]
- 4.Garraway LA, Lander ES. Lessons from the cancer genome. Cell. 2013;153:17–37. doi: 10.1016/j.cell.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 5.Bennardo N, Cheng A, Huang N, Stark JM. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. 2008;4:e1000110. doi: 10.1371/journal.pgen.1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sparks JL, Chon H, Cerritelli SM, Kunkel TA, Johansson E, Crouch RJ, Burgers PM. RNase H2-initiated ribonucleotide excision repair. Mol Cell. 2012;47:980–6. doi: 10.1016/j.molcel.2012.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin JR, Zeman MK, Chen JY, Yee MC, Cimprich KA. SHPRH and HLTF act in a damage-specific manner to coordinate different forms of postreplication repair and prevent mutagenesis. Mol Cell. 2011;42:237–49. doi: 10.1016/j.molcel.2011.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aguilera A, Garcia-Muse T. R loops: from transcription byproducts to threats to genome stability. Molecular cell. 2012;46:115–124. doi: 10.1016/j.molcel.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 9.Chan YA, Hieter P, Stirling PC. Mechanisms of genome instability induced by RNA-processing defects. Trends Genet. 2014;30:245–53. doi: 10.1016/j.tig.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cerritelli SM, Crouch RJ. Ribonuclease H: the enzymes in eukaryotes. FEBS J. 2009;276:1494–505. doi: 10.1111/j.1742-4658.2009.06908.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stirling PC, Chan YA, Minaker SW, Aristizabal MJ, Barrett I, Sipahimalani P, Kobor MS, Hieter P. R-loop-mediated genome instability in mRNA cleavage and polyadenylation mutants. Genes & development. 2012;26:163–175. doi: 10.1101/gad.179721.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li X, Manley JL. Inactivation of the SR protein splicing factor ASF/SF2 results in genomic instability. Cell. 2005;122:365–378. doi: 10.1016/j.cell.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 13.Gomez-Gonzalez B, Garcia-Rubio M, Bermejo R, Gaillard H, Shirahige K, Marin A, Foiani M, Aguilera A. Genome-wide function of THO/TREX in active genes prevents R-loop-dependent replication obstacles. The EMBO journal. 2011 doi: 10.1038/emboj.2011.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mischo HE, Gomez-Gonzalez B, Grzechnik P, Rondon AG, Wei W, Steinmetz L, Aguilera A, Proudfoot NJ. Yeast Sen1 helicase protects the genome from transcription-associated instability. Molecular cell. 2011;41:21–32. doi: 10.1016/j.molcel.2010.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Santos-Pereira JM, Herrero AB, Garcia-Rubio ML, Marin A, Moreno S, Aguilera A. The Npl3 hnRNP prevents R-loop-mediated transcription-replication conflicts and genome instability. Genes Dev. 2013;27:2445–58. doi: 10.1101/gad.229880.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Balk B, Maicher A, Dees M, Klermund J, Luke-Glaser S, Bender K, Luke B. Telomeric RNA-DNA hybrids affect telomere-length dynamics and senescence. Nat Struct Mol Biol. 2013;20:1199–205. doi: 10.1038/nsmb.2662. [DOI] [PubMed] [Google Scholar]
- 17.Ginno PA, Lim YW, Lott PL, Korf IF, Chedin F. GC skew at the 5′ and 3′ ends of human genes links R-loop formation to epigenetic regulation and transcription termination. Genome research. 2013 doi: 10.1101/gr.158436.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roy D, Zhang Z, Lu Z, Hsieh CL, Lieber MR. Competition between the RNA transcript and the nontemplate DNA strand during R-loop formation in vitro: a nick can serve as a strong R-loop initiation site. Mol Cell Biol. 2010;30:146–59. doi: 10.1128/MCB.00897-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Britton S, Dernoncourt E, Delteil C, Froment C, Schiltz O, Salles B, Frit P, Calsou P. DNA damage triggers SAF-A and RNA biogenesis factors exclusion from chromatin coupled to R-loops removal. Nucleic Acids Res. 2014;42:9047–62. doi: 10.1093/nar/gku601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kakarougkas A, Ismail A, Chambers AL, Riballo E, Herbert AD, Kunzel J, Lobrich M, Jeggo PA, Downs JA. Requirement for PBAF in transcriptional repression and repair at DNA breaks in actively transcribed regions of chromatin. Mol Cell. 2014;55:723–32. doi: 10.1016/j.molcel.2014.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tresini M, Warmerdam DO, Kolovos P, Snijder L, Vrouwe MG, Demmers JA, van IWF, Grosveld FG, Medema RH, Hoeijmakers JH, Mullenders LH, Vermeulen W, Marteijn JA. The core spliceosome as target and effector of non-canonical ATM signalling. Nature. 2015;523:53–8. doi: 10.1038/nature14512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Francia S, Michelini F, Saxena A, Tang D, de Hoon M, Anelli V, Mione M, Carninci P, d’Adda di Fagagna F. Site-specific DICER and DROSHA RNA products control the DNA-damage response. Nature. 2012;488:231–5. doi: 10.1038/nature11179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Keskin H, Shen Y, Huang F, Patel M, Yang T, Ashley K, Mazin AV, Storici F. Transcript-RNA-templated DNA recombination and repair. Nature. 2014;515:436–9. doi: 10.1038/nature13682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang FT, Yu K, Balter BB, Selsing E, Oruc Z, Khamlichi AA, Hsieh CL, Lieber MR. Sequence dependence of chromosomal R-loops at the immunoglobulin heavy-chain Smu class switch region. Mol Cell Biol. 2007;27:5921–32. doi: 10.1128/MCB.00702-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sollier J, Stork CT, Garcia-Rubio ML, Paulsen RD, Aguilera A, Cimprich KA. Transcription-coupled nucleotide excision repair factors promote R-loop-induced genome instability. Mol Cell. 2014;56:777–85. doi: 10.1016/j.molcel.2014.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tian M, Alt FW. Transcription-induced cleavage of immunoglobulin switch regions by nucleotide excision repair nucleases in vitro. J Biol Chem. 2000;275:24163–72. doi: 10.1074/jbc.M003343200. [DOI] [PubMed] [Google Scholar]
- 27.Bhatia V, Barroso SI, Garcia-Rubio ML, Tumini E, Herrera-Moyano E, Aguilera A. BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature. 2014;511:362–5. doi: 10.1038/nature13374. [DOI] [PubMed] [Google Scholar]
- 28.Michl J, Zimmer J, Tarsounas M. Interplay between Fanconi anemia and homologous recombination pathways in genome integrity. EMBO J. 2016;35:909–23. doi: 10.15252/embj.201693860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hatchi E, Skourti-Stathaki K, Ventz S, Pinello L, Yen A, Kamieniarz-Gdula K, Dimitrov S, Pathania S, McKinney KM, Eaton ML, Kellis M, Hill SJ, Parmigiani G, Proudfoot NJ, Livingston DM. BRCA1 Recruitment to Transcriptional Pause Sites Is Required for R-Loop-Driven DNA Damage Repair. Mol Cell. 2015;57:636–47. doi: 10.1016/j.molcel.2015.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karran P. DNA double strand break repair in mammalian cells. Current opinion in genetics & development. 2000;10:144–150. doi: 10.1016/s0959-437x(00)00069-1. [DOI] [PubMed] [Google Scholar]
- 31.Schlacher K, Wu H, Jasin M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell. 2012;22:106–16. doi: 10.1016/j.ccr.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sawyer SL, Tian L, Kahkonen M, Schwartzentruber J, Kircher M, Majewski J, Dyment DA, Innes AM, Boycott KM, Moreau LA, Moilanen JS, Greenberg RA University of Washington Centre for Mendelian G Consortium FC. Biallelic mutations in BRCA1 cause a new Fanconi anemia subtype. Cancer Discov. 2015;5:135–42. doi: 10.1158/2159-8290.CD-14-1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Howlett NG, Taniguchi T, Olson S, Cox B, Waisfisz Q, De Die-Smulders C, Persky N, Grompe M, Joenje H, Pals G, Ikeda H, Fox EA, D’Andrea AD. Biallelic inactivation of BRCA2 in Fanconi anemia. Science. 2002;297:606–9. doi: 10.1126/science.1073834. [DOI] [PubMed] [Google Scholar]
- 34.Rickman KA, Lach FP, Abhyankar A, Donovan FX, Sanborn EM, Kennedy JA, Sougnez C, Gabriel SB, Elemento O, Chandrasekharappa SC, Schindler D, Auerbach AD, Smogorzewska A. Deficiency of UBE2T, the E2 Ubiquitin Ligase Necessary for FANCD2 and FANCI Ubiquitination, Causes FA-T Subtype of Fanconi Anemia. Cell Rep. 2015;12:35–41. doi: 10.1016/j.celrep.2015.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Walden H, Deans AJ. The Fanconi anemia DNA repair pathway: structural and functional insights into a complex disorder. Annu Rev Biophys. 2014;43:257–78. doi: 10.1146/annurev-biophys-051013-022737. [DOI] [PubMed] [Google Scholar]
- 36.Schwab RA, Nieminuszczy J, Shah F, Langton J, Lopez Martinez D, Liang CC, Cohn MA, Gibbons RJ, Deans AJ, Niedzwiedz W. The Fanconi Anemia Pathway Maintains Genome Stability by Coordinating Replication and Transcription. Mol Cell. 2015;60:351–61. doi: 10.1016/j.molcel.2015.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garcia-Rubio ML, Perez-Calero C, Barroso SI, Tumini E, Herrera-Moyano E, Rosado IV, Aguilera A. The Fanconi Anemia Pathway Protects Genome Integrity from R-loops. PLoS Genet. 2015;11:e1005674. doi: 10.1371/journal.pgen.1005674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tuduri S, Crabbe L, Conti C, Tourriere H, Holtgreve-Grez H, Jauch A, Pantesco V, De Vos J, Thomas A, Theillet C, Pommier Y, Tazi J, Coquelle A, Pasero P. Topoisomerase I suppresses genomic instability by preventing interference between replication and transcription. Nature cell biology. 2009;11:1315–1324. doi: 10.1038/ncb1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alzu A, Bermejo R, Begnis M, Lucca C, Piccini D, Carotenuto W, Saponaro M, Brambati A, Cocito A, Foiani M, Liberi G. Senataxin associates with replication forks to protect fork integrity across RNA-polymerase-II-transcribed genes. Cell. 2012;151:835–46. doi: 10.1016/j.cell.2012.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Poli J, Gerhold CB, Tosi A, Hustedt N, Seeber A, Sack R, Herzog F, Pasero P, Shimada K, Hopfner KP, Gasser SM. Mec1, INO80, and the PAF1 complex cooperate to limit transcription replication conflicts through RNAPII removal during replication stress. Genes Dev. 2016;30:337–54. doi: 10.1101/gad.273813.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wahba L, Gore SK, Koshland D. The homologous recombination machinery modulates the formation of RNA-DNA hybrids and associated chromosome instability. eLife. 2013;2:e00505. doi: 10.7554/eLife.00505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wickramasinghe VO, Venkitaraman AR. RNA Processing and Genome Stability: Cause and Consequence. Mol Cell. 2016;61:496–505. doi: 10.1016/j.molcel.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chan YA, Aristizabal MJ, Lu PY, Luo Z, Hamza A, Kobor MS, Stirling PC, Hieter P. Genome-wide profiling of yeast DNA:RNA hybrid prone sites with DRIP-chip. PLoS Genet. 2014;10:e1004288. doi: 10.1371/journal.pgen.1004288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ginno PA, Lott PL, Christensen HC, Korf I, Chedin F. R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Molecular cell. 2012;45:814–825. doi: 10.1016/j.molcel.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wahba L, Amon JD, Koshland D, Vuica-Ross M. RNase H and multiple RNA biogenesis factors cooperate to prevent RNA:DNA hybrids from generating genome instability. Molecular cell. 2011;44:978–988. doi: 10.1016/j.molcel.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.El Hage A, French SL, Beyer AL, Tollervey D. Loss of Topoisomerase I leads to R-loop-mediated transcriptional blocks during ribosomal RNA synthesis. Genes & development. 2010;24:1546–1558. doi: 10.1101/gad.573310. [DOI] [PMC free article] [PubMed] [Google Scholar]