

Abstract
Many bacterial regulatory genes appear to be dispensable, as they can be deleted from the genome without loss of bacterial functionalities. In Helicobacter pylori, the hp1043 gene, also known as hsrA, is one of the transcriptional regulator that is essential for cell viability. This gene could not be deleted, nor the amount of protein modulated, supporting the hypothesis that HP1043 could be involved in the regulation of crucial cellular processes. Even though detailed structural data are available for the HP1043 protein, its targets are still ill-defined. Using Chromatin Immunoprecipitation-sequencing (ChIP-seq), one of the most powerful approaches to characterize protein-DNA interactions in vivo, we were able to identify genome-wide several new HP1043 binding sites. Moreover, in vitro DNA binding assays enabled precise mapping of the HP1043 binding sites on the new targets, revealing the presence of a conserved nucleotide sequence motif. Intriguingly, a significant fraction of the newly identified binding sites overlaps promoter regions controlling the expression of genes involved in translation. Accordingly, when protein translation was blocked, a significant induction of almost all HP1043 target genes was detected. These observations prompted us to propose HP1043 as a key regulator in H. pylori, likely involved in sensing and in coordinating the response to environmental conditions that provoke an arrest of protein synthesis. The essential role of HP1043 in coordinating central cellular processes is discussed.
Helicobacter pylori is a major pathogen, highly widespread among the human population, and it has been recognized as class I carcinogen by World Health Organization. It is considered the primary cause of severe gastrointestinal diseases such as peptic ulcer, gastric adenocarcinoma and MALT lymphoma1,2. The ability of this Gram-negative bacterium to colonize the harsh stomach niche and to establish a persistent infection depends on the coordinated expression of housekeeping genes as well as virulence factors that allow the pathogen to adapt to environmental conditions and to counteract host-defence mechanisms. A peculiar feature of the small-sized (1.66 Mb) H. pylori genome is the relative scarcity of genes encoding regulators of transcription. To date, only 17 transcriptional regulators have been identified and characterized to different extents3. Besides 3 σ factors (the housekeeping σ80 and the alternative RNA polymerase sigma subunits σ54/σ28, both involved in the transcription of flagellar genes) and 4 transcriptional repressors involved in metal homeostasis (Fur and NikR) or stress response (HrcA and HspR), H. pylori employs several two-component systems. Two-component systems are composed of a histidine-kinase and a response regulator. Upon signal perception, the sensor kinase catalyses its auto-phosphorylation and then transfers the phosphoryl group to a partner response regulator, a specific DNA binding protein that modulates transcription of target genes. Two-component systems are employed by H. pylori to coordinate gene expression in crucial cellular processes like chemotaxis (CheA/CheY)4, copper resistance (CrdS/CrdR)5, flagellar regulation (FlgS/FlgR)6, and acid acclimation (ArsS/ArsR)7. Intriguingly, H. pylori genome harbours 2 genes, named hp1043 (HPG27_RS02035) and hp1021 (HPG27_RS02145), encoding for two so-called “orphan response regulators”, as their partner sensor kinases are missing. HP1043, in particular, appears to be essential for cell viability. The hp1043 gene, in fact, could not be deleted unless a second gene copy was integrated into the H. pylori chromosome8. The HP1043 regulator belongs to the OmpR family with a highly degenerate receiver sequence incapable of being phosphorylated. Different biochemical and structural studies suggest that HP1043 could exert its function in a phosphorylation-independent manner and it could be classified as belonging to a new response regulator family8,9. NMR-spectroscopy and X-ray crystallography suggested that HP1043 exists as a symmetric dimer, with two functional domains, an N-terminal regulatory domain and C-terminal DNA-binding domain. The dimer appears to be stable in solution in the un-phosphorylated state10. Even though detailed structural data are available for HP1043, the target genes bound and regulated by this regulator are still ill-defined. Specifically, to date only three HP1043 genomic binding sites have been characterized at the molecular level. In vitro DNA binding studies demonstrated that HP1043 binds its own promoter and the promoter region of tlpB, a gene encoding a methyl-accepting chemotaxis protein11. Moreover, in a recent study we demonstrated binding of HP1043 to the promoter region of cncR1, a small regulatory RNA of H. pylori G27 involved in the opposite modulation of motility and adhesion to host cells12. The impossibility of generating a knock-out mutant for hp1043 gene, or even of modulating the amount of HP1043 protein in the cell, has hampered the detailed characterization of its regulatory function11. Initially proposed as a regulator of cell cycle-related functions11, two recent studies attempted a link of HP1043 to homeostatic stress control (and named it HsrA for homeostatic stress regulator) of the bacterial cell and to a role in oxidative stress defence and nitrogen metabolism13,14. While gel mobility shift experiments support direct binding of HP1043 (HsrA) to the porGDAB promoter region, the regulation of other genes by HP1043 was inferred solely by the finding of a putative binding sequence13,15.
In the present study we report the identification of HP1043 (HsrA) DNA targets in vivo and the characterization of selected binding sites in vitro. The combination of recent advances in deep sequencing coupled with well-established Chromatin Immunoprecipitation (ChIP) protocols for bacterial transcription factors, establishes ChIP-seq as one of the most powerful approaches to identify genome-wide all the in vivo binding sites of a regulator of interest. The ChIP-seq analysis here presented allowed us to identify several new in vivo HP1043 binding sites. Moreover, in vitro protein-DNA binding assays enabled precise mapping of the HP1043 binding sites on new targets, whose analysis revealed the presence of a conserved nucleotide sequence motif. Interestingly, a significant fraction of the newly identified motifs overlaps promoters associated to genes involved in the process of translation. Accordingly, a stress signal leading to the arrest of protein synthesis, resulted in a significant induction of almost all HP1043 target genes. These observations prompted us to propose HP1043 (HsrA) as a key regulator in H. pylori, likely involved in sensing and in coordinating the response to environmental conditions that provoke an arrest of protein synthesis.
Results
Genome-wide binding of HP1043 (HsrA)
To date, the exact positions of only three HP1043 genomic binding targets, located on the promoter regions of hp1043, tlpB and cncR1 genes, have been characterized in details and they are represented in Fig. 1. To identify genome-wide all the HP1043 targets and thus obtain an unbiased overview of HP1043 regulatory role in H. pylori, we applied a ChIP-seq approach in vivo. In particular, liquid-grown H. pylori G27 wild type cells were cross-linked, sonicated and HP1043 protein-DNA complexes were immunoprecipitated (IP) with a specific polyclonal α-HP1043 antibody; at the same time a control sample (Input), deriving from sonicated but non-immunoprecipitated DNA, was prepared and used to estimate the background. A minimum of 2 million reads, with optimal mapping performances (>98.5%), for each sample and biological replicate were obtained (Supplementary Table S1) and used to generate the genome-wide binding profiles visualized in Fig. 2A. Combining Homer2 peak calling and Irreproducible Discovery Rate (IDR) procedure, we identified a set of 37 highly reproducible peaks (Fig. 2A and Table 1). These putative binding sites were the most enriched in the IP sample and highly reproducible among the two biological replicates (see Methods for a details). Afterwards, the peaks identified were annotated according to the H. pylori G27 RefSeq annotation (GCF_000021165.1), and cross-mapped to the transcription start sites and ncRNAs defined in strain 2669516 (see Methods). Specifically, binding sites centred between position −150 and +50 with respect to a transcription start site were considered associated to a promoter region, while those centred outside of this positional range were classified as internal or intergenic on the basis of the peak central position (Table 1). According to this classification, 89% of the identified HP1043 binding sites (33/37) were associated to promoters, hence in a canonical position to exert a regulatory function, and about 1/3 of them were bidirectional promoters. Three out of 37 (8%) of the peaks mapped in internal regions, while only one single peak (3%) was classified as intergenic (Fig. 2B). Interestingly, several strongly enriched peaks were located in proximity of tRNAs genes (bolded in Table 1, Supplementary Table S2).
Figure 1. Schematic representation of the hp1043 locus in H. pylori G27 along with the previously reported HP1043 binding sites.
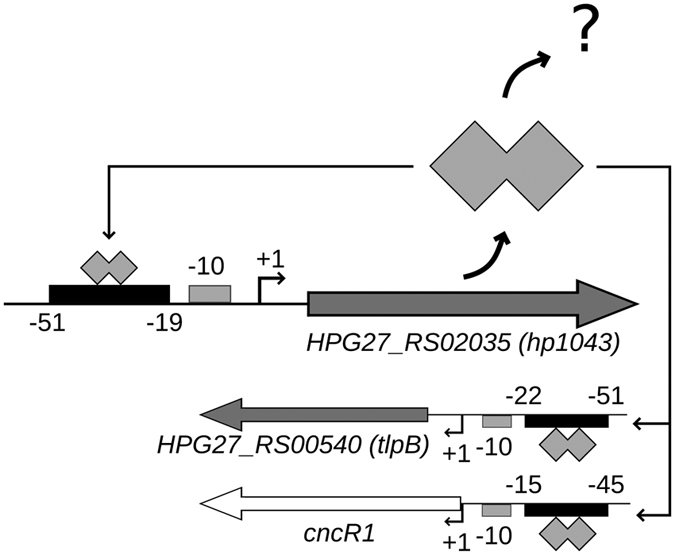
The binding sites of HP1043 are represented with black boxes, positions are relative to the transcription start sites. The transcriptional regulator, represented by a polyhedral shape, binds the promoter region of three target genes: hp1043 and tlpB11, and cncR112. Protein coding genes are depicted with grey block arrows, the small non-coding cncR1 RNA gene with a white block arrow. Transcription start sites are indicated with a bent arrow marked +1, the −10 region with grey boxes.
Figure 2. Genome-wide binding analysis of HP1043.

(Panel A): genome-wide binding of HP1043 determined by ChIP-seq. The base count data, deriving from sequencing reads alignment on H. pylori G27 genome, of immunoprecipitated sample (labelled “ChIP”, light blue track) and control sample (labelled “Input”, black track) are represented; the position of the HP1043 binding sites, identified by peak-calling analysis, are highlighted by vertical bars and labelled according to the peak number. (Panel B): pie chart summarizing the positional analysis of the HP1043 binding sites relative to annotated genes. (Panel C): analysis of the functional categories of genes associated with novel HP1043 binding sites, identified by ChIP-seq.
Table 1. Location of putative HP1043 binding sites.
| Peak_name | start:end | Score | Class | Nearest genes |
|---|---|---|---|---|
| HsrA_Peak_1 | 342452:342626 | 33634,2 | bidirectional promoter | nadE < tRNA-Arg > ilvC |
| HsrA_Peak_2 | 1570125:1570299 | 29290,7 | promoter | > tRNA-Phe |
| HsrA_Peak_3 | 1476498:1476672 | 27064 | promoter | 16SrRNA< |
| HsrA_Peak_4$ | 1:126 | 26419 | promoter | > HPG27_RS07980 |
| HsrA_Peak_5 | 448262:448436 | 24526,6 | promoter | tRNA-Pro < |
| HsrA_Peak_6 | 1212453:1212627 | 22077 | promoter | fldA < |
| HsrA_Peak_7 | 1276504:1276678 | 21217,1 | promoter | atpE< |
| HsrA_Peak_8 | 1194610:1194784 | 20592,6 | promoter | 16SrRNA< |
| HsrA_Peak_9 | 1140440:1140614 | 20533 | bidirectional promoter | galE<tRNA-Ile |
| HsrA_Peak_10 | 1234366:1234540 | 20239,5 | bidirectional promoter | nupC<tRNA-Met > HPG27_RS05875 |
| HsrA_Peak_11 | 1648608:1648782 | 17967,3 | promoter | > flgG2,flgG |
| HsrA_Peak_12 | 1198357:1198531 | 17780,6 | promoter | rpS16,rpsP < |
| HsrA_Peak_13 | 1411804:1411978 | 17187,4 | bidirectional promoter | asRNA_HPG27_RS06840< > HPG27_RS06835 (pseudogene) |
| HsrA_Peak_14 | 1268342:1268516 | 16391,8 | promoter | secE < |
| HsrA_Peak_15 | 96262:96436 | 16366,7 | bidirectional promoter | fabD < tRNA-Ser |
| HsrA_Peak_16 | 1275920:1276094 | 15715,5 | intergenic | tRNA-Leu«tRNA-Leu |
| HsrA_Peak_17 | 1135651:1135825 | 15701,4 | bidirectional promoter | nrdB,nrdF< > tRNA-Leu |
| HsrA_Peak_18 | 395023:395197 | 15492,7 | promoter | tRNA-Ser < |
| HsrA_Peak_19 | 829599:829773 | 14406,8 | promoter | as_HPG27_RS03935< |
| HsrA_Peak_20 | 1290665:1290839 | 14058,4 | bidirectional promoter | hemN2,hemN< > cytc553 |
| HsrA_Peak_21 | 1576835:1577009 | 13402,5 | promoter | isoA< |
| HsrA_Peak_22 | 536813:536987 | 13358,6 | promoter | cncR1_Hpnc2630< |
| HsrA_Peak_23 | 1573020:1573194 | 13228,3 | internal | dnaA< |
| HsrA_Peak_24 | 1362385:1362559 | 12858 | promoter | rpl36,rpmJ< |
| HsrA_Peak_25 | 94349:94523 | 11431,6 | promoter | rpoD< |
| HsrA_Peak_26 | 1163325:1163499 | 11422,2 | promoter | > asRNA_HPG27_RS05530 (Hpnc3560) |
| HsrA_Peak_27 | 79140:79314 | 11103,6 | promoter | tRNA-Val< |
| HsrA_Peak_28 | 1099169:1099343 | 10857,3 | promoter | > rps1,rpsA |
| HsrA_Peak_29 | 16420:16594 | 10304,9 | bidirectional promoter | putative_SRP_RNA < > HPG27_RS00110 |
| HsrA_Peak_30 | 1270429:1270603 | 10176,2 | promoter | > hetA |
| HsrA_Peak_31 | 1318842:1319016 | 9864 | bidirectional promoter | bioC < > secG |
| HsrA_Peak_32 | 1196544:1196718 | 9212,8 | promoter | rpl19,rplS< |
| HsrA_Peak_33 | 722928:723102 | 8986,8 | internal | > flgR |
| HsrA_Peak_34 | 478010:478184 | 8089,2 | promoter | > hofC |
| HsrA_Peak_35 | 4434:4608 | 7929,2 | internal | tRNA-Lys < |
| HsrA_Peak_36 | 31591:31765 | 7905,6 | bidirectional promoter | HPG27_RS00165< > uspA |
| HsrA_Peak_37 | 1480392:1480566 | 7634,1 | promoter | > rpl34,rpmH |
$Located on G27 plasmid; Bold, feature included in the peak; >/<, target gene strand.
Among HP1043 target genes, we found the small RNA cncR1, previously demonstrated to be bound by this regulator12, while the other three known targets (i.e. hp1043 itself, tlpB11 and porG14), were not present in the top scoring list. However, two regions mapping nearby the promoters of hp1043 and porG were identified as enriched by the peak caller and are present in the full list of 107 peaks identified by Homer2 (Supplementary Table S3). These peaks were not included in the top list only because of the stringent criteria adopted to evaluate the peak calling reproducibility. This observation suggests that we may find some authentic and reliable HP1043 binding sites also among some of the less reproducible ChIP-seq peaks. For example, inspection of the latter peaks allowed to pinpoint an enriched region, present in both replicas, positioned on the tlpB promoter. Then, to obtain a general view of HP1043 regulatory function, each promotorial binding site was associated to the downstream operon. A functional enrichment analysis was then performed to determine if the genes included in the operons belonged to specific functional categories. For this purpose, protein coding genes were classified using Clusters of Orthologous Groups of proteins (COGs) database17, while tRNAs and rRNAs were included to the translation category. According to this analysis, it turned out that a significant fraction (30%) of genes, associated to a HP1043 promotorial binding site and likely controlled by the regulator, is involved in the process of protein translation (Fig. 2C). Specifically, the promoters of several tRNA and rRNA genes, as well as of genes encoding ribosomal proteins (HPG27_RS05725/rps16, HPG27_RS06525/rpl36, HPG27_RS05215/rps1, HPG27_RS05705/rpl19, HPG27_RS07170/rpl34) or involved in rRNA and ribosome maturation and assembly (HPG27_RS05770/ybeY endoribonuclease and HPG27_RS06295/frr ribosome recycling factor) were targeted in vivo by HP1043. Strikingly, in the same analysis we identified HP1043 binding sites associated to genes involved in other fundamental cellular processes like energy production and conversion (flavodoxin, HPG27_RS05775/fldA; ATP synthase C chain, HPG27_RS06075/atpE; cytochrome c553, HPG27_RS06145/cytc553), as well as RNA transcription (HPG27_RS00460/rpoD, coding for the RNA polymerase housekeeping σ80 factor18). In this respect, we noticed that one more RNA polymerase gene, HPG27_RS06505/rpoA, encoding the RNA polymerase α subunit, is included in an operon likely controlled by HP1043. This last observation was manually curated because in the last version of H. pylori G27 genome annotation (GCF_000021165.1) this gene is mis-annotated to pseudogene. Overall, from the above data we can infer that HP1043 may represents a key regulator in H. pylori, involved in the control of crucial cellular processes, such as transcription and translation.
Validation of novel HP1043 targets
To confirm HP1043 binding sites identified by ChIP-seq, a subset of them was selected and used as probes for in vitro binding to a purified recombinant HP1043 protein in DNase I footprinting experiments. Among the 37 putative highly confident HP1043 targets (Table 1) we selected 7 representative genomic regions: 6 binding sites mapping within promoters and 1 site falling inside a coding sequence. We also included a DNA probe covering the promoter of the HPG27_RS06315/nuoA gene. This putative HP1043 binding site was present in the total peak list and close to the ranking position of hp1043, but excluded from the top list. Similarly to some other peaks not included in the top list of Table 1, this binding site exhibits a broad region of enrichment encompassing the promoter, even if the peak centre appears mis-positioned with respect to the core promoter region. Thus, to assess if the peaks having this profile represent bona-fide HP1043 binding sites with a lower binding affinity rather than artifacts of the ChIP-seq experiment, we decided to validate further by DNase I footprinting one of them (Fig. 3). Consistent with ChIP-seq findings, all the 8 genomic regions selected were directly targeted in vitro by the purified recombinant HP1043 protein (all probes showed a clear area of DNase I protection), apparently with different relative affinities (some probes showed protection in the presence of higher amounts of HP1043 protein). Probes bound in vitro with higher relative affinity are reported in Fig. 3, panel A and B, while probes bound with lower relative affinity are shown in panel C and D. Increasing concentrations of HP1043 determined the appearance of a region of protection (black box) flanked at least on one side by a single DNase I hyper-sensitive band (black arrowhead). HP1043 binding to these 4 targets protected a DNA region of about 30 bp and the protection was saturated at 1.7 μM protein concentration (Fig. 3, panel A, lanes 2, 7, 12 and panel B, lane 2); these were considered high affinity binding sites. HP1043 binding to the promoters of isoA, rpoD, rpl36 and of nuoA genes (Fig. 3, panel C and D) appears at higher protein concentrations and with a slightly different binding pattern. Here the protection was saturated at a protein concentration of 13.3 μM, and again protected a region of about 30 bp flanked, only in some cases, by one or more bands of hyper-sensitivity to DNase I digestion. These were considered low affinity binding sites. Overall, the above in vitro footprinting assays confirm in vivo binding data. Notably, HP1043 binds in vitro exactly to the same positions of the enriched regions derived from ChIP-seq data analysis for all the top scoring peaks (Fig. 3A, B, C lower part of each panel). Moreover, the footprinting experiment reported in Fig. 3B (lanes 1 to 5) validates HP1043 intra-cistronic binding within the flgR coding sequence, further supporting ChIP-seq analysis and suggesting the existence of a minority of non-canonical binding sites, apparently not associated with regulatory functions.
Figure 3. In vitro binding of purified recombinant HP1043 protein on selected genomic regions identified by ChIP-seq.

In panel A high affinity promoter binding sites are shown, in panel B footprinting validation of an intragenic binding site is reported, in panel C low affinity promoter binding sites are represented and in panel D footprint validation of a binding site not included in the top scoring peak list. Radiolabelled DNA probes, harbouring the genomic regions of interest, were incubated with increasing concentrations of purified HP1043 (0, 1.7, 3.3, 6.6, 13.3 μM of dimeric HP1043 from left to right in each experiment) and subjected to DNase I digestion. Purified DNA fragments were separated on a polyacrylamide denaturing gel along with a G + A sequence reaction ladder (Panel C and data not shown) to map the binding sites. On the left of each autoradiograph, a schematic representation of the genomic region is drawn, with block arrows depicting coding sequences (black arrows or blocks) or putative sRNA (white arrow). Bent arrows represent the transcriptional start sites identified in H. pylori strain G27 by primer extension analyses (Supplementary Fig. 3S), white boxes the −10 promoter sequence. Notably, the initiation of RNA transcription at the analysed promoters is conserved between strain G27 and 26695. Black vertical lines on the right of each autoradiograph represent regions of protection from DNase I digestion, while black arrowheads highlight DNase I hyper-sensitive sites. Numbers refer to the positions with respect to the transcription start site (or with respect to the ATG translational start codon for the internal binding site on HPG27_RS03405). For the binding site mapped in the intergenic region between HPG27_RS00110 and putative SRP_RNA genes, HP1043 binding positions are reported with respect to the transcriptional start site of both genes (with no brackets for putative SRP_RNA, with brackets for HPG27_RS00110). Below each autoradiograph, a magnification of base-count data, deriving from ChIP-seq (as in Fig. 2A) of the genomic regions of interest, is reported together with a schematic representation of genes and features of the locus.
In summary, we confirmed HP1043 binding to 8 new sites identified in in vivo ChIP-seq experiments and defined in vitro the nucleotide sequences bound by the protein.
Molecular characterization of HP1043 (HsrA) binding to DNA
The DNase I footprinting data indicated that the newly identified HP1043 binding sites overlap the core promoter region and map, in all cases, immediately upstream of the −10 box (Fig. 3, panels A, C and D). This observation parallels with the positions of HP1043 binding sites on the three previously characterized target sites (Fig. 1 11,12). To get a detailed picture of the promoter sequences encompassing HP1043 binding sites, the core promoter sequences of the newly and previously validated HP1043 targets were analysed using the GLAM2 software19. Results are shown in Fig. 4A. The HP1043 target promoters appear to be characterized by a −10 hexamer typical of the housekeeping σ80-dependent transcription, preceded, in some cases, by an extended TG motif (Fig. 4A, upper part). Moreover, a sequence resembling the −35 box is lacking in these promoters, apparently replaced by two conserved AT-rich motifs separated by a spacer region (Fig. 4A, lower part). Intriguingly, the regions protected by HP1043 binding in DNase I footprinting assays (highlighted in Fig. 4A, upper part) encompass, in almost all cases (except in rpoD), these two conserved AT-rich motifs. To determine a possible consensus motif recognized by the HP1043 protein, we performed the same analysis using only the regions protected by HP1043 in the in vitro footprint experiments as input sequences. In this way GLAM2 outlined a highly conserved motif, reported in Fig. 4B. Specifically, the motif appears to be constituted by two direct repeats (TTTAAG) separated by a 5 bp-spacer, in which the first highly conserved hexamer is followed by a second less conserved one. Notably, the spacer length is conserved, positioning the centre of the two direct repeats at 11 bp distance that corresponds to 1 helical turn, suggesting that a dimer of HP1043 recognizes the DNA on the same face of the double helix. The conserved sequence motif (underlined and in bold in Fig. 4B, upper part) is either centred within the experimentally mapped binding sites (secE, cyt553, cncR1 and nuoA), or shifted towards one side of the protected region (isoA, rpoD, hp1043 and tlpB). Possibly, this is due to the intrinsic low level of resolution in mapping protein binding sites with DNase I footprinting assays. In fact, some regions of the DNA probes used for these assays naturally lack bands of digestion even in the sample without protein and this introduces a degree of uncertainty in defining the precise boundaries of the binding regions. Hence, to precisely map HP1043 (HsrA) binding sites and to further characterize the connection between the identified consensus motif and the intimate contacts of HP1043 with target DNA, hydroxyl-radical footprinting assays were carried out on selected targets, previously probed by DNase I footprintings (Fig. 3). Results are reported in Fig. 5. On all the promoter probes tested, HP1043 binding resulted in a periodic pattern of three short protected tracts of 3/4 nucleotides in length, separated by two non-protected regions of 5/6 nucleotides. Intriguingly, for all binding sites the central protection centers exactly within the spacer that separates the conserved direct repeats, while the other two protected DNA tracts fall immediately upstream and downstream of the direct repeats of the consensus binding motif (Fig. 5). It is worth mentioning that regions protected in hydroxyl-radical footprinting experiments reflect limited accessibility of radical ions to the DNA minor groove and, for this reason, these protected regions do not necessarily represent the portions of the probe directly contacted by the protein. Considering that HP1043 footprint regions surround the conserved direct repeats (Fig. 5), our data suggest that HP1043 could interact with the TTTAAG repeats in the DNA major groove narrowing the adjacent minor grooves which results in the protection observed in vitro by hydroxyl-radical footprinting. Interestingly, hydroxyl-radical footprinting analysis allowed the identification of a peculiar organization on rpoD promoter, which harbours multiple HP1043 binding sites. In fact, on this promoter, hydroxyl-radical footprinting assay (Fig. 5, lanes 26–30), besides the binding site already mapped with DNase I footprinting (Fig. 3C, lanes 6–10), revealed the existence of a distal HP1043 binding site, spanning positions −45 to −68 with respect to the initiation of RNA transcription. Moreover, the analysis of the DNA sequence of this distal binding site, revealed an HP1043 binding sequence highly similar to the consensus binding motif described above. The rpoD gene represents the first example of an HP1043 target with multiple binding sites, suggesting a complex mechanism of transcriptional regulation. In conclusion, hydroxyl-radical probing allowed to further refine the positions of HP1043 binding, providing the demonstration that the direct repeats of the proposed consensus motif are recognized by HP1043, likely through a major groove read-out mechanism.
Figure 4.

Characterization of HP1043 bound promoters (A) and definition of HP1043 consensus binding motif (B). Nucleotide sequence motifs were identified by using GLAM2 software19, forcing strand specific alignment and maintaining all the default parameters to obtain the weblogos. Panel A, nucleotide sequence alignment (upper part) and weblogo (lower part) relative to 10 promoter regions bound by HP1043. Specifically, DNA sequences spanning from the transcriptional start site (+1) to position −70 of the 7 genes analysed by footprinting (Fig. 3A, C, D) were aligned together with the same DNA regions upstream tlpB, cncR1 and hp1043 genes. Nucleotides matching the consensus −10 box and the extended TG motif are highlighted in black, while DNA regions protected by HP1043 binding, as defined by DNase I footprintings (Fig. 3), are highlighted in red. Names reported on the left of each sequence refer to the new H. pylori G27 genome annotation, while names on the right of each sequence report the common gene name and, in brackets, the name referred to the H. pylori 26695 genome annotation. Panel B, nucleotide sequence alignment of the 10 protected regions in DNase I footprintings (Fig. 3), mapping in the promoters of the same genes reported in panel A. Aligned sequences and the resulting sequence logo are shown in the upper and lower part of panel B, respectively. The sequences of the two direct repeats are represented in bold and underlined; numbers flanking each sequence refer to the coordinates of the DNase footprinting protected regions (shown also in Fig. 3). In both sequence logos (lower part of both A and B panels) the height of each letter represents the relative conservation of each base.
Figure 5. Hydroxyl-radical footprinting analysis of HP1043 binding to selected genomic regions.

Radiolabelled DNA probes, harbouring the genomic regions of interest, were incubated with increasing concentrations of purified HP1043 dimer (0, 0.8, 1.7, 3.3 μM of dimeric HP1043 from left to right in each experiment) and subjected to hydroxyl-radical digestion. Purified DNA fragments were separated on a polyacrylamide denaturing gel along with a G + A sequence reaction ladder to map the binding sites. On the bottom of each autoradiograph, the nucleotide sequence of the HP1043 binding sites mapped is reported: grey-highlighted nucleotides depict the hexameric direct repeats of the binding motif, while the black dots indicate the nucleotides protected in hydroxyl-radical footprintings on the labelled DNA strands. Symbols are detailed in the legend to Fig. 3.
Genes directly targeted by HP1043 are up-regulated upon translational arrest
ChIP-seq data indicated that HP1043 binds in vivo regulatory regions of several genes involved in translation (Fig. 2C). Hence, we hypothesized that the regulative function of HP1043 could be boosted by a signal affecting protein synthesis. To verify this hypothesis, in a preliminary survey, H. pylori G27 wild type cells were liquid-grown to mid-exponential phase, and HP1043 mRNA abundance was monitored at different time points after blocking protein synthesis by the addition of a sub-lethal concentration of tetracycline, which prevents aminoacyl tRNA attachment to ribosomal acceptor site20. Quantitative Real-Time PCR (qRT-PCR) assay revealed that the hp1043 transcript amounts significantly increase upon tetracycline treatment, reaching a maximum induction upon 60 min of treatment (data not shown). Similar results were obtained upon treatment with chloramphenicol, which blocks ribosomes on mRNA during translation21. These preliminary results prompted us to further investigate the impact of translational stress on the HP1043 (HsrA) regulon. Total RNA collected from H. pylori wild-type cells exposed for 60 min to tetracycline and from a control culture were used to generate strand-specific cDNA libraries for RNA-sequencing analysis. Overall, differential gene expression outlined 1065 out of the 1592 total number of H. pylori genes to be deregulated upon antibiotic treatment. Of these, 543 genes were upregulated, representing 34% of the H. pylori genes (Fig. 6A, column X). However, focusing on the panel of genes belonging to all the operons controlled in vivo by HP1043, we observed that 52% of them were upregulated after tetracycline treatment (Fig. 6A, column Y), with a much more significant enrichment (68%) of upregulated HP1043 direct targets among the genes leading each operon (Fig. 6A, column Z). Several cellular pathways were affected by this treatment, supporting the notion that block of translation is a major challenge for bacterial cells, which respond with a wide transcriptional reprogramming of most key cellular processes. It is worth-noting that genes coding for rRNA and non-coding RNA were not included in the analysis because the first were depleted by the RNA sample preparation and the second are not annotated in strain G27.
Figure 6.

RNA-seq (A) and Real Time PCR (B) analyses of transcript level variations of novel HP1043 targets in response to translation arrest. Panel A): relative percentage of the genes upregulated after tetracycline treatment considering all annotated H. pylori G27 genes (column X, with exclusion of rRNA genes, depleted during sample preparation), the genes in operons under direct control of HP1043 (column Y with exclusion of 2 rRNA genes and of the genes absent in H. pylori annotation: 4 antisense RNA, and 2 small RNA), and the genes leading the operons under direct control of HP1043 (column Z with the exclusion of the genes indicated for column Y), respectively. The reported p-value was calculated using Fisher test. Panel B): effect of tetracycline treatment on transcript amounts of a selection of genes associated with an HP1043 binding site assessed by real time (qRT-PCR) analysis. Total RNA was extracted from cells treated or not treated with 0.5 μg/ml tetracycline for 60 min and reverse transcribed to cDNA. Transcript levels of a selection of genes not associated (panel B, left graph) or associated (panel B, central graph) with an HP1043 binding site were quantified by qRT-PCR, using the housekeeping 16 S rRNA gene as control. The same analysis was carried out on some tRNA genes (panel B, right graph) associated with HP1043 binding. Statistically significant differences were assessed by Student’s t-test (Error bars indicate the standard deviation deriving from three independent biological samples, each analysed in duplicate technical replicates). Symbols: *p value < 0.05; **p value < 0.01; ***p value < 0.001; ns, p value > 0.05, not significant.
To validate RNA-seq data, the differential mRNA levels of a subset of HP1043 targets were assayed upon tetracycline exposure by qRT-PCR analysis. Three negative controls (frpB1, fecA1 and nixA i.e. three genes not targeted by HP1043) were included in the analysis and remained essentially unchanged upon antibiotic challenge (Fig. 6B, left histogram). Results reported in Fig. 6B (central histogram) show that transcript levels of selected HP1043 target genes (hp1043, rpoD, secE, rpl36, cytc553) increased upon tetracycline treatment, with fold variations ranging from 3- to 7-fold. Moreover, as shown in Fig. 6B (right histogram), a similar increasing trend upon translational arrest was observed for a selection of tRNA genes. These data suggest a possible involvement of the orphan response regulator HP1043 (HsrA) in the transcriptional response of H. pylori to environmental conditions or signals that promote arrest of protein synthesis.
Discussion
Two-component signal transduction systems typically regulate bacterial cellular functions in response to environmental conditions through a phosphorylation-dependent process. The human pathogen H. pylori relies on such regulatory systems to control important cellular functions such as motility, chemotaxis, acid acclimation and copper resistance22. The orphan response regulator HP1043, also known as HsrA, is proven to be essential for cell growth and shows no requirement for the well-known phosphorelay scheme to be functional. In the present study, we have set up chromatin immunoprecipitation with α-HP1043 antibody followed by deep sequencing (ChIP-seq). This approach led to the identification of several new HP1043 genomic targets (Fig. 2A). To our knowledge, this is the first study that provides a genome-wide analysis of the HP1043 binding in vivo. Specifically, 37 genomic HP1043 binding sites were identified (Table 1), the majority of which are associated to a promoter region (Fig. 2B). A predominant fraction of genes associated to HP1043 binding encodes for proteins involved in crucial cellular functions, such as protein synthesis (tRNAs and ribosomal proteins coding genes), gene transcription (RNA polymerase subunits), and energy metabolism (operon containing genes coding for the NADH-ubiquinone oxidoreductase whole complex, as well as the ATP synthase C-chain gene promoter) (Fig. 2C, Table 1). Thus, it seems that HP1043 plays a key role for the fitness of the bacterium, which is a prerequisite for a successful infection. Moreover, our findings suggest that HP1043 regulator might represent a central regulatory switch mechanism that H. pylori exploits to modulate its metabolism and growth behaviour. In this respect, it is interesting to note the connection between HP1043 regulation and the arrest of translation (Fig. 6), a major challenge for bacterial cells.
Recent works by Olekhnovich and colleagues14,15 suggested an involvement of HP1043 in directly regulating a list of about 70 genes encoding for proteins with disparate functions. However, it is worth mentioning that this proposed regulon was defined by searching against the H. pylori genome with a consensus binding motif defined by aligning two binding sites only15. In our study, we have defined the HP1043 regulon by identifying in vivo several new direct targets and noticed that many previously proposed binding sites appear not bound in our experimental conditions.
The list of 37 HP1043 genomic binding sites (Table 1) derives from a stringent peak-calling analysis, that takes into consideration the reproducible high fold enrichment of the immunoprecipitated DNA regions with respect to the input DNA in two independent biological replicates, thus representing high-confidence candidates. The identification of this list of bona fide HP1043 targets likely prevented from the inclusion of some false positives, but at the same time it may have determined the exclusion of several real binding sites characterized by low affinity binding levels and/or low reproducibility among replicates. In this respect, HP1043 binding on its own promoter, a known target previously characterized at the molecular level11, was not included in the top list of highly significant binding sites. Moreover, we have shown that a putative HP1043 target, mapping upstream of the transcription start site of the HPG27_RS06315 gene, not included in the top list, is indeed an authentic HP1043 binding site (Fig. 3D). Hence, besides the new binding sites identified, it can be hypothesized that the HP1043 regulon may include additional members, not pinpointed by our analysis. Intriguingly, 8% of the newly identified genomic binding sites are not associated to promoters, mapping within protein coding sequences (Figs 2B and 3B). This observation poses some questions about the functional role of HP1043 binding to these internal sites. Even though we cannot exclude the existence of alternative and still ill-defined mechanisms of transcriptional regulation exerted by HP1043 bound inside coding sequences, possibly some HP1043 intracistronic binding sites could be associated with still-unknown internal promoters driving the transcription of intracistronic or antisense transcripts. Alternatively, these sites are not associated to regulation of transcription. The advent of the ‘omics’ revolution allowed the observation of binding sites not associated to regulation of several regulators, like H. pylori Fur repressor and E. coli CRP activator23,24. Even though it has recently been proposed that regulators with this behaviour may have evolved from nucleoid associated proteins25, validation of this hypothesis needs further investigation.
The alignment of promoter regions harbouring HP1043 binding sites (Fig. 4A) revealed that genes controlled by this regulator are transcribed by a putative vegetative σ80-dependent promoter with a conserved −10 box. Moreover, the −35 hexamer, typical of housekeeping promoters appeared to be lacking, consistent with previous observations in the H. pylori 26695 strain16. Intriguingly, in the subset of promoters here analysed, the −35 motif is replaced by two conserved AT-rich motifs separated by a spacer region (Fig. 4A, lower panel), overlapping HP1043 binding site. The position of HP1043 binding sites, just upstream the −10 hexamer, is typical of activators of transcription26,27. For example the binding sites of Bacillus subtilis PhoP transcriptional activator, belonging to the large OmpR/PhoB subfamily of response regulators to which HP1043 has previously been associated10, are typically centred between positions −17 to −66 of the activated promoters28. Accordingly, we speculate that HP1043 acts as an activator of transcription, boosting the activity of weak promoters controlling crucial genes involved in key cellular functions. Binding of HP1043 upstream of the −10 box would facilitate the contacts between the regulator and RNA polymerase, thereby stimulating initiation of transcription. This hypothesis might also be partly supported by the data summarized in Fig. 6B.
To identify sequence specific determinants for HP1043 binding, 10 nucleotide sequences protected in footprinting analyses were aligned using the GLAM2 computer program. A highly conserved motif (Fig. 4B) was identified and then proved to be bound by HP1043 through hydroxyl-radical footprintings (Fig. 5). The conserved binding motif is composed of two direct repeats (one highly conserved repeat followed by a second less conserved repeat) separated by a 5-bp spacer conserved in length. The HP1043 binding motif appears to be located on the coding strand in almost all promoters analysed. The only exception is represented by hp1043 (Fig. 4B), in which the conserved direct repeat maps on the non-coding DNA strand. In a previous study, a portion of hp1043 promoter was used to characterize HP1043 binding to DNA through electrophoretic mobility shift assay carried out on wild-type and mutated probes10. In particular, deletions of the second less conserved repeat, as well as single base mutations of highly conserved A and G of the first repeat, significantly impaired protein binding to DNA, supporting the identified HP1043 consensus binding motif.
In vitro characterization of HP1043 binding to selected promoters (Figs 3 and 5) revealed the existence of high affinity and low affinity binding sites. However, the comparison of the conserved direct repeats of these targets did not suggest any evident sequence feature responsible for the discriminative HP1043 binding capacity. Further characterization of the HP1043 DNA binding motif is crucial to address this point.
The above mentioned conserved spacing between the direct repeats puts the centre of the two direct repeats at 11 bp distance that corresponds to 1 helical turn, suggesting that a dimer of HP1043 recognizes the DNA on the same face of the double helix. The structure of HP1043 determined by NMR and X-ray crystallography10 supports this hypothesis, revealing a symmetrical dimer with two functional domains: the regulatory (dimerization) domain and the DNA-binding (transactivation) domain. In particular, the DNA-binding domains in the dimer appeared to be spaced by a distance compatible with one helix turn, allowing the interaction of each DNA-binding domain with one repeat of the conserved motif. Considering that the HP1043 regulatory domains form a symmetric dimer and that they are connected to their respective DNA-binding domains through a short 2-residues linker, it is conceivable that HP1043 forms a symmetric dimer in a head-to-head orientation with DNA. Consequently, HP1043 should be expected to contact a binding site made by an inverted repeat. Surprisingly, our sequence conservation analysis led to the identification of a binding motif characterized by two direct repeats (Fig. 4B). This observation can partially be explained considering the different conservation of the two hemi-sites. Specifically, the HP1043 binding motif appears to be constituted by a first highly conserved hexamer followed by a second less conserved repeat. The different degree of conservation between the two hemi-sites could account for a different specificity of DNA recognition of the two HP1043 monomers. A DNA recognition mechanism like this has been proposed for HpNikR, an H. pylori transcriptional regulator of nickel homeostasis29. It has been shown that a dimer of symmetrical dimers of NikR, expected to contact inverted repeats, has more affinity for binding sites that deviate from a perfect inverted repeat architecture29. Accordingly, it has been proposed that during the interaction between NikR and DNA target sequence, the more conserved hemi-site acts as a recognition site, while the second less conserved repeat acts as a structural (stabilizing) binding site29. The DNA recognition mechanism of HP1043 could be similar, with the primary recognition event taking place only on the highly conserved repeat, thereby stabilized by a weaker interaction between the second repeat and the other DNA binding domain. Another possibility is that, upon DNA recognition, the HP1043 dimer undergoes a structural reorganization, allowing a prototypical interaction between a direct repeat motif and a proper oriented dimer. Further experiments will disentangle this apparent paradox in HP1043-DNA docking mechanism.
Methods
Bacterial strains and culture conditions
Bacterial strains used in this study are listed in Table 2. H. pylori G27 wild type cells were revitalized from glycerol stocks on Brucella broth agar plates containing 5% fetal calf serum (FCS) in a 9% CO2–91% air atmosphere at 37 °C and 95% humidity in a water-jacketed incubator (Thermo Forma Scientific). Liquid cultures were performed in Brucella broth medium supplemented with 5% FCS in glass flasks. E. coli cells were grown on Luria–Bertani (LB) agar plates or LB liquid broth; when required, ampicillin was added to the medium to achieve a final concentration of 100 μg/ml.
Table 2. Bacterial strains and plasmids.
| Bacterial strains/plasmids | Description | Source/Reference |
|---|---|---|
| Strain | ||
| H. pylori G27 wild type | Clinical isolate, wild type | 42 |
| E. coli DH-5α | supE44 ΔlacU169 (ϕ80 lacZΔM15) hsdR17 recA1 endA1 gyrA96 thi-1 relA1 | 43 |
| Plasmid | ||
| pGEM-T-Easy | Cloning vector; Ampr. | Promega |
| pGEM0703 | pGEM-T-Easy derivative, containing a 437 bp DNA fragment corresponding to the region from 722.774 to 723.211 of H. pylori G27 genome amplified by PCR with oligonucleotides 0703FPF/0703FPR. This region corresponds to a portion of the coding sequence of HPG27_RS03405 (HP0703 according to 26695 annotation). | This work |
| pGEMp1203 | pGEM-T-Easy derivative, containing a 302 bp DNA fragment corresponding to the region from 1.268.274 to 1.268.576 of H. pylori G27 genome amplified by PCR with oligonucleotides 1203FPF/1203FPR. This region encompasses the putative promoter region of HPG27_RS06000 (HP1203 according to 26695 annotation). | This work |
| pGEMp1227 | pGEM-T-Easy derivative, containing a 308 bp DNA fragment corresponding to the region from 1.290.538 to 1.290.846 of H. pylori G27 genome amplified by PCR with oligonucleotides 1227FPF/1227FPR. This region encompasses the putative promoter region of HPG27_RS06145 (HP1227 according to 26695 annotation). | This work |
| pGEMsRNA17_18 | pGEM-T-Easy derivative, containing a 426 bp DNA fragment corresponding to the region from 16.304 to 16.730 of H. pylori G27 genome amplified by PCR with oligonucleotides sRNA17_18FPF/sRNA17_18FPR. This region encompasses the intergenic region between putative SRP_RNA and HPG27_RS00110. | This work |
| pGEMA1.4 | pGEM-T-Easy derivative, containing a 199 bp DNA fragment corresponding to the region from 1.576.846 to 1.577.045 of H. pylori G27 genome amplified by PCR with oligonucleotides A1.4FPF/A1.4FPR. This region encompasses the putative promoter region of isoA toxin/antitoxin system. | This work |
| pGEMp0088 | pGEM-T-Easy derivative, containing a 275 bp DNA fragment corresponding to the region from 94.285 to 94.560 of H. pylori G27 genome amplified by PCR with oligonucleotides 0088FPF/0088FPR. This region encompasses the putative promoter region of HPG27_RS00460 (HP0088 according to 26695 annotation). | This work |
| pGEMp1296 | pGEM-T-Easy derivative, containing a 477 bp DNA fragment corresponding to the region from 1.362.205 to 1.362.682 of H. pylori G27 genome amplified by PCR with oligonucleotides 1296FPF/1296FPR. This region encompasses the putative promoter region of HPG27_RS06525 (HP1297 according to 26695 annotation). | This work |
| pGEMp1260 | pGEM-T-Easy derivative, containing a 242 bp DNA fragment corresponding to the region from 1.321.773 to 1.322.015 of H. pylori G27 genome amplified by PCR with oligonucleotides 1260FPF/1260FPR. This region encompasses the putative promoter region of HPG27_RS06315 (HP1260 according to 26695 annotation). | This work |
| pGEM0703HY | pGEM-T-Easy derivative, containing a 117 bp DNA fragment corresponding to the region from 722.983 to 723.100 of H. pylori G27 genome amplified by PCR with oligonucleotides 0703HYF/0703HYR. This region corresponds to a portion of the coding sequence of HPG27_RS03405 (HP0703 according to 26695 annotation). | This work |
| pGEMp1203HY | pGEM-T-Easy derivative, containing a 148 bp DNA fragment corresponding to the region from 1.268.391 to 1.268.539 of H. pylori G27 genome amplified by PCR with oligonucleotides 1203HYF/1203HYR. This region encompasses the putative promoter region of HPG27_RS06000 (HP1203 according to 26695 annotation) | This work |
| pGEMp1227HY | pGEM-T-Easy derivative, containing a 146 bp DNA fragment corresponding to the region from 1.290.664 to 1.290.810 of H. pylori G27 genome amplified by PCR with oligonucleotides 1227HYF/1227HYR. This region encompasses the putative promoter region of HPG27_RS06145 (HP1227 according to 26695 annotation) | This work |
| pGEMp0088HY | pGEM-T-Easy derivative, containing a 136 bp DNA fragment corresponding to the region from 94.431 to 94.567 of H. pylori G27 genome amplified by PCR with oligonucleotides 0088HYF/0088HYR. This region encompasses the putative promoter region of HPG27_RS00460 (HP0088 according to 26695 annotation). | This work |
| pGEMpsRNA17_18HY | pGEM-T-Easy derivative, containing a 129 bp DNA fragment corresponding to the region from 16.516 to 16.645 of H. pylori G27 genome amplified by PCR with oligonucleotides sRNA17_18HYF/sRNA17_18HYR. This region encompasses the intergenic region between putative SRP_RNA and HPG27_RS00110. | This work |
| pGEMpA1.4HY | pGEM-T-Easy derivative, containing a 189 bp DNA fragment corresponding to the region from 1.576.888 to 1.577.077 of H. pylori G27 genome amplified by PCR with oligonucleotides A1.4HYF/A1.4HYR. This region encompasses the putative promoter region of isoA toxin/antitoxin system. | This work |
DNA manipulations
DNA manipulations were performed as described by Sambrook et al.30. All restriction and modification enzymes were used according to the manufacturers’ instructions (New England Biolabs). Preparations of plasmid DNA were carried out with NucleoBond Xtra Midi plasmid purification kit (Macherey-Nagel).
Overexpression and purification of recombinant HP1043 protein
Recombinant N-terminal His-tagged HP1043 protein was overexpressed in E. coli and affinity-purified as previously described11. Purified HP1043 was dialysed against two changes of 1 × 1043 Footprinting Buffer (1 × 1043 FPB: 10 mM Tris-HCl pH 7.5; 50 mM NaCl; 10 mM MgCl2; 1 mM DTT; 0.01% Igepal CA-630; 10% glycerol) for DNA-binding assays or against two changes of 1X PBS (137 mM NaCl; 2.7 mM KCl; 10 mM NaH2PO4; 1.8 mM KH2PO4; pH 7.4) for animal immunization. Protein concentration was determined by Bradford colorimetric assay (Bio-Rad) and the purity of the protein preparations was analysed by SDS-PAGE.
Chromatin Immunoprecipitation (ChIP) with α-HP1043 polyclonal antibody
Available α-HP1043 polyclonal antibody from immunized rabbits12 were purified by 3 sequential precipitations with 35% saturated (NH4)2SO4 and subsequent resuspension in water. H. pylori G27 wild type cultures were liquid-grown to an OD600 of 1.0, harvested, crosslinked, sonicated and immunoprecipitated as previously described12. Briefly, protein-DNA complexes were chemically crosslinked with 1% formaldehyde, then DNA was shared by extensive sonication with Bioruptor (Diagenode). HP1043 covalently linked to target DNA fragments were immunoprecipitated by incubating the crosslinked whole cell extracts with the polyclonal α–HP1043 antibody and then capturing the resulting complexes with Protein-G-conjugated sepharose beads. Cross-linking was reverted for 6 h at 65 °C, with occasional mixing. DNA was extracted once with phenol-chloroform and further extracted with chloroform. After the addition of 1% glycogen, DNA was ethanol precipitated and suspended in 50 μl double-distilled H2O, as previously described12.
ChIP-seq library preparation and sequencing
Illumina libraries were prepared, for each biological replicate either from 5 ng of immunoprecipitated-DNA (IPs) or from 5 ng of the Input-DNA following the Illumina TruSeq ChIP-seq DNA sample preparation protocol; then each library was sequenced on a MiSeq Illumina sequencer and 51 bp single stranded reads were produced.
ChIP-seq data analysis
Bowtie 2 (v2.2.6)31 was used to align raw reads, produced from ChIP sequencing experiments, to H. pylori G27 genome. End-to-end mapping was performed and non-deterministic option was specified to force a single assignment of multi-mapping reads to the best scoring region (if present) or a random attribution in the case of regions with identical scores. High quality reads were then selected requiring: for uniquely mapping reads MAPQ (mapping quality) greater than 30 and alignment score greater than −10 while for multi-mapping reads alignment score was set equal or greater than −10. The quality of ChIP-Seq data was evaluated following ENCODE quality metrics (https://code.google.com/archive/p/phantompeakqualtools/) and the numerical values obtained are provided in Supplementary Table S1. The cross-correlation analysis resulted in good NSC and RSC values, obtained using the code from Dr. Kundaje at Stanford University (https://code.google.com/archive/p/phantompeakqualtools/) cited by ENCODE consortium (http://genome.ucsc.edu/ENCODE/encodeTools.html). Moreover, we obtained average PBC scores. Irreproducible Discovery Rate procedure (IDR v 2.0.2) following ENCODE guidelines32, and using Homer (v4.7.2)33 as peak caller, was performed to measure sample reproducibility and to identify consistent peaks. Homer parameters were set according to the authors’ indication for IDR calculation (-P. 1 –LP. 1 –poisson. 1), -L was set to three. The “findPeaks score” column was selected as ranking column for IDR calculations, as suggested by Homer authors. The pool of two independent input preparations was used as background for the analysis, as suggested by IDR procedure. Peaks were manually annotated to the genes having transcription start site within −150/ + 50 bp from the middle of the peak (promoter peaks), to the genes containing the middle of peak (intragenic) or to the two genes surrounding the intergenic regions containing the peak (intergenic), according to the current version of H. pylori G27 RefSeq annotation (GCF_000021165.1). Transcription start sites were derived by blasting the 50 bp before the initiation of transcription found by Sharma et al.16 for 26695 and controlling their coherence with our RNA-seq signals. A transcriptional start site was automatically cross-mapped when fragments matched with at least 90% identity and at least with 45/51 nucleotides in length. To further define peaks annotation, for the regions where HP1043 was bound, we considered also the fragments matching with an identity of at least 80% and an overlap of 35/51 nucleotides, if showing an increase of transcription (according to our RNAseq data) in correspondence to the cross-mapped transcriptional start site. To obtain COG34 annotation for the putative protein coding targets the protein accession number of those genes were submitted to CDD35 online database and COG alphanumeric code was converted to function according to the official COG classification. Raw data are publicly available at Sequence Reads Archive under accession number BioProject PRJNA327549.
DNase I footprinting
Genomic regions harbouring the putative HP1043 binding sites identified by ChIP-seq analysis were PCR-amplified with oligonucleotides listed in Supplementary Table S4 and cloned in pGEM-T-Easy vector (Table 2). DNA fragments obtained by digestion with the appropriate restriction enzymes were 5′-end labelled with [γ-32P]-ATP and T4 polynucleotide kinase and gel purified. Approximately, 15 fmol of labelled probe were used for each footprinting reaction. DNase I footprinting experiments were performed as previously described36. Briefly, the labelled probes were incubated with increasing amounts of purified recombinant HP1043 protein in 1 × 1043 FPB containing 300 ng of sonicated salmon sperm DNA as non-specific competitor in a final volume of 50 μl for 20 min at room temperature. The partial digestion of the labelled probe was carried out using 0.01U of DNase I (Novagen) diluted in 1 × 1043 FPB supplemented with 5 mM CaCl2 and 1 mM DTT for 90 s at room temperature. Reactions were stopped with 140 μl of STOP buffer (192 mM NaOAc pH 5.2; 32 mM EDTA pH 8.0; 0.14% SDS; 64 μg/μl sonicated salmon sperm DNA), phenol-chloroform extracted and ethanol precipitated. Samples were resuspended in 10 μl of Formamide Loading Buffer (FLB: 95% formamide; 10 mM EDTA; 0.02% bromophenol blue; 0.02% xylene cyanol), denatured at 100 °C for 3 min, fractionated on a 8 M urea-6% polyacrylamide sequencing gel and auto-radiographed.
Hydroxyl-radical footprinting
Genomic regions of interest were 5′-end labelled and purified as described above for DNase I footprintings. Hydroxyl-radical footprinting assays were performed as previously described37. Briefly, the labelled probes were incubated with increasing amounts of HP1043 protein in 1 × 1043 FPB0 (10 mM Tris-HCl pH 7.5; 50 mM NaCl; 10 mM MgCl2; 1 mM DTT; 0.01% Igepal CA-630) containing 300 ng of sonicated salmon sperm DNA as non-specific competitor in a final volume of 30 μl for 20 min at room temperature. Partial digestion of the labelled probe was performed by the simultaneous addition of 2 μl each of the following solutions: 2 μl of Fe:EDTA solution (125 mM Fe (NH4)2 (SO4)2; 250 mM EDTA pH 8.0), 2 μl of 0.1 M DTT and 2 μl 1% H2O2. After a 2-min incubation, cutting reaction was stopped with the addition of 25 μl of STOP solution (600 mM NaOAc pH 5.2; 100 ng/μl sonicated salmon sperm DNA; 4% glycerol), phenol-chloroform extracted and ethanol precipitated. Samples were resuspended in 6 μl of FLB, denatured at 100 °C for 3 min, fractionated on 8 M urea-8.4% polyacrylamide sequencing gel and auto-radiographed.
RNA isolation
H. pylori cultures were liquid-grown until mid-exponential phase (OD600 = 0.8) then treated with 0.5 μg/ml of tetracycline for 60 min. Bacterial cells were harvested and total RNA was extracted with TRI-reagent (Sigma-Aldrich), according to manufacturer’s protocol.
RNA-seq: library preparation, sequencing and analyses
Ribosomal RNAs were depleted starting from 1 μg of total RNA from each of the conditions analysed by using the RiboZero Gram negative kit (Epicentre, Illumina). Strand specific RNA-seq libraries were prepared by using the ScriptSeqTM v2 RNAseq library preparation kit (Epicentre, Illumina) starting from 50 ng of previously rRNA depleted RNA from each biological replicate and for all the conditions analysed. Then each library was sequenced on a MiSeq Illumina sequencer and 76 bp reads were produced. Bowtie 2 (v2.2.6)31 was used to align raw reads to H. pylori G27 genome with the same parameters used for ChIP-seq data. High quality reads were selected requiring: for uniquely mapping reads MAPQ (mapping quality) greater than 30 and alignment score greater than −15; for multi-mapping reads alignment score was set equal or greater than −15 rRNA depletion, strand specificity and gene coverage were evaluated using BEDTools (v2.20.1*)38 and SAMtools (v0.1.19)39 to verify the library preparation and sequencing performances (see Supplementary Table S1). Strand specific reads overlapping to the genes annotated in H. pylori G27 RefSeq annotation (GCF_000021165.1) for at least 50% of their length were considered to produce the raw-counts of each sample. Only rRNAs were excluded as they were depleted during the library preparation procedure. The R package DESeq2 (v1.4.5)40 was then used to normalize the counts and to individuate differentially expressed features showing BH adjusted p-value lower than 0.01. DESeq2 uses one of the most common strategy to normalize data after sequencing, which is the median-of-ratio method. In brief for each non variable gene the ratio of the expression level between samples is calculated, then the median ratio across all expressed genes is used as the normalization scale.
Raw data are publicly available at Sequence Reads Archive under accession number BioProject PRJNA327549.
qRT-PCR analysis
Synthesis of cDNA and qRT-PCR analysis were carried out as previously described41. Briefly, after removal of contaminating genomic DNA through DNase I digestion, 1 μg of DNA-free total RNA was mixed with 50 ng of random hexamers (Invitrogen), dNTPs (1 mM each), AMV-Reverse Transcriptase (Promega) and incubated for 1 h at 37 °C for cDNA synthesis. For qRT-PCR analyses, 2 μl of the diluted (1:10) cDNA samples were mixed with 5 μl of 2X Power Up SYBR Green master Mix (ThermoFisher Scientific) and oligonucleotides specific for the genes of interest (Supplementary Table S4) at 400 nM concentration in a final volume of 10 μl. qRT-PCR was performed using the following cycling protocol: 95 °C for 2 min, then 40 cycles consisting of a denaturation for 5 s at 95 °C followed by 30 s at 60 °C (annealing and extension steps). For each real time experiment, the specificity of the reaction was checked by including a melting profile at the end of the run. Data were analysed using the ∆∆Ct method, using the 16 S rRNA gene as internal reference. qRT-PCR of 16 S rRNA on samples from cells untreated or treated with tetracycline gave overlapping amplification curves, indicating that the amount of 16 S rRNA was not changed during the time-course experiment.
Additional Information
How to cite this article: Pelliciari, S. et al. Insight into the essential role of the Helicobacter pylori HP1043 orphan response regulator: genome-wide identification and characterization of the DNA-binding sites. Sci. Rep. 7, 41063; doi: 10.1038/srep41063 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
We would like to thank Giada Caredda and Maria Vurchio (Institute of Biomedical Technologies, National Research Council, Milan) for technical and administrative support. This work was supported by Grants from the Italian Ministry of Education and University (2010P3S8BR_003 to VS, and 2010P3S8BR_002 to CP) and by a grant from the University of Bologna to VS. SP is the recipient of a fellowship from the PhD program in Cellular and Molecular Biology of the University of Bologna.
Footnotes
Author Contributions The experiments were conceived and designed by A. Danielli, C. Peano, D. Roncarati, V. Scarlato and performed by S. Pelliciari, E. Pinatel, C. Peano, A. Vannini. Data analysis was carried out by S. Pelliciari, E. Pinatel, A. Vannini, S. Puccio. The paper was written by D. Roncarati and V. Scarlato with contributions by C. Peano, G. De Bellis,. E. Pinatel. All authors reviewed the manuscript.
References
- Salama N. R., Hartung M. L. & Muller A. Life in the human stomach: persistence strategies of the bacterial pathogen Helicobacter pylori. Nat Rev Micro 11, 385–399 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gisbert J. P. & Calvet X. Review article: Common misconceptions in the management of Helicobacter pylori-associated gastric MALT-lymphoma. Aliment. Pharmacol. Ther. 34, 1047–1062 (2011). [DOI] [PubMed] [Google Scholar]
- Scarlato V., Delany I., Spohn G. & Beier D. Regulation of transcription in Helicobacter pylori: simple systems or complex circuits? Int. J. Med. Microbiol. 291, 107–17 (2001). [DOI] [PubMed] [Google Scholar]
- Foynes S. et al. Helicobacter pylori possesses two CheY response regulators and a histidine kinase sensor, CheA, which are essential for chemotaxis and colonization of the gastric mucosa. Infect. Immun. 68, 2016–2023 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waidner B., Melchers K., Stähler F. N., Kist M. & Bereswill S. The Helicobacter pylori CrdRS two-component regulation system (HP1364/HP1365) is required for copper-mediated induction of the copper resistance determinant CrdA. J. Bacteriol. 187, 4683–4688 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niehus E. et al. Genome-wide analysis of transcriptional hierarchy and feedback regulation in the flagellar system of Helicobacter pylori. Mol. Microbiol. 52, 947–961 (2004). [DOI] [PubMed] [Google Scholar]
- Pflock M. et al. Characterization of the ArsRS regulon of Helicobacter pylori, involved in acid adaptation. 188, 3449–3462 (2006). [DOI] [PMC free article] [PubMed]
- Schär J., Sickmann A. & Beier D. Phosphorylation-independent activity of atypical response regulators of Helicobacter pylori. J. Bacteriol. 187, 3100–3109 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller S., Pflock M., Schär J., Kennard S. & Beier D. Regulation of expression of atypical orphan response regulators of Helicobacter pylori. Microbiol. Res. 162, 1–14 (2007). [DOI] [PubMed] [Google Scholar]
- Hong E. et al. Structure of an atypical orphan response regulator protein supports a new phosphorylation-independent regulatory mechanism. J. Biol. Chem. 282, 20667–20675 (2007). [DOI] [PubMed] [Google Scholar]
- Delany I., Spohn G., Rappuoli R. & Scarlato V. Growth phase-dependent regulation of target gene promoters for binding of the essential orphan response regulator HP1043 of Helicobacter pylori. J. Bacteriol. 184, 4800–4810 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannini A., Roncarati D. & Danielli A. The cag-pathogenicity island encoded CncR1 sRNA oppositely modulates Helicobacter pylori motility and adhesion to host cells. Cell. Mol. Life Sci. 1–18, doi: 10.1007/s00018-016-2151-z (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer S. et al. Novel function assignment to a member of the essential HP1043 response regulator family of epsilon-proteobacteria. Microbiol. (United Kingdom) 159, 880–889 (2013). [DOI] [PubMed] [Google Scholar]
- Olekhnovich I. N., Vitko S., Valliere M. & Hoffmana P. S. Response to metronidazole and oxidative stress is mediated through homeostatic regulator hsra (hp1043) in Helicobacter pylori. J. Bacteriol. 196, 729–739 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olekhnovich I. N. et al. Mutations to essential orphan response regulator HP1043 of Helicobacter pylori result in growth-stage regulatory defects pathogenesis. Infect. Immun. 81, 1439–1449 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma C. M. et al. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature 464, 250–255 (2010). [DOI] [PubMed] [Google Scholar]
- Tatusov R. L., Koonin E. V. & Lipman D. J. A Genomic Perspective on Protein Families. Science (80-.). 278, 631–637 (1997). [DOI] [PubMed] [Google Scholar]
- Beier D., Spohn G., Rappuoli R. & Scarlato V. Functional analysis of the Helicobacter pylori principal sigma subunit of RNA polymerase reveals that the spacer region is important for efficient transcription. Mol. Microbiol. 30, 121–134 (1998). [DOI] [PubMed] [Google Scholar]
- Frith M. C., Saunders N. F. W., Kobe B. & Bailey T. L. Discovering sequence motifs with arbitrary insertions and deletions. PLoS Comput. Biol. 4 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choprat A. N. Tetracycline Analogs Whose Primary Target Is Not the Bacterial Ribosome. Antimicrob Agents Chemother 38, 637–640 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisunov G. Y., Evsyutina D. V., Arzamasov A. a., Butenko I. O. & Govorun V. M. Ribosomal profiling of Mycoplasma gallisepticum. ActaNaturae 7, 107–112 (2015). [PMC free article] [PubMed] [Google Scholar]
- Danielli A. & Scarlato V. Regulatory circuits in Helicobacter pylori: Network motifs and regulators involved in metal-dependent responses. FEMS Microbiology Reviews 34, 738–752 (2010). [DOI] [PubMed] [Google Scholar]
- Danielli A. et al. In vivo dissection of the Helicobacter pylori fur regulatory circuit by genome-wide location analysis. J. Bacteriol. 188, 4654–4662 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grainger D. C., Hurd D., Harrison M., Holdstock J. & Busby S. J. W. Studies of the distribution of Escherichia coli cAMP-receptor protein and RNA polymerase along the E. coli chromosome. Proc. Natl. Acad. Sci. USA 102, 17693–17698 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visweswariah S. S. & Busby S. J. W. Evolution of bacterial transcription factors: How proteins take on new tasks, but do not always stop doing the old ones. Trends Microbiol. 23, 463–467 (2015). [DOI] [PubMed] [Google Scholar]
- Busby S. & Ebright R. H. Promoter structure, promoter recognition, and transcription activation in prokaryotes. Cell 79, 743–6 (1994). [DOI] [PubMed] [Google Scholar]
- Browning D. F. & Busby S. J. W. Local and global regulation of transcription initiation in bacteria. Nat. Rev. Microbiol. 14, 638–650 (2016). [DOI] [PubMed] [Google Scholar]
- Eder S., Liu W. & Hulett F. M. Mutational analysis of the phoD promoter in Bacillus subtilis: Implications for PhoP binding and promoter activation of Pho regulon promoters. J. Bacteriol. 181, 2017–2025 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dosanjh N. S., West A. L. & Michel S. L. J. Helicobacter pylori NikR’s interaction with DNA: A two-tiered mode of recognition. Biochemistry 48, 527–536 (2009). [DOI] [PubMed] [Google Scholar]
- Sambrook J., Fritsch E. & Maniatis T. Molecular Cloning: A Laboratory Manual. (Cold Spring Harbor Laboratory Press, 1989). [Google Scholar]
- Langmead B. & Salzberg S. L. Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landt S. et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 22, 1813–1831 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S. et al. Simple Combinations of Lineage-Determining Transcription Factors Prime cis-Regulatory Elements Required for Macrophage and B Cell Identities. Mol. Cell 38, 576–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov R. L. et al. The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4, 41 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A. et al. CDD: NCBI’s conserved domain database. Nucleic Acids Res. 43, D222–D226 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roncarati D., Danielli A. & Scarlato V. CbpA acts as a modulator of HspR repressor DNA binding activity in Helicobacter pylori. J. Bacteriol. 193, 5629–5636 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agriesti F. et al. FeON-FeOFF: the Helicobacter pylori Fur regulator commutates iron-responsive transcription by discriminative readout of opposed DNA grooves. Nucleic Acids Res. 42, 3138–3151 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan A. R. & Hall I. M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M. I., Huber W. & Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelliciari S., Vannini A., Roncarati D. & Danielli A. The allosteric behavior of Fur mediates oxidative stress signal transduction in Helicobacter pylori. Front. Microbiol. 6, 1–10 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Z. et al. Analysis of expression of CagA and VacA virulence factors in 43 strains of Helicobacter pylori reveals that clinical isolates can be divided into two major types and that CagA is not necessary for expression of the vacuolating cytotoxin. Infect. Immun. 63, 94–98 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D. Studies on transformation of Escherichia coli with plasmids. J Mol Biol 166, 557–580 (1983). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.