

Abstract
To determine the optimum duration of follow-up for the assessment of drug efficacy against Plasmodium falciparum malaria, 96 trial arms from randomized controlled trials (RCTs) with follow-up of 28 days or longer that were conducted between 1990 and 2003 were analyzed. These trials enrolled 13,772 patients, and participating patients comprised 23% of all patients enrolled in RCTs over the past 40 years; 61 (64%) trial arms were conducted in areas where the rate of malaria transmission was low, and 58 (50%) trial arms were supported by parasite genotyping to distinguish true recrudescences from reinfections. The median overall failure rate reported was 10% (range, 0 to 47%). The widely used day 14 assessment had a sensitivity of between 0 and 37% in identifying treatment failures and had no predictive value. Assessment at day 28 had a sensitivity of 66% overall (28 to 100% in individual trials) but could be used to predict the true failure rate if either parasite genotyping was performed (r2 = 0.94) or if the entomological inoculation rate was known. In the assessment of drug efficacy against falciparum malaria, 28 days should be the minimum period of follow-up.
Clinical trials of antimalarial drugs have been conducted for over 100 years (7, 71). The number of trials and their size have increased markedly in recent years; half of all published randomized trials have been conducted in the past 7 years, and the average number of patients included in trials has tripled (34). The design of trials testing the efficacy of drugs against Plasmodium falciparum malaria was first standardized by the World Health (WHO) between 1965 and 1973. To assess the efficacy of the 4-aminoquinolines (chloroquine and amodiaquine), a 28-day period of follow-up was proposed and generally adopted (70). During the introduction of mefloquine (which has a terminal elimination half-life of approximately 2 weeks in cases of malaria) it was observed that some recrudescences can occur after this 28-day period, and so longer follow-up periods were employed during some of the initial evaluations of this drug (16). In recent years, particularly following a review and report in 1996 by the WHO, a shorter period of follow-up (14 days) was suggested for the evaluation of antimalarial drugs in settings where the rate of malaria transmission was high (72). This has been used widely both in low- and high-transmission settings. Thus, in recent years there has been an increase both in the number of clinical trials with a short period of follow-up and also in periods of follow-up longer than 28 days, in southeast Asia, where mefloquine and other new antimalarials have been evaluated (14, 17, 34, 36, 53, 61-64).
In the past, accurate assessments of the efficacy of drugs against P. falciparum malaria required that the patient should not be exposed again to malarial infection during the follow-up period, as recrudescence and reinfection could not be distinguished reliably on clinical or parasitological grounds. This limited the conduct of trials to areas where the disease was not endemic or to in-hospital facilities. The introduction of molecular genotyping methods has allowed recrudescent infections to be distinguished reliably from newly acquired infections, and thus trials can now be conducted in areas where the disease is endemic—even those areas where the rate of malaria transmission is high (4, 13). This has allowed large community-based trials with extended follow-up to be conducted in malaria-affected communities and led to a recent considerable increase in both the number and size of antimalarial drug studies. It is increasingly accepted that cure of malaria infection should be the objective of treatment, as treatment failure (i.e., recrudescence) is associated with increased morbidity even in areas where transmission is high and reinfection is inevitable (37). Recrudescences encourage the spread of resistance. As resistance worsens, treatment failures manifest progressively earlier than after initial treatment (69), until parasitemia is no longer cleared (WHO resistance level R2) and then no longer responds at all to the antimalarial drug (WHO resistance level R3). As a consequence, mortality rises. Critical to the accurate definition of drug-resistant malaria is the length of follow-up. Various times have been adopted, ranging from 7 to 63 days, but 14 and 28 days are the durations used most commonly. We present a review of antimalarial drug trials that we have conducted since 1990 with follow-up periods of 28 days or longer to assess the optimum duration of observation for the accurate characterization of antimalarial drug efficacy in vivo.
MATERIALS AND METHODS
Data were obtained retrospectively from antimalarial drug trials in uncomplicated falciparum malaria that have been conducted or coordinated where individual patient follow-up was carried out for at least 28 days. We particularly sought those trials that were randomized and in which recrudescence could be reliably distinguished from new infection by PCR genotyping (i.e., true treatment failure rates were known). In the majority of studies, genotyping involved comparison by PCR of variable blocks within merozoite surface protein 1 (MSP1), merozoite surface protein 2 (MSP2), and glutamate-rich protein (GLURP) (7, 13). The objectives of this study were (i) to define a reference (true) parasitological failure rate, (ii) to compare the day 14 and day 28 failure rates with this true failure rate to establish the usefulness of either in predicting true failure, and (iii) to compare the widely used day 14 to the day 28 failure rates. The sources of the trials analyzed are given in Table 1.
TABLE 1.
Sources of trials included in this study
| Organization and location(s) of trial | No. of trial arms | Reference(s) or source |
|---|---|---|
| Shoklo Malaria Research Unit, Mae Sot, Tak, Thailand | 50 | 22-24, 29-33, 35, 36, 41-44, 54, 57, 58, 61-64 |
| F. Nosten, unpublished data | ||
| Epicentre, Paris, France | 9 | |
| Uganda | 5 | 21 |
| Laos | 1 | 15 |
| Liberia | 3 | 5, 6 |
| J. P. Guthmann, unpub- lished data | ||
| South East African Combination Antimalarial Therapy | 4 | K. Barnes, unpublished data |
| Mozambique | 1 | |
| South Africa | 3 | |
| Vietnam | 6 | 17 |
| T. T. Hien, unpublished data | ||
| Laos | 4 | 27, 28 |
| Thailand | 7 | 48, 49, 60 |
| Medecins sans Frontières- Holland, Yangon, Myanmar | 10 | 53, 54 |
| F. Smithuis, unpublished data | ||
| Tropical Diseases Research, WHO, Geneva, Switzerland | 26 | |
| Peru | 2 | 2 |
| São Tomé | 2 | 2 |
| Senegal | 2 | 1 |
| Uganda | 3 | 2 |
| Kenya | 5 | 1, 2 |
| Malawi | 3 | 2 |
| Burkina Faso | 2 | 2 |
| Côte d'Ivoire | 2 | 2 |
| Gabon | 2 | 1 |
| The Gambia | 3 | 66 |
Definition of endpoints.
The unit of analysis was an arm in a antimalarial drug trial. For those studies with 63 days of follow-up, we considered the day 63, PCR-adjusted treatment failure rate as the reference or the true failure rate, because recrudescence in a nonpregnant patient after this time is very unlikely (52).
We defined early failures as cases where parasitemia had not fallen by more than 75% at 48 h after starting treatment (original WHO classification, R3) (70). Where a precise 48-h value was not available, the day 3 value was used instead.
Failures at any later time were defined by positive (i.e., detectable on blood smear) parasitemia. The numbers of failures at 7, 14, 28, 42, and 63 days were compared. Patients who deteriorated clinically between days 2 and 7 with persistent or rising parasitemia were pooled with the day 7 failures.
In each trial, at each time point, we documented the number of patients examined and the number of failures that had occurred. We documented the number of samples that were PCR genotyped, the number of those classified as recrudescent infections, and the number of samples which could not be classified reliably in the PCR analysis (i.e., were considered indeterminate). Failures before or on day 7 (i.e., without parasite clearance) were not genotyped, as they were assumed to have derived only from the initial symptomatic infection. The PCR results were used to estimate the number of true failures in each individual trial after day 7 with the following formula: estimated number of true treatment failures after day 7 = number of patients with recurrent parasitemia × [(number of recrudescences by PCR)/(number of samples for which PCR analysis was done − number of indeterminate samples)].
A number of explanatory variables characterizing each trial arm were also recorded. These were country where the study was conducted, estimated contemporaneous entomological inoculation rate (EIR), year when the study started, the drugs used and their total doses, total number of patients, mean or median age of the patients, standard deviation of age, range of age, geometric mean parasitemia at day 0, range of parasitemias at day 0, the mean or median parasite clearance time, and mean or median fever clearance time. The study areas were divided arbitrarily into low-transmission (estimated EIR, ≤2 infectious bites/person/year), medium-transmission (estimated EIR, 2 to 10 infectious bites/person/year), and high-transmission (estimated EIR, >10 infectious bites/person/year) settings.
Data from the published literature.
We also reviewed all randomized controlled trials published in the past 40 years in which parasitological failure rates at both day 14 and day 28 or later were quoted (34), excluding those published trials which were included in this more-detailed evaluation.
Statistical analysis of failure rates.
Data were analyzed with STATA, version 8.0 (Stata Corp., College Station, Tex.).
Data were provided as count data, i.e., the number of observed patients for each time interval and the number of patients with treatment failures, and then transformed into survival time data. Patients lost to follow-up were treated as censored at the last examination. Patients who missed an examination at time t but were still parasite negative when they returned at time t + 1 examination were assumed to have been negative at time t. The estimated numbers of true failures were rounded to the nearest integer. New infections or samples indeterminate by PCR were treated as censored.
The trials differed in design. In several trials, where PCR genotyping was performed, it was not conducted on every treatment failure. The detailed analyses were therefore done only of data from trials in which PCR results were available at least for failures occurring at ≤14 days and also at ≤28 days following the start of treatment. The numbers of trials and corresponding numbers of patients for the different periods of follow-up are shown in Fig. 1. Cumulative failure rates were estimated individually by the Kaplan-Meier method for each trial arm. The cumulative failure rate estimates at days 14, 28, and 63 were compared within trial arms. The relationship between the day 63 failure rate and failure rate estimates at day 14 or day 28 was examined by linear regression, which also explored associations with the individual trial characteristics.
FIG. 1.

Summary of trial arms and patient numbers.
The longer a patient is followed, the more likely a new malaria infection is to occur. This cumulative probability distribution is determined by the malaria transmission intensity. We modeled the relationship between the cumulative proportion of true failures detected in these studies and the duration of follow-up with a three-parameter logistic function
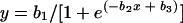 |
(1) |
where b1, b2, and b3 are the parameters, x is duration of follow-up, and y is the cumulative proportion of true failures detected.
The relationship between the proportion of all recurrent parasitemias that were recrudescences and the true failure rate was modeled using the negative exponential growth function
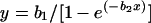 |
(2) |
where b1 and b2 are parameters, x is the true failure rate, and y is the proportion of all recurrent parasitemias which were recrudecences.
RESULTS
Data were obtained from 116 antimalarial drug trial arms which had enrolled 16,281 patients and which were carried out between 1990 and 2003 (Table 1). Of these, 96 trial arms (13,772 patients; 85% of all patients) came from randomized controlled trials (Fig. 1). This comprises 23% of all patients enrolled in published randomized trials in uncomplicated malaria caused by P. falciparum over the past 40 years (34).
Table 2 summarizes the trials' characteristics. Overall, 67% of studies (treatment arms) were located in Asia; 90% of studies were carried out on at least 60 patients, while 20% had 200 or more patients enrolled. More than half, or 73 of the studies (63%; 9,959 patients) of the trials were conducted in areas of where the rate of transmission of malaria was low, and only 14 (12%; 2,255 patients) trials came from stable high-transmission areas. Studies from high-transmission areas were never longer than 28 days. All 17 studies with 63 days of follow-up came from low-transmission areas. Overall, 21 studies (19%) were carried out with children only (<15 years of age), and 20 studies (17%) included only adults (>14 years of age).
TABLE 2.
Characteristics of trials included in the study
| Characteristica | No. (%) of treatment arms | Median (range) |
|---|---|---|
| No. of patients | 116 | 129 (18-533) |
| Continent | ||
| Africa | 36 (31) | |
| Asia | 78 (67) | |
| South America | 2 (2) | |
| Estimated entomological inocu- lation rate (no. of infectious bites/person/year) | ||
| Low (<2) | 73 (63) | |
| Medium (2-10) | 29 (25) | |
| High (>10) | 14 (12) | |
| Drug terminal elimination half-life (days) | ||
| Short (<1) | 21 (18) | |
| Medium (1-7) | 38 (33) | |
| Long (>7) | 57 (49) | |
| Mean age (yr) | 116 | 18 (1.3-33) |
| Geometric mean parasitemia at admission/μl | 114 | 10,070 (670-347,400) |
| Mean FCT (h) | 84 | 31 (19-62) |
| Mean PCT (h) | 85 | 62 (24-228) |
FCT, fever clearance time; PCT, parasite clearance time.
Patients were treated with a wide selection of drug regimens: (i) artesunate or artemether alone or in combination with antibiotics (clindamycin and doxycycline), (ii) chloroquine alone or in combination with artesunate or quinine, (iii) halofantrine, (iv) artemether-lumefantrine, (v) mefloquine alone or in combination with artesunate or artemether, (vi) quinine alone or in combination with antibiotics (clindamycin, rifampin, tetracycline, and doxycycline), (vii) sulfadoxine-pyrimethamine alone or in combination with artesunate, (viii) dihydroartemisinin-piperaquine, or (ix) atovaquone-proguanil alone or in combination with artesunate.
As slowly eliminated antimalarial drugs suppress reinfections and delay the appearance of recrudescences (69), the treatments were grouped into three categories according to the antimalarial drug terminal elimination half-life or, in case of combination treatment, according to the longest half-life of the component drugs (Table 3). Overall, 29% of trials used drugs with short half-lives (<1 day), 18% used drugs with medium half-lives (1 to 7 days), and 53% used drugs with long half-lives (>7 days).
TABLE 3.
Categorization of antimalarial treatment groups by antimalarial drug terminal elimination half-life
| Treatment duration | Drug half-life | Therapy |
|---|---|---|
| Short | 0-24 h | Artesunate or artemether alone or in combination with antibiotics |
| Quinine alone or in combination with antibiotics (clindamycin, rifampin, tetracycline) | ||
| Medium | 1-7 days | Halofantrine |
| Artemether-lumefantrine | ||
| Atovaquone-proguanil alone or in combination with an artemisinin derivative | ||
| Sulfadoxine-pyrimethamine alone or in combination with artesunate | ||
| Long | >7 days | Chloroquine alone or in combination with artesunate or quinine |
| Mefloquine alone or in combination with an artemisinin derivative | ||
| Dihydroartemisinin-piperaquine |
PCR genotyping corrected failure rates.
PCR genotyping was performed on data from 64 trial arms, 54 of which had PCR results for failures occurring at or before day 14 and at or before day 28. In addition, four trial arms had no failures observed during the 28-day follow-up period. Thus, accurate assessments for up to 28 days after the start of the treatment were available for 58 trial arms. Among these 58 trial arms, data from 26 trial arms had also PCR genotyping done for any failures occurring before day 42 of follow-up, and 17 arms had PCR results for failures occurring at any time up to 63 days follow-up. For failures occurring before day 14, the true failure rate was the same as the PCR-corrected value in 22 of 30 trials (73%) (i.e., all of the observed failures were true recrudescences), while this proportion decreased for assessments at later times, in 32 of 51 (day 28), 6 of 20 (day 42), and 3 of 11 (day 63) trials.
(i) Studies with 63-day follow-up (17 trial arms).
The day 14 assessment missed between 64 and 100% of all treatment failures in these studies (i.e., sensitivity was from 0 to 36%). The day 28 assessment missed between 8 and 84% (median, 40%) of failures, with the higher values coming from trials of slowly eliminated drugs with low levels of resistance (i.e., trials with few failures). Pooling all the results between the studies, the day 14 assessment missed 177 of 196 true failures (90%; overall sensitivity, 10%), and the day 28 assessment missed 67 of 196 failures (34%, overall sensitivity, 66%). As can be seen in Fig. 2, the day 14 assessment missed all failures following treatment with rapidly eliminated drugs in these studies.
FIG. 2.

Underestimation of the true treatment failure rate. Box plots show the median percentage (25 to 75% interquartile range; range) of true treatment failures not detected by day 14 (black box) and corresponding proportions of failures not detected by day 28 (white box) for antimalarial drugs with short (A), medium (B) and long (C) elimination half-lives. The true failure rate was considered the PCR genotype-confirmed failure rate at day 63.
(ii) Studies with 42-day follow-up (26 trial arms).
Compared with the day 42 assessment results, a median of 86% of failures were missed by day 14 assessment (range, 20 to 100%), and 18% (range, 0 to 100%) were missed by the day 28 assessment. When results from all 26 study arms with day 42 assessments were pooled, it was found that the day 14 assessment missed 159 (75%; overall sensitivity, 25%), and the day 28 assessment missed 32 (15%; overall sensitivity, 85%) of the 212 failures detected. When the overall failure rate was <10% (15 studies), the day 28 median underassessment was 26% (range, 0 to 100%), and when the failure rate exceeded 10% (11 studies), the day 28 assessment missed 15% (range, 0 to 40%).
(iii) Studies with 28-day follow-up (58 trial arms).
Observed failure rates at day 28 were nearly always much higher than rates at day 14. Of those trials with PCR-corrected results, 50% had failure rates at day 28 that were at least 2.6 times higher than the corresponding rate at day 14 (range, 1 to 22 times). The day 14 median underassessment of failure rate assessed at day 28 was 71%. In 50% of trials, more than 70% of failures occurred between days 14 and 28 (range, 0 to 100%). In the four trial arms with rapidly eliminated drugs, none of the failures was detected at day 14. A median of 9% of failures were detected at day 14 in trials with drugs with half-lives of 1 to 7 days (23 studies), and median of 34% of failures were detected in trials with slowly eliminated drugs (25 studies).
Figure 3 shows scatter plots of failure rates at days 14 and 28, for low-, medium-, and high-transmission areas. Although the observed failure rates were higher in the high-transmission area (reflecting the current use of ineffective drugs), the marked differences between day 14 and 28 rates remain. A 100% treatment success rate assessed at day 14 was associated with a true failure rate of up to 46% in these studies. Figure 4 plots the failure rate assessed at day 14 against the failure rate at maximum follow-up (assessed at 63 days for 17 trial arms, at 42 days for 15 trial arms, and at 28 days for 36 trial arms). As explained above, for those trials with a maximum follow-up of 28 days the discrepancy between the failure rate estimated at 14 days and estimated true failure rate is underestimated.
FIG. 3.

Relationship of malaria transmission intensity to underestimation of failure rate. Index plots of day 14 (•) and the corresponding day 28 failure rates (○) for the malaria transmission categories in order, from left to right, of increasing day 14 failure rates. PCR genotype-corrected results are shown. Each pair of dots corresponds to one trial arm. (A) Low transmission intensity. Estimated EIR, <2 infectious bites/person/year. (B) Medium transmission intensity. EIR, 2 to 10 infectious bites/person/year. (C) High transmission intensity. EIR, >10 infectious bites/person/year.
FIG. 4.

The relationship between the day 14 parasitological assessment and the failure rate at the maximum period of follow-up (maximum observed failure rate). Each circle represents a trial result in which the PCR genotype-corrected maximum failure rate was known at the end of follow-up. Black circles (•) denote results for trial arms with 63 or 42 days of follow-up, and the hollow circles (○) correspond to arm trials with 28 days of follow-up. Note that a failure rate at day 14 cannot exceed the maximum failure rate (lower triangle). A day 14 failure rate (vertical dotted line) of 25% has been recommended as the threshold for antimalarial drug policy change.
Cumulative estimates of therapeutic failure and time.
Figure 5 shows the relationship between the cumulative proportion of failures observed and the duration of follow-up for each of three treatment groups (short, medium, and long terminal elimination half-lives of antimalarial drugs, respectively). The relationships are sigmoid. There was a decrease in the slope of the linear portion of the sigmoid curve with an increase in the antimalarial drug half-life, although there was little difference between the curves for drugs with short or medium half-lives. The fitted relationships reach estimated maximum sensitivities at 35, 42, and >63 days of follow-up for short, medium, and long half-life antimalarial drugs, respectively.
FIG. 5.

Relationship between the proportion of antimalarial treatment failures detected and the duration of follow-up for (A) short half-life, (B) medium half-life, and (C) long half-life antimalarial drugs. PCR genotype-corrected results are shown.
Predicting the failure rate.
There was no evident relationship in these studies between the failure rate observed at day 14 and the degree of underestimation of the true failure rate. The differences were too large and unpredictable. The discrepancy was consistently high and reached 100% across the whole range of values of the true failure rates observed. For the failure rate at day 28, a pattern of decreased differences for higher values of the true failure rate was observed. Thus, whereas the day 14 assessment could not be used to predict the true failure rate, there was a linear relationship between the day 28 assessment and the day 63 rate in these trials (Fig. 6). Fitting the regression line estimated this relationship as follows: true failure rate = 3.49 + (1.09 × failure rate at day 28).
FIG. 6.

Relative utility of (A) day 14 assessment and (B) day 28 assessment in predicting true failure rate. PCR genotype-corrected results are shown.
The elimination half-life of the drug did not affect the results, nor did other trial characteristics. It should be noted that this relationship was obtained from data only in low-transmission areas, and these results cannot be extrapolated to areas of higher malaria transmission. Table 4 lists the predicted true failure rates for the different day 28 failure rates based on the above model.
TABLE 4.
Estimated true failure rate from trial arms with PCR genotyping and 63-day follow-up (n = 17)a
| Day 28 failurerate (%) | Predicted true failure rate (%) |
|---|---|
| 2 | 5 |
| 5 | 8 |
| 10 | 14 |
| 15 | 19 |
| 20 | 25 |
| 25 | 30 |
| 30 | 36 |
| 40 | 47 |
| 50 | 57 |
Predicted true failure rate = y; observed failure rate at day 28 = x. The true failure rate was calculated as follows: y = 3.49 + 1.09x.
The relationship between newly acquired and recrudescent infections.
The occurrence of a new patent infection during follow-up is related to the duration of follow-up, the elimination half-life of the drug, and the drug susceptibility of the newly acquired infection. The longer the follow-up, the more likely it is that a new infection will occur. As failure rates rise, an increasing proportion of recurrent parasitemias are recrudescences (i.e., treatment failures). The following exponential growth function was found to provide the best fit for the relationship between the proportion of recurrent parasitemias which were recrudescent infections and the true failure rate (based on studies with PCR genotyping and 63-day follow-up):
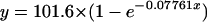 |
(3) |
where x is the true failure rate as a percentage and y is the proportion (expressed as a percentage) of recurrent parasitemias which were recrudescences (Fig. 7).
FIG. 7.

Relationship between the proportion of recurrent infections during a 63-day follow-up period which are true recrudescences and the overall true failure rate in low-transmission settings. PCR genotype-corrected data are shown.
The cumulative number of documented infections at any time is equal to the sum of the recrudescent infections (treatment failures) and new infections. The number of recrudescent infections was independent of the transmission intensity in this large series, although most of the data derived from low-transmission settings. The number of new infections depends on the transmission intensity. Assuming that new infections cannot become patent within 2 weeks of starting antimalarial treatment and assuming a constant level of transmission, then when the EIR is an average value of 1 infectious bite per person per year, it can be estimated that 13% of patients would have acquired a new infection during the 63-day follow-up, for an EIR of 2, 27% for an EIR of 3, 40% for an EIR of 4, 54% for an EIR of 5, and so on. In the absence of residual suppressive drug levels and at these relatively low levels of transmission, most or all of these infections are expected to become patent. But at higher levels of transmission, acquired immunity plays an increasingly important role in contributing to self-cure, and the number of acquired infections which would become patent cannot be estimated so confidently. Using the above calculations, we estimated the number of patients with recurrent parasitemia (recrudescences plus new infections) for given failure rates (Table 5).
TABLE 5.
Estimated percentages of patients with reappearance of parasites (recrudescence or new infection) by day 63a
| EIR | Treatment failure rate (%) | % of patients with parasites by day 63 |
|---|---|---|
| 0.5 | 5 | 11 |
| 10 | 16 | |
| 20 | 25 | |
| 30 | 35 | |
| 1 | 5 | 18 |
| 10 | 22 | |
| 20 | 31 | |
| 30 | 39 | |
| 2 | 5 | 30 |
| 10 | 34 | |
| 20 | 41 | |
| 30 | 49 |
Percentages are based on the assumption that no suppression of newly acquired infections has occurred.
Data from trials without PCR genotyping.
There were 58 trial arms without adequate genotyping data which enrolled 7,456 patients. The median (range) failure rates at day 14 were 5% (0 to 97%), and the rates at day 28 were 21% (1 to 96%). The median (interquartile range; range) day 14/day 28 failure rate ratio as a percentage was 23% (3 to 50%; 0 to 100%). There were 24 trials with 42-day follow-up; the median day 14/day 42 failure rate ratio (i.e., sensitivity) was 11% (0 to 29; 0 to 61%). There were 14 trials with 63-day follow up; the median day 14/day 63 failure rate ratio was 9% (0 to 10; 0 to 32%). The median day 28/day 42 ratio for 24 studies was 50% (41 to 72; 24 to 100%), and the median day 28/day 63 ratio for 14 studies was 25% (15 to 58; 8 to 69%).
Data from the published literature.
We identified a further 21 treatment arms comprising a total of 3,259 patients where both day 14 and day 28 failure rates were reported (11, 12, 18, 19, 38, 40, 51, 55, 56); 9 treatment arms were with drugs in the medium half-life category, and 12 treatment arms were with drugs in the long half-life category. The median (range) day 28 failure rate was 25% (0 to 66%). The majority of studies with long periods of follow-up were of long half-life drugs. The day 14 assessment detected 47% (0 to 76%) of the day 28 failures. There were only five trials with >42 days of follow-up, and in these the day 14 assessment detected a median of 33% (0 to 62%), and the day 28 assessment was 70% (0 to 88%) of the overall failures. This is comparable with the results from the studies with genotyping: for day 14 assessment, 14% (0 to 80%) and for day 28 assessment, 82% (0 to 100%). It was also comparable with the results from the trials without genotyping: for day 14 assessment, 11% (0 to 61%) and day 28 assessment, 50% (24 to 100%). The median (range) proportion (as a percentage) of day 28 failures detected at day 14 (i.e., the day 14/day 28 ratio) was 31% (0 to 73%) for day 28 failure rates of <25% (9 studies), and 51% (10 to 76%) for day 28 failure rates of ≥25% (P = 0.020; Mann-Whitney test).
DISCUSSION
These trials, which comprised approximately 23% of all patients enrolled in randomized trials of the drug treatment of falciparum malaria in the past 40 years, confirm that a follow-up period of 14 days is insufficient to characterize the therapeutic response to antimalarial treatment. The day 14 assessment commonly missed all treatment failures. The degree of underestimation was so large and so variable that it was effectively useless as a predictor of the true failure rate, irrespective of the transmission setting. The day 14 assessment was introduced by the WHO for use in settings of intense levels of disease transmission (72) because of the operational difficulties in conducting longer follow-up in trials and problems distinguishing recrudescences from reinfections, and because it was considered that clinical response should be the main criterion upon which national malaria treatment policy was based. Recently, a WHO consultation group produced a review of the methods for monitoring antimalarial resistance (73), which emphasizes the distinction between the methods of assessment in areas of high malaria transmission and those where transmission is at a low or moderate level, and it puts more weight than previously on parasitological measures. As before, the “primary intent of the recommended protocol(s) is the monitoring of drug efficacy over time for strictly programmatic purposes” (73). The aim is to ensure a “minimal evidence base from which Ministries of Health can develop informed treatment guidelines and policies” (73). The meaning of these terms is unclear. The clinical trial data presented here, derived from a very large number of controlled trials, indicate that if the day 14 assessment is used this evidence base cannot include a true assessment, or even an approximation, of the treatment failure rate. A test with diagnostic sensitivity of between 0 and 37% should never be used. Only when an antimalarial drug is failing so badly that a significant proportion of all infections do not clear the parasitemia at all (e.g., R2 or R3 early treatment failures), does the day 14 failure rate approximate the true failure rate. At this stage mortality is rising, and it is well past the time when the drug should be replaced. This is reflected in previously published trials by the significantly higher proportion of failures detected by the 14-day assessment when the day 28 failure rate exceeded 25% (51 versus 31% of the failures detected by day 28).
The WHO has recommended that antimalarial treatment policy should be changed when the day 14 failure rate exceeds 25%. For Africa, four “policy states,” defined by the day 14 failure rate, have been proposed: Grace (0 to 4%), Alert (5 to 14%), Action (15 to 24%), and Change (≥25%). But a day 14 failure rate of 25% is compatible with a true failure rate of over 60% (Fig. 4). This is surely unacceptable. By the time day 14 failure rates exceed 25%, a significant proportion of infections have early, potentially lethal, treatment failures. A 100% success rate assessed at day 14 was associated with a true failure rate, based on parasite genotyping and 63-day follow-up, as high as 46%. These very large differences contribute to the current widespread confusion over treatment efficacy with researchers pointing to in vivo and in vitro evidence for high-level resistance, and donors and policy makers claiming that drugs such as chloroquine and sulfadoxine-pyrimethamine still “work well” based on 14 day tests—despite rising mortality (20, 25, 59). The day 28 assessment was much better than the day 14 assessment and allowed prediction of the true failure rate in low-transmission settings. Incorporation of epidemiological information (an estimate of EIR) allowed prediction of the true failure rates even if parasite genotyping (to distinguish reinfection from recrudescence) was not available. As there are no data with follow-up longer than 28 days in higher transmission settings, we cannot estimate the degree to which the 28-day assessment underestimates the treatment failure rate in such areas. Immunity certainly contributes to the therapeutic response. There has been a tendency to assume that the contribution of immunity to antimalarial drug efficacy malaria is uniform across patients of a particular age, but this is unlikely to be true. Severe malaria represents an unusual response to an unfamiliar parasite (68). This is why there may be high rates of dangerous early failures in antimalarial drug trials from high-transmission settings with overall failure rates which are deemed satisfactory (40).
Modeling the data from these trials suggested that a 42-day follow-up captures nearly all treatment failures after treatment with antimalarial drugs which have terminal elimination half-lives of less than 1 week. The increased immunity in high-transmission areas may well prevent late recrudescences, but clearly more information on these relationships in higher-transmission settings is needed. These data indicate clearly that the day 14 assessment is useful only in identifying treatments which should already have been replaced. It should no longer be used. The traditional day 28 assessment is clearly much better and does provide useful information, although it still underestimates the true failure rate by as much as 40%. Underestimation is greater when failure rates are low or when slowly eliminated antimalarials are used.
The main determinants of the interval to recrudescence are the susceptibility of the infecting parasite population, the number of parasites present when antimalarial treatment starts, and the pharmacokinetic properties of the antimalarial drug in the treated patient (69). Immunity is also a factor (10, 26, 74). The distribution and range of intervals are determined by these variables. At low levels of resistance parasite densities fall below the level of detection in the blood, and the patient improves clinically. But the infection is not eradicated from the body in all patients, and when drug levels have fallen below the MIC of the drug for the infecting parasite population, the infection reexpands (67). Following a 7-day course of a rapidly eliminated drug such as quinine or an artemisinin derivative, the time to recrudescence averages 3 weeks and results from efficient parasite multiplication after the fourth drug exposed cycle (i.e., after 8 days) (50, 69). In the presence of residual, partially effective concentrations of a slowly eliminated antimalarial, the rate of parasite population reexpansion is reduced, and the interval to recrudescence is often longer than 4 weeks (52). Recrudescence is associated usually with a return of illness, failure to recover from or worsening of anemia, increased gametocyte carriage (and thus transmissibility) that fuels resistance, and a higher risk of failure with subsequent treatment (45-47). Even in high-transmission settings where reinfection is inevitable within a short time, therapeutic failure is associated with increased anemia, a major cause of childhood death, and eventually, as resistance worsens, directly increased mortality (37). Thus, recrudescence is associated with significant adverse effects both for the individual and the community. The objective of treatment must be to cure the infection.
These data provide information which will help with the design of antimalarial drug trials. It is evident from the results shown in Fig. 5 that the more slowly eliminated the antimalarial drug is, the longer the suppression of the eventual recrudescence will be. For relatively sensitive infections treated with slowly eliminated compounds, a significant proportion of recrudescences occur after 42 days, and this is probably the minimum duration of follow-up acceptable for assessing such treatments accurately. Follow-up for more than 8 weeks is better if possible, but there is a risk of significant numbers of drop-outs and reduced precision in the failure rate estimates. By contrast, follow-up for 28 days captures the majority of failures with drugs such as quinine, artemisinin derivatives, lumefantrine, atovaquone, halofantrine, and sulfadoxine-pyrimethamine. A 42-day follow-up is optimal for these drugs.
The current approach to monitoring antimalarial drug resistance relies upon having multiple sentinel sites which conduct in vivo and, in some cases, in vitro monitoring. The aim is to provide an early warning system so Ministries of Health can respond to increasing drug resistance in a timely fashion. This aim is undermined considerably by the common use of 14-day assessments, because such assessments detect only very high levels of resistance, and thus the health consequences of worsening antimalarial resistance are considerably underestimated. Furthermore, as policy change is commonly very slow to respond to such information and because once the decision is made full implementation of a malaria treatment policy usually takes at least 18 to 24 months (3), the situation has often deteriorated further before more-effective treatment is substituted. Several things have changed for the better in recent years (39); there is more interest in and more financial support for provision of effective antimalarial drugs, the deleterious effects of resistance on individual and public health are better appreciated, parasite genotyping provides distinction of recrudescence from reinfection allowing the conduct of community-based trials, and there are better molecular markers of antimalarial drug resistance (4, 8, 9, 13, 46, 65). For antifol, chloroquine, atovaquone, and mefloquine resistance, the molecular markers are now good enough to be used to predict likely therapeutic responses, and the markers can be calibrated and their predictive values further refined against clinical trial results with adequate periods of follow-up. These require only a small blood sample to be taken and dried on filter paper. A revision of the surveillance approach is needed. Doing fewer trials, but doing them properly, combined with more widespread application of molecular markers (8) could well provide a more sensitive, and thus more effective, early-warning system that is less expensive and more informative.
Acknowledgments
We are very grateful to our colleagues who provided us with clinical trial data. The following principal investigators and their respective teams and partners contributed to the WHO/TDR database of clinical trials: L. von Seidlein, P. Kremsner, C. Obonyo, M. Loolpapit, G. Priotto, V. Gill, P. Brasseur, G. Malenga, B. Sirima, M. Henry, and T. Ruebush.
M. Adjuik was funded by WHO/TDR under the supervision of P. Garner and A. Babiker. This study was part of the Wellcome Trust-Mahidol University-Oxford Tropical Medicine Research Programme funded by the Wellcome Trust of Great Britain.
The views expressed in this paper are those of the authors and not of their institutions.
REFERENCES
- 1.Adjuik, M., P. Agnamey, A. Babiker, S. Borrman, P. Brasseur, M. Cisse, F. Cobelens, S. Diallo, J. F. Faucher, P. Garner, S. Gikunda, P. G. Kremsner, S. Krishna, B. Lell, M. Loolpapit, P. B. Matsiegui, M. A. Missinou, J. Mwanza, F. Ntoumi, P. Olliaro, P. Osimbo, P. Rezbach, E. Some, and W. R. Taylor. 2002. Amodiaquine-artesunate versus amodiaquine for uncomplicated Plasmodium falciparum malaria in African children: a randomized, multicentre trial. Lancet 359:1365-1372. [DOI] [PubMed] [Google Scholar]
- 2.Adjuik, M., A. Babiker, P. Garner, P. Olliaro, W. Taylor, N. White, et al. 2004. Artesunate combinations for treatment of malaria: meta-analysis. Lancet 363:9-17. [DOI] [PubMed] [Google Scholar]
- 3.Bloland, P. B., and M. Ettling. 1999. Making malaria-treatment policy in the face of drug resistance. Ann. Trop. Med. Parasitol. 93:5-23. [DOI] [PubMed] [Google Scholar]
- 4.Brockman, A., R. E. Paul, T. J. Anderson, I. Hackford, L. Phaiphun, S. Looareesuwan, F. Nosten, and K. P. Day. 1999. Application of genetic markers to the identification of recrudescent Plasmodium falciparum infections on the northwestern border of Thailand. Am. J. Trop. Med. Hyg. 60:14-21. [DOI] [PubMed] [Google Scholar]
- 5.Checchi, F., R. Durand, S. Balkan, B. T. Vonhm, J. Z. Kollie, P. Biberson, E. Baron, J. Le Bras, and J. P. Guthmann. 2002. High Plasmodium falciparum resistance to chloroquine and sulfadoxine-pyrimethamine in Harper, Liberia: results in vivo and analysis of point mutations. Trans. R. Soc. Trop. Med. Hyg. 96:664-669. [DOI] [PubMed] [Google Scholar]
- 6.Checchi, F., S. Balkan, B. T. Vonhm, M. Massaquoi, P. Biberson, P. Eldin de Pecoulas, P. Brasseur, and J. P. Guthmann. 2002. Efficacy of amodiaquine for uncomplicated Plasmodium falciparum malaria in Harper, Liberia. Trans. R. Soc. Trop. Med. Hyg. 96:670-673. [DOI] [PubMed] [Google Scholar]
- 7.Covell, G., G. R. Coatney, J. W. Field, and J. Singh. 1955. Chemotherapy of malaria. W. H. O. monograph series 27. World Health Organization, Geneva, Switzerland. [PubMed]
- 8.Djimde, A., O. K. Doumbo, R. W. Steketee, and C. V. Plowe. 2001. Application of a molecular marker for surveillance of chloroquine-resistant falciparum malaria. Lancet 358:890-891. [DOI] [PubMed] [Google Scholar]
- 9.Djimde, A., O. K. Doumbo, J. F. Cortese, K. Kayentao, S. Doumbo, Y. Diourte, A. Dicko, X. Z. Su, T. Nomura, D. A. Fidock, T. E. Wellems, C. V. Plowe, and D. Coulibaly. 2001. A molecular marker for chloroquine-resistant falciparum malaria. N. Engl. J. Med. 344:257-263. [DOI] [PubMed] [Google Scholar]
- 10.Djimde, A. A., O. K. Doumbo, O. Traore, A. B. Guindo, K. Kayentao, Y. Diourte, S. Niare-Doumbo, D. Coulibaly, A. K. Kone, Y. Cissoko, M. Tekete, B. Fofana, A. Dicko, D. A. Diallo, T. E. Wellems, D. Kwiatkowski, and C. V. Plowe. 2003. Clearance of drug-resistant parasites as a model for protective immunity in Plasmodium falciparum malaria. Am. J. Trop. Med. Hyg. 69:558-563. [PubMed] [Google Scholar]
- 11.Dorsey, G., Njama, M. R. Kamya, A. Cattamanchi, D. Kyabayinze, S. G. Staedke, A. Gasasira, and P. J. Rosenthal. 2002. Sulfadoxine/pyrimethamine alone or with amodiaquine or artesunate for treatment of uncomplicated malaria: a longitudinal randomized trial. Lancet 360:2031-2038. [DOI] [PubMed] [Google Scholar]
- 12.Driessen, G. J., S. van Kerkhoven, B. J. Schouwenberg, G. Bonsu, and J. P. Verhave. 2002. Sulphadoxine/pyrimethamine: an appropriate first-line alternative for the treatment of uncomplicated falciparum malaria in Ghanaian children under 5 years of age. Trop. Med. Int. Health 7:577-583. [DOI] [PubMed] [Google Scholar]
- 13.Farnert, A., A. P. Arez, H. A. Babiker, H. P. Beck, A. Benito, A. Bjorkman, M. C. Bruce, D. J. Conway, K. P. Day, L. Henning, O. Mercereau-Puijalon, L. C. Ranford-Cartwright, J. M. Rubio, G. Snounou, D. Walliker, J. Zwetyenga, and V. E. de Rosario. 2001. Genotyping of Plasmodium falciparum infections by PCR: a comparative multicentre study. Trans. R. Soc. Trop. Med. Hyg. 95:225-232. [DOI] [PubMed] [Google Scholar]
- 14.Fontanet, A. L., B. D. Johnston, A. M. Walker, Y. Bergqvist, U. Hellgren, and W. Rooney. 1994. Falciparum malaria in eastern Thailand: a randomised trial of the efficacy of a single dose of mefloquine. Bull. W. H. O. 72:73-81. [PMC free article] [PubMed] [Google Scholar]
- 15.Guthmann, J. P., S. Kasparian, R. Phetsouvanh, N. Nathan, M. Garcia, S. Phompida, A. Brockman, M. Gastellu, and D. Legros. 2002. The efficacy of chloroquine for the treatment of acute, uncomplicated, Plasmodium falciparum malaria in Laos. Ann. Trop. Med. Parasitol. 96:553-557. [DOI] [PubMed] [Google Scholar]
- 16.Harinasuta, T., D. Bunnag, and W. H. Wernsdorfer. 1983. A phase II clinical trial of mefloquine in patients with chloroquine-resistant falciparum malaria in Thailand. Bull. W. H. O. 61:299-305. [PMC free article] [PubMed] [Google Scholar]
- 17.Hien, T. T., P. P. Mai, C. Dolecek, P. Phuong, N. T. Dung, N. T. Truong, N. Thanh, L. H. Thai, D. T. H. An, N. T. H. Quyen, N. J. White, and J. J. Farrar. 2004. Dihydroartemisinin-piperaquine against multidrug resistant falciparum malaria in Viet Nam: randomized clinical trial. Lancet 363:18-22. [DOI] [PubMed] [Google Scholar]
- 18.Hung, L. N., P. J. de Vries, T. D. Le, L. Bich, P. L. Ho, N. H. Tran, et al. 1997. Single dose artemisinin-mefloquine versus mefloquine alone for uncomplicated falciparum malaria. Trans. R. Soc. Trop. Med. Hyg. 91:191-194. [DOI] [PubMed] [Google Scholar]
- 19.Kofoed, P. E., F. Lopez, P. Johansson, A. Sandstrom, K. Hedegaard, P. Aaby, and L. Rombo. 2002. Treatment of children with Plasmodium falciparum malaria with chloroquine in Guinea-Bissau. Am. J. Trop. Med. Hyg. 67:28-31. [DOI] [PubMed] [Google Scholar]
- 20.Korenromp, E. L., B. G. Williams, E. Gouws, C. Dye, and R. W. Snow. 2003. Measurement of trends in childhood malaria mortality in Africa: an assessment of progress toward targets based on verbal autopsy. Lancet Infect. Dis. 3:349-358. [DOI] [PubMed] [Google Scholar]
- 21.Legros, D., K. Johnson, P. Houpikian, M. Makanga, J. K. Kabakyenga, A. O. Talisuna, and W. R. Taylor. 2002. Clinical efficacy of chloroquine or sulfadoxine-pyrimethamine in children under five from south-western Uganda with uncomplicated falciparum malaria. Trans. R. Soc. Trop. Med. Hyg. 96:199-201. [DOI] [PubMed] [Google Scholar]
- 22.Luxemburger, C., F. ter Kuile, F. Nosten, G. Dolan, J. H. Bradol, L. Phaipun, T. Chongsuphajaisiddhi, and N. J. White. 1994. Single day mefloquine-artesunate combination in the treatment of multi-drug resistant falciparum malaria. Trans. R. Soc. Trop. Med. Hyg. 88:213-217. [DOI] [PubMed] [Google Scholar]
- 23.Luxemburger, C., F. Nosten, D. Raimond, T. Chongsuphajaisiddhi, and N. J. White. 1995. Oral artesunate in the treatment of uncomplicated hyperparasitemic falciparum malaria. Am. J. Trop. Med. Hyg. 53:522-525. [DOI] [PubMed] [Google Scholar]
- 24.Luxemburger, C., R. N. Price, F. Nosten, F. ter Kuile, T. Chongsuphajaisiddhi, and N. J. White. 1996. Mefloquine in infants and young children. Ann. Trop. Paediatr. 16:281-286. [DOI] [PubMed] [Google Scholar]
- 25.Marsh, K. 1998. Malaria disaster in Africa. Lancet 352:1965-1967. [DOI] [PubMed] [Google Scholar]
- 26.Mayxay, M., K. Chotivanich, S. Pukrittayakamee, P. Newton, S. Looareesuwan, and N. J. White. 2001. Contribution of humoral immunity to the therapeutic response in falciparum malaria. Am. J. Trop. Med. Hyg. 65:918-923. [DOI] [PubMed] [Google Scholar]
- 27.Mayxay, M., P. N. Newton, M. Khanthavong, P. Tiengkham, R. Phetsouvanh, S. Phompida, A. Brockman, and N. J. White. 2003. Chloroquine versus sulfadoxine-pyrimethamine for treatment of Plasmodium falciparum malaria in Savannakhet province, Lao People's Democratic Republic: an assessment of national antimalarial drug recommendations. Clin. Infect. Dis. 37:1021-1028. [DOI] [PubMed] [Google Scholar]
- 28.Mayxay, M., R. Phetsouvanh, S. Phompida, P. N. Newton, M. Khanthavong, B. Vannachone, A. Brockman, and N. J. White. 2003. A randomised comparison of oral chloroquine and sulfadoxine-pyrimethamine for the treatment of uncomplicated Plasmodium falciparum malaria in Laos. Trans. R. Soc. Trop. Med. Hyg. 97:343-344. [DOI] [PubMed] [Google Scholar]
- 29.McGready, R., T. Cho, L. Hkirijaroen, J. Simpson, T. Chongsuphajaisiddhi, N. J. White, and F. Nosten. 1998. Quinine and mefloquine in the treatment of multidrug-resistant Plasmodium falciparum malaria in pregnancy. Ann. Trop. Med. Parasitol. 92:643-653. [DOI] [PubMed] [Google Scholar]
- 30.McGready, R., A. Brockman, T. Cho, D. Cho, M. van Vugt, C. Luxemburger, T. Chongsuphajaisiddhi, N. J. White, and F. Nosten. 2000. Randomized comparison of mefloquine-artesunate versus quinine in the treatment of multi-drug resistant falciparum malaria in pregnancy. Trans. R. Soc. Trop. Med. Hyg. 94:689-693. [DOI] [PubMed] [Google Scholar]
- 31.McGready, R., T. Cho T, N. K. Keo, K. L. Thwai, L. Villegas, S. Looareesuwan, N. J. White, and F. Nosten. 2001. Artemisinin antimalarials in pregnancy: a prospective treatment study of 539 episodes of multidrug-resistant Plasmodium falciparum. Clin. Infect. Dis. 33:2009-2016. [DOI] [PubMed] [Google Scholar]
- 32.McGready, R., T. Cho, Samuel, L. Villegas, A. Brockman, M. van Vugt, S. Looareesuwan, N. J. White, and F. Nosten. 2001. Randomized comparison of quinine-clindamycin versus artesunate in the treatment of falciparum malaria in pregnancy. Trans. R. Soc. Trop. Med. Hyg. 95:651-656. [DOI] [PubMed] [Google Scholar]
- 33.McGready, R., K. L. Thwai, T. Cho, Samuel, S. Looareesuwan, N. J. White, and F. Nosten. 2002. The effects of quinine and chloroquine antimalarial treatments in the first trimester of pregnancy. Trans. R. Soc. Trop. Med. Hyg. 96:180-184. [DOI] [PubMed] [Google Scholar]
- 34.Myint, H. Y., P. Tipmanee, F. Nosten, S. Pukrittayakamee, N. P. J. Day, S. Looareesuwan, and N. J. White. 2004. A systematic overview of published antimalarial drug trials. Trans. R. Soc. Trop. Med. Hyg. 98:73-81. [DOI] [PubMed] [Google Scholar]
- 35.Nosten, F., F. ter Kuile, T. Chongsuphajaisiddhi, C. Luxemburger, H. K. Webster, M. Edstein, L. Phaipun, K. L. Thew, and N. J. White. 1991. Mefloquine-resistant falciparum malaria on the Thai-Burmese border. Lancet 337:1140-1143. [DOI] [PubMed] [Google Scholar]
- 36.Nosten, F., C. Luxemburger, F. O. ter Kuile, C. Woodrow, J. Pa Eh, T. Chongsuphajaisiddhi, and N. J. White. 1994. Treatment of multi-drug resistant Plasmodium falciparum malaria with 3-day artesunate-mefloquine combination. J. Infect. Dis. 170:971-977. [DOI] [PubMed] [Google Scholar]
- 37.Nosten, F., and E. Ashley. 2004. The detection and treatment of Plasmodium falciparum malaria: time for change. J. Postgrad. Med. 50:35-39. [PubMed] [Google Scholar]
- 38.Pillai, D. R., A. C. Labbe, V. Vanisaveth, B. Hongvangthong, S. Pomphida, S. Inkathone, K. Zhong, and K. C. Kain. 2001. Plasmodium falciparum malaria in Laos: chloroquine treatment outcome and predictive value of molecular markers. J. Infect. Dis. 183:789-795. [DOI] [PubMed] [Google Scholar]
- 39.Plowe, C. 2003. Monitoring antimalarial drug resistance: making the most of the tools at hand. J. Exp. Biol. 206:3745-3752. [DOI] [PubMed] [Google Scholar]
- 40.Plowe, C. V., J. G. Kublin, F. K. Dzinjalamala, D. S. Kamwendo, R. A. Mukadam, P. Chimpeni, M. E. Molyneux, and T. E. Taylor. 2004. Sustained clinical efficacy of sulphadoxine-pyrimethamine for uncomplicated falciparum malaria in Malawi after 10 years as first line treatment: five year prospective study. Br. Med. J. 328:545-548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Price, R. N., F. Nosten, C. Luxemburger, A. Kham, A. Brockman, T. Chongsuphajaisiddhi, and N. J. White. 1995. Artesunate versus artemether in combination with mefloquine for the treatment of multidrug-resistant falciparum malaria. Trans. R. Soc. Trop. Med. Hyg. 89:523-527. [DOI] [PubMed] [Google Scholar]
- 42.Price, R. N., F. Nosten, C. Luxemburger, M. van Vugt, L. Phaipun, T. Chongsuphajaisiddhi, and N. J. White. 1997. Artesunate/mefloquine treatment of multi-drug resistant falciparum malaria. Trans. R. Soc. Trop. Med. Hyg. 91:574-577. [Erratum: 92:122, 1998.] [DOI] [PubMed] [Google Scholar]
- 43.Price, R. N., C. Luxemburger, M. van Vugt, F. Nosten, A. Kham, J. Simpson, S. Looareesuwan, T. Chongsuphajaisiddhi, and N. J. White. 1998. Artesunate and mefloquine in the treatment of uncomplicated multi-drug resistant hyperparasitaemic falciparum malaria. Trans. R. Soc. Trop. Med. Hyg. 92:207-211. [DOI] [PubMed] [Google Scholar]
- 44.Price, R. N., M. van Vugt, F. Nosten, C. Luxemburger, A. Brockman, L. Phaipun, T. Chongsuphajaisiddhi, and N. J. White. 1998. Artesunate versus artemether for the treatment of recrudescent multi-drug resistant falciparum malaria. Am. J. Trop. Med. Hyg. 59:883-888. [DOI] [PubMed] [Google Scholar]
- 45.Price, R. N., F. Nosten, C. Luxemburger, L. Phaipun, F. O. ter Kuile, M. van Vugt, T. Chongsuphajaisiddhi, and N. J. White. 1999. Risk factors for gametocyte carriage in uncomplicated falciparum malaria. Am. J. Trop. Med. Hyg. 60:1019-1023. [DOI] [PubMed] [Google Scholar]
- 46.Price, R. N., C. Cassar, A. Brockman, M. Duraisingh, M. van Vugt, N. J. White, F. Nosten, and S. Krishna. 1999. The pfmdr1 gene is associated with a multidrug resistance phenotype in Plasmodium falciparum from the western border of Thailand. Antimicrob. Agents Chemother. 43:2943-2949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Price, R. N., J. Simpson, F. Nosten, C. Luxemburger, L. Hkirjaroen, F. O. ter Kuile, T. Chongsuphajaisiddhi, and N. J. White. 2001. Factors contributing to anemia in uncomplicated falciparum malaria. Am. J. Trop. Med. Hyg. 65:614-622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pukrittayakamee, S., A. Chantra, S. Vanijanonta, R. Clemens, S. Looareesuwan, and N. J. White. 2000. Therapeutic responses to quinine and clindamycin in multidrug-resistant falciparum malaria. Antimicrob. Agents Chemother. 44:2395-2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pukrittayakamee, S., S. Prakongpan, S. Wanwimolruk, R. Clemens, S. Looareesuwan, and N. J. White. 2003. Adverse effect of rifampin on quinine efficacy in uncomplicated falciparum malaria. Antimicrob. Agents Chemother. 47:1509-1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pukrittayakamee, S., S. Wanwimolruk, K. Stepniewska, A. Jantra, S. Huyakorn, S. Looareesuwan, and N. J. White. 2003. Quinine pharmacokinetic pharmacodynamic relationships in uncomplicated falciparum malaria. Antimicrob. Agents Chemother. 47:3458-3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schellenberg, D., E. Kahigwa, C. Drakeley, A. Malende, A., J. Wigayi, C. Msokame, J. J. Aponte, M. Tanner, H. Mshinda, C. Menendez, and P. L. Alonso. 2002. The safety and efficacy of sulfadoxine-pyrimethamine, amodiaquine, and their combination in the treatment of uncomplicated Plasmodium falciparum malaria. Am. J. Trop. Med. Hyg. 67:17-23. [DOI] [PubMed] [Google Scholar]
- 52.Simpson, J. A., E. R. Watkins, R. N. Price, L. Aarons, D. E. Kyle, and N. J. White. 2000. Mefloquine pharmacokinetic-pharmacodynamic models: implications for dosing and resistance. Antimicrob. Agents Chemother. 44:3414-3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Smithuis, F. M., J. B. van Woensel, E. Nordlander, W. S. Vantha, and F. O. ter Kuile. 1993. Comparison of two mefloquine regimens for the treatment of Plasmodium falciparum malaria on the northeastern Thai-Cambodian border. Antimicrob. Agents Chemother. 37:1977-1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smithuis, F., I. van der Broek, I. Katterman, M. K. Kyaw, A. Brockman, S. Lwin, and N. J. White. 2004. Optimising operational use of artesunate-mefloquine; a randomised comparison of four treatment regimens. Trans. R. Soc. Trop. Med. Hyg. 98:182-192. [DOI] [PubMed] [Google Scholar]
- 55.Sowunmi, A., F. A. Fehintola, A. A. Adedeji, A. G. Falade, C. O. Falade, O. O. Akinyinka, and A. M. Oduola. 2000. Comparative efficacy of chloroquine plus chlorpheniramine alone and in a sequential combination with sulfadoxine-pyrimethamine, for the treatment of acute, uncomplicated, falciparum malaria in children. Ann. Trop. Med. Parasitol. 94:209-217. [DOI] [PubMed] [Google Scholar]
- 56.Sowunmi, A., A. I. Ayede, A. G. Falade, V. N. Ndikum, C. O. Sowunmi, A. A. Adedeji, C. O. Falade, T. C. Happi, and A. M. Oduola. 2001. Randomized comparison of chloroquine and amodiaquine in the treatment of acute, uncomplicated, Plasmodium falciparum malaria in children. Ann. Trop. Med. Parasitol. 95:549-558. [DOI] [PubMed] [Google Scholar]
- 57.ter Kuile, F., F. Nosten, M. Thieren, C. Luxemburger, M. D. Edstein, T. Chongsuphajaisiddhi, L. Phaipun, H. K. Webster, and N. J. White. 1992. High dose mefloquine in the treatment of multidrug resistant falciparum malaria. J. Infect. Dis. 166:1393-1400. [DOI] [PubMed] [Google Scholar]
- 58.ter Kuile, F. O., G. Dolan, F. Nosten, M. D. Edstein, C. Luxemburger, L. Phaipun, T. Chongsuphajaisiddhi, H. K. Webster, and N. J. White. 1993. Halofantrine versus mefloquine in the treatment of multi-drug resistant falciparum malaria. Lancet 341:1044-1049. [DOI] [PubMed] [Google Scholar]
- 59.Trape, J. F., G. Pison, M. P. Preziosi, C. Enel, A. Desgrees du Lou, V. Delaunay, B. Samb, E. Lagarde, J. F. Molez, and F. Simondon. 1998. Impact of chloroquine resistance on malaria mortality. C. R. Acad. Sci. III, Sci. Vie 321:689-697. [DOI] [PubMed] [Google Scholar]
- 60.Vanijanonta, S., A. Chantra, N. Phophak, D. Chindanond, R. Clemens, and S. Pukrittayakamee. 1996. Therapeutic effects of chloroquine in combination with quinine in uncomplicated falciparum malaria. Ann. Trop. Med. Parasitol. 90:269-275. [DOI] [PubMed] [Google Scholar]
- 61.van Vugt, M., A. Brockman, B. Gemperli, C. Luxemburger, I. Gathman, C. Royce, T. Slight, S. Looareesuwan, N. J. White, and F. Nosten. 1998. Randomised comparison of artemether-benflumetol and artesunate-mefloquine in the treatment of multi-drug resistant falciparum malaria. Antimicrob. Agents Chemother. 42:135-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.van Vugt, M., P. Wilairatana, B. Gemperli, I. Gathmann, L. Phaipun, A. Brockman, C. Luxemburger, N. J. White, F. Nosten, and S. Looareesuwan. 1999. Efficacy of six doses or artemether-benflumetol in the treatment of multi-drug resistant falciparum malaria. Am. J. Trop. Med. Hyg. 60:936-942. [DOI] [PubMed] [Google Scholar]
- 63.van Vugt, M., S. Looareesuwan, P. Wilairatana, R. McGready, L. Villegas, I. Gathmann, R. Mull, A. Brockman, N. J. White, and F. Nosten. 2000. Artemether-lumefantrine for the treatment of multidrug resistant falciparum malaria. Trans. R. Soc. Trop. Med. Hyg. 94:545-548. [DOI] [PubMed] [Google Scholar]
- 64.van Vugt, M., E. Leonardi, L. Phaipun, T. Slight, K. L. Thway, R. McGready, A. Brockman, L. Villegas, S. Looareesuwan, N. J. White, and F. Nosten. 2002. Treatment of uncomplicated multidrug-resistant falciparum malaria with artesunate-atovaquone-proguanil. Clin. Infect. Dis. 35:1498-1504. [DOI] [PubMed] [Google Scholar]
- 65.Viriyakosol, S., N. Siripoon, C. Petcharapirat, P. Petcharapirat, W. Jarrar, S. Thaithong, K. N. Brown, and G. Snounou. 1995. Genotyping of Plasmodium falciparum isolates by the polymerase chain reaction and potential uses in epidemiological studies. Bull. W. H. O. 73:85-95. [PMC free article] [PubMed] [Google Scholar]
- 66.von Seidlein, L., P. Milligan, M. Pinder, K. Bojang, C. Anyalebechi, R. Gosling, R. Coleman, J. I. Ude, A. Sadiq, M. Duraisingh, D. Warhurst, A. Alloueche, G. Targett, K. McAdam, B. Greenwood, G. Walraven, P. Olliaro, and T. Doherty. 2000. Efficacy of artesunate plus pyrimethamine-sulphadoxine for uncomplicated malaria in Gambian children: a double blind, randomised, controlled trial. Lancet 355:352-357. [DOI] [PubMed] [Google Scholar]
- 67.White, N. J. 1997. Assessment of the pharmacodynamic properties of antimalarial drugs in vivo. Antimicrob. Agents Chemother. 41:1413-1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.White, N. J. 1999. Antimalarial drug resistance and mortality in falciparum malaria. Trop. Med. Int. Health 4:469-470. [DOI] [PubMed] [Google Scholar]
- 69.White, N. J. 2002. The assessment of antimalarial drug efficacy. Trends Parasitol. 18:458-464. [DOI] [PubMed] [Google Scholar]
- 70.World Health Organization. 1973. Chemotherapy of malaria and resistance to antimalarials. Report of a W. H. O. Scientific Group. W.H.O. Tech. Rep. Ser. 529:30-35. [PubMed] [Google Scholar]
- 71.World Health Organization. 1986. The chemotherapy of malaria. W. H. O. monograph series 27. World Health Organization, Geneva, Switzerland.
- 72.World Health Organization. 1996. Assessment of therapeutic efficacy of antimalarial drugs for uncomplicated falciparum malaria in areas with intense transmission. W.H.O./MAL/96.1077. World Health Organization, Geneva, Switzerland.
- 73.World Health Organization. 2002. Monitoring antimalarial drug resistance. Report of a W.H.O. consultation, W.H.O./CDS/CSR/EPH/2002.17. World Health Organization, Geneva, Switzerland.
- 74.Yorke, W., and J. W. S. MacFie. 1924. Observations on malaria made during treatment of general paralysis. Trans. R. Soc. Trop. Med. Hyg. 18:33-39. [Google Scholar]