

Abstract
There is considerable need for new modeling approaches in the study of combined antimicrobial effects. Current methods based on the Loewe additivity and Bliss independence models are associated with implicit assumptions about the interacting system. To circumvent these limitations, we propose an alternative approach to the quantification of pharmacodynamic drug interaction (PDI). Pilot time-kill studies were performed with 108 CFU of Pseudomonas aeruginosa/ml at baseline with meropenem or tobramycin alone. The studies were repeated with 25 concentration combinations of meropenem (0 to 64 mg/liter) and tobramycin (0 to 32 mg/liter) in a five-by-five array. The data were modeled with a three-dimensional response surface using effect summation as the basis of null interaction. The interaction index (Ii) is defined as the ratio of the volumes under the planes (VUP) of the observed and expected surfaces: VUPobserved/VUPexpected. Synergy and antagonism are defined as Ii values of <1 and >1, respectively. In all combinations, an enhanced killing effect was seen compared to that of either drug at the same concentration. The most significant synergism was observed between 1 and 5 mg/liter of meropenem and between 1 and 4 mg/liter of tobramycin; seven out of nine combinations had a >2-log drop compared to the more potent agent. The Ii was found to be 0.76 (95% confidence interval, 0.65 to 0.91) for the concentration ranges of the agents. The results corroborate previous data indicating that meropenem is synergistic with an aminoglycoside when used in combination against P. aeruginosa. Our parametric approach to quantifying PDI appears robust and warrants further investigations.
The ability to describe combined drug effects objectively is one of the major challenges in anti-infective pharmacology and is of paramount importance in the study of combination therapy. The pharmacodynamic drug interaction (PDI) of antimicrobials is typically described by qualitative terms, such as synergy, additivity (indifference), or antagonism. It is increasingly recognized that these standard approaches lack the sensitivity to capture various kinds of important information, such as the variability of the interacting system, extent of interaction, and emergence of resistance. Consequently, it is difficult to compare different combinations in a rational and robust manner.
When attempting to evaluate pharmacodynamic interaction (synergism and antagonism), one typically constructs an expected null interaction model, which predicts the effect of multiple drugs in the absence of interaction. Without a physically and theoretically founded null interaction model, it would be difficult to make any reasonable evaluations of the combined action of multiple pharmacological agents. Two main metrics which are widely adopted exist for pharmacodynamic interactions, each presenting a consistent and well-supported perspective on the expected effect of noninteracting agents. They are the Loewe additivity and Bliss independence models. Each of these methodologies provides a framework for defining null interaction, but both methods may be associated with multiple implicit assumptions of the interacting system (2). Each method is specialized in some fashion and only applicable after certain additional considerations are addressed.
The Loewe additivity theory states that one drug cannot interact with itself; two drugs which do not interact should effectively combine as dilutions of the same agent. However, it does not directly address well-accepted phenomena, such as target site saturation, which could be indications of a drug's self-interaction. The fact that drugs do interact with themselves is the simplest explanation of nonlinear concentration-effect relationships. At very low drug concentrations, the effect of the drug is negligible since there is no significant drug-target interaction. As the concentration of drug increases, the likelihood of drug molecules binding to the target site also increases, resulting in a proportional increase in effect. However, at very high drug concentrations, the drug molecules compete for the same target site; therefore, the effect mediated via target site activation or inhibition will be less than proportional to the increase in drug concentration. As a result, an overall sigmoid (nonlinear) concentration-effect relationship is commonly observed. In the case of two agents with disparate and nonlinear concentration-effect relationships, a high performance expectation may be imposed on the system (a proportional increase in effect is anticipated even at high drug concentrations). As a result, the Loewe additivity model may give rise to unexpected results when the concentration-effect relationships are nonlinear, unless the concentration-effect relationships are identical (in the case of a drug interacting with itself).
The Bliss independence theory was derived from the probability theory; its validity extends to those cases where two agents are expected to be wholly transparent to one another (1). Traditionally the effect of an agent is expressed as a percentage of the maximal effect (or growth inhibition), and the maximal effect is defined as the effect observed with an infinite amount (or the absence) of an agent. There are two assumptions implicit in this traditional approach. Firstly, it is implied that both drugs under investigation have the same maximal effect. And secondly, once the maximal effect has been reached by the first drug, the addition of the second drug will not contribute further to the overall observed effect. Both assumptions arose from the agents in the combination having different target sites on the same metabolic pathway. Once the substrate in an essential metabolic pathway (or enzyme system) is maximally activated or inhibited, a further increase in the amount of agent(s) will not translate to an enhanced pharmacological response. However, in clinical practice the agents in combination therapy are often intentionally chosen to have different mechanisms of action. By targeting multiple metabolic pathways simultaneously, it is hoped that the combined effect will be more than that which can be achieved maximally with one agent. Consequently, the assumptions of the Bliss independence model may be questionable. Since the effect observed with an infinite amount of one agent may not truly be the maximal effect (combination therapy may achieve a greater effect), it should not be set as a reference for comparison to other drug effects.
With the increasing prevalence of multidrug-resistant pathogens, such as human immunodeficiency virus, mycobacteria, and gram-negative bacteria (e.g., Pseudomonas aeruginosa), there is a growing need for more robust, informative models that can identify optimal (or effective) treatment combinations for these pathogens. To this end, we propose an alternative approach to describing the nature and quantifying the PDI of antimicrobials. To illustrate our approach, we examined the combined effect of meropenem and tobramycin against a standard strain of P. aeruginosa. (This study was presented in part at the Interscience Conference on Antimicrobial Agents and Chemotherapy, Chicago, Ill., 14 to 17 September 2003.)
MATERIALS AND METHODS
Antimicrobial agent.
Meropenem powder was supplied by AstraZeneca (Wilmington, Del.). Tobramycin powder was purchased from Sigma (St. Louis, Mo.). A stock solution of each agent at 1,024 mg/liter in sterile water was prepared, aliquoted, and stored at −20°C. Prior to each susceptibility test, an aliquot of the drug was thawed and diluted to the desired concentrations with cation-adjusted Mueller-Hinton II broth (Ca-MHB; BBL, Sparks, Md.).
Microorganism.
P. aeruginosa ATCC 27853 (American Type Culture Collection, Rockville, Md.) was used in the study. The bacterium was stored at −70°C in Protect storage vials (Key Scientific Products, Round Rock, Tex.). Fresh isolates were subcultured twice on 5% blood agar plates (Hardy Diagnostics, Santa Maria, Calif.) for 24 h at 35°C prior to each experiment.
Susceptibility studies.
Meropenem and tobramycin MICs and minimal bactericidal concentrations (MBCs) for P. aeruginosa ATCC 27853 were determined in Ca-MHB by using a broth macrodilution method as described by NCCLS (15). The final concentration of bacteria in each broth macrodilution tube was approximately 5 × 105 CFU/ml of Ca-MHB. Serial twofold dilutions of drugs were used. The MIC was defined as the lowest concentration of drug that resulted in no visible growth after 24 h of incubation at 35°C in ambient air. Samples (50 μl) from clear tubes and the cloudy tube with the highest drug concentration were plated on Mueller-Hinton agar (MHA) plates (Hardy Diagnostics). The MBC was defined as the lowest concentration of drug that resulted in killing of ≥99.9% of the initial inoculum. The drug carryover effect was assessed by visual inspection of the distribution of colonies on medium plates. The studies were conducted in duplicate and repeated at least once on a separate day.
Pilot studies.
Time-kill studies were conducted with meropenem and tobramycin at different and escalating concentrations. Six concentrations of each agent, normalized to 0 (control), 0.25, 1, 4, 16, and 64 times the MIC of the respective agent, were used. An overnight culture of the isolate was diluted 30-fold with prewarmed Ca-MHB and incubated further at 35°C until reaching late-log-phase growth. The bacterial suspension was diluted with Ca-MHB according to optical density; 9 ml of the suspension was transferred to 50-ml sterile conical flasks, each containing 1 ml of a drug solution at 10 times the target concentration. The final concentration of the bacterial suspension in each flask was approximately 108 CFU/ml. The high inoculum was used to simulate the bacterial load in severe infection and to allow the resistant subpopulation(s) to be present at the baseline. The experiment was conducted for 24 h in a shaker water bath set at 35°C.
Samples were obtained from each flask in triplicate at 24 h to characterize the effect of various drugs and their exposures on the total bacterial population. Prior to being cultured quantitatively, the bacterial samples (0.5 ml) were centrifuged at 10,000 × g for 15 min and reconstituted with sterile normal saline to their original volumes in order to minimize the drug carryover effect. Total bacterial populations were quantified by spiral plating 10× serial dilutions of the samples onto MHA plates. The medium plates were incubated in a humidified incubator (35°C) for 18 to 24 h, and the bacterial density from each sample was determined by the use of CASBA-4 colony scanner software (Spiral Biotech, Bethesda, Md.). The killing effect at 24 h was described using an inhibitory sigmoid Emax model, weighted by the inverse of the observation variances.
Optimal design.
The optimal drug concentrations for capturing parameter estimates describing the killing effect most precisely at 24 h were estimated using ADAPT II (D. Z. D'Argenio and A. Schumitzky, ADAPT II user's manual, Biomedical Simulations Resource, University of Southern California). D-optimality was employed to optimize the determinant of the inverse Fisher information matrix (5, 9). Assay variance was based on the best-fit relationship between the mean and variance of the observations. Both linear and quadratic (up to cubic) relationships were explored. Clinically achievable concentration (and supra-MIC) ranges were used for meropenem (1 to 64 mg/liter) and tobramycin (1 to 32 mg/liter), respectively.
Interaction study.
Time-kill studies were repeated twice using 25 concentration combinations in a five-by-five array. In addition to the total bacterial population, subpopulations with reduced susceptibility were quantified by culturing onto cation-adjusted MHA plates (BBL) supplemented with the exposed agents at a concentration of three times the MIC. Since the drug susceptibility testing was performed with twofold dilutions of the agents, and a one-tube difference (twofold in drug concentration) is commonly regarded as an acceptable interday variation in susceptibility, quantitative cultures on drug-supplemented medium plates (at three times the MIC) would enable the presence of the bacterial subpopulation(s) with reduced susceptibility to be detected reliably. The medium plates were incubated in a humidified incubator (35°C) for 18 to 24 h (total population) and up to 72 h (subpopulations with reduced susceptibility). Total bacterial density data at 24 h (in duplicate) were modeled using a three-dimensional surface. Effect summation was used as the definition of additivity (null interaction) (2) as follows:
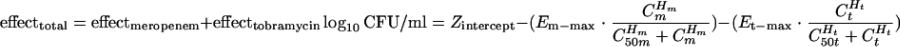 |
where Zintercept is the bacterial density at 24 h in the absence of drug, Em-max/Et-max is the maximal effect of meropenem/maximal effect of tobramycin, Cm/Ct is the concentration of meropenem/concentration of tobramycin, Hm/Ht is the sigmoidicity of meropenem/sigmoidicity of tobramycin, and C50m/C50t is the concentration of meropenem needed to achieve 50% of the maximal effect/concentration of tobramycin needed to achieve 50% of the maximal effect.
The volumes under the planes (VUP) of the observed and expected surfaces were computed by interpolation and double integration, respectively (Maple 7; Maplesoft, Waterloo, Ontario, Canada). The interaction index was defined as VUPobserved/VUPexpected. Synergy and antagonism were defined as interaction index values of <1 and >1, respectively. The 95% confidence interval (CI) of VUPobserved was computed with mean data points ± 1.96 standard deviations.
RESULTS
Susceptibility.
The MIC/MBCs of meropenem and tobramycin for P. aeruginosa ATCC 27853 were both found to be 1/1 mg/liter.
Pilot studies.
Meropenem and tobramycin exhibited different concentration-killing profiles (Fig. 1). Killing of meropenem appeared to have been maximized at a concentration of <10 mg/liter, while no such ceiling effect was observed with tobramycin. These observations are consistent with previous in vitro studies reporting the partially concentration-dependent bactericidal activity of the β-lactams and concentration-dependent killing of the aminoglycosides (4). A good model fit was obtained for both agents (r2 = 0.99). The optimal concentrations for capturing the most precise parameter estimates describing the killing effect of meropenem at 24 h were 1, 2, 5, and 64 mg/liter. On the other hand, the optimal concentrations for capturing the most precise parameter estimates describing the killing effect of tobramycin at 24 h were 1, 1.5, 4, and 32 mg/liter. The expected (null interactive) surface response was as shown in Fig. 2A.
FIG. 1.

Twenty-four-hour killing data and optimal concentrations to capture killing at 24 h for meropenem (A) and tobramycin (B).
FIG. 2.

Expected (null interactive) surface as computed by double integration of time-kill data (from Fig. 1); concentration ranges: meropenem, 0 to 64 mg/liter, and tobramycin, 0 to 32 mg/liter (A), and meropenem, 0 to 5 mg/liter, and tobramycin, 0 to 4 mg/liter (B).
Interaction study.
To facilitate understanding of the data analysis, we plotted the expected combined effect of the agent combination using a three-dimensional response surface (Fig. 2). Using two horizontal axes (x and y) representing the individual drug concentrations and a vertical axis (z) representing the effect of the drug combination, the null interactive surface was essentially a collection of all points which represent the anticipated additive effect of the two drugs used in combination. The actual effects of two drugs in combination were determined when used at various concentration combinations. The observed effect associated with each concentration combination was represented by a datum point in three-dimensional space, as shown in Fig. 3A. The observed experimental data were also superimposed on the expected response surface for easy visual inspection, as shown in Fig. 4A. The spatial orientation of this point in relation to the null interactive surface could be effectively used to describe the nature and extent of interaction. If the datum point was below the null interactive surface (observed killing is more than anticipated killing), this would suggest synergy. On the other hand, if the datum point was above the null interactive surface (observed killing is less than anticipated killing), this would signify antagonism. In addition, the distance between the experimental datum point and the null interactive plane was used as an index of interaction. The further the distance, the greater the extent of interaction, as shown in Fig. 4B.
FIG. 3.

Experimental (observed) data (A) and the response surface as computed by interpolation (B)
FIG. 4.

Comparison of the expected surface (shown in Fig. 2B) and observed data points (shown in Fig. 3A) (A); extent of synergy (predicted effect minus observed effect) exhibited by different concentration combinations (B).
In all combinations, an enhanced killing effect was seen compared to either drug at the same concentration. The most significant synergism was observed between 1 and 5 mg of meropenem/liter and 1 to 4 mg of tobramycin/liter; seven out of nine combinations had a >2-log drop compared to the more potent agent. The VUPexpected in these concentration ranges was found to be 90.2 (Fig. 2B), while the VUPobserved was found to be 68.5 (95% CI, 58.9 to 82.4) (Fig. 3B). The interaction index was found to be 0.76 (95% CI, 0.65 to 0.91).
Resistance selection.
Resistant isolates were found on medium plates supplemented with either meropenem or tobramycin from flasks exposed to one agent only (data not shown). No viable bacteria were recovered from medium plates supplemented with meropenem and/or tobramycin at three times the MIC with any agent combination.
DISCUSSION
The emergence of antimicrobial resistance among various pathogens is a rapidly spreading problem threatening our therapeutic armamentarium (6, 16). Many clinicians are concerned that common infections may be untreatable in the near future. P. aeruginosa is an important pathogen associated with serious nosocomial infections. It is also associated with multiple mechanisms of resistance to various antibiotics (efflux pumps, β-lactamase production, porin channel deletion, multifunctional group transferases, target site alteration, etc.) (13). Treatment of pseudomonal infections often represents a challenge to clinicians. Given that the drug development process takes many years, it is imperative that the utility of available antimicrobial agents be preserved through judicious and optimal use. There are very few agents in the advanced stage of development designed to target multidrug-resistant gram-negative bacteria, and we are at risk of going back to the preantibiotic era in the event of an outbreak (11). Consequently, it is especially important that we preserve the clinical utility of presently available agents against this bacterium. Combination therapy is commonly used clinically, in view of the drugs' synergistic activity and different mechanisms of resistance. It is hoped that a synergistic pharmacodynamic combination would provide enhanced bactericidal effect to prevent the emergence of resistance.
Although numerous studies have investigated the PDI of antimicrobial agents, many of the methods used are unsatisfactory. The checkerboard method and the fractional inhibitory concentration (FIC) index make up one of the most widely used methodologies in studying the in vitro interaction of antimicrobial agents. The FIC index is defined as the sum of all fractional inhibitory concentrations of antimicrobial agents in a combination.
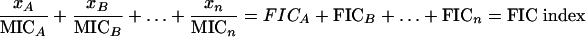 |
Here xn is the concentration of drug n required to inhibit microbial growth in a drug combination, and MICn is the MIC of drug n for the microorganism in the absence of another agent. Although the checkerboard method examines a range of concentrations of the two drugs, it relies on a subjective (visually determined) endpoint of growth inhibition and does not provide any information on the extent of bacterial growth or kill over time. The drugs in combination are considered to be synergistic, additive (no interaction), and antagonistic if the FIC indexes are ≤0.5, 1.0, and >4.0, respectively. Despite its wide acceptance and adoption in numerous recent studies (8, 14, 17, 18, 20), the basis of null interaction of the FIC index is Loewe additivity. It has been pointed out previously that the underlying assumption of the linear concentration-effect relationship may not be valid; thus, the relevance of the results may be questionable (10).
Standard in vitro time-kill studies are also widely used. While they provide objective information on the extent of killing, they are equally unsatisfactory in examining two drugs alone and in combination to determine drug interaction. In most studies, the effect of the two agents is investigated at a specific concentration combination. The rationale for the choice of the drug concentration (peak, trough, or the average drug concentration at steady state) investigated is often not fully explained or justified. Thus, it is difficult to extrapolate the results to clinical situations where serum drug concentrations fluctuate over time. In addition, the threshold magnitude for the definition of drug interaction (synergy is commonly defined as a more than 2-log kill with the combination than with the more potent single agent in the combination) is empirically chosen. Under this metric, the interaction between two agents is characterized by ordinal categories (synergistic, additive, and antagonistic). The extent of interaction among different agent combinations in the same category cannot be compared on a statistical basis, since all synergistic combinations are deemed equally potent. Furthermore, the results from the checkerboard method and time-kill studies may not correlate with each other (3, 12), and their predictions have not been validated in clinical studies.
Somewhat similar to our approach, attempts have also been made to develop fully parameterized response surfaces which would allow analysis of concentration response data for two drug combinations (7). An interaction index was derived when two cytotoxic agents were used in combination against murine leukemia cells in an in vitro system. The experimental data were analyzed by use of an empirically derived mathematical model using Loewe additivity as the basis of null interaction. Based upon the limitations of the Loewe additivity model, we feel that such approaches may be relevant only in situations where the underlying assumptions of the Loewe additivity model can be justified. Since the interaction term is empirically derived, it may be difficult to appreciate the relevance of the extent of drug interaction.
Recognizing the shortcomings of the current methods, herein we have proposed an alternative method for characterizing PDI. In order to circumvent the limitations of the Loewe additivity model, we used effect summation as the basis of null interaction. This allows a robust evaluation to be performed even with nonlinear concentration-effect relationships. We simply expressed the anticipatory combined effect as the sum of the effects of individual agents. The mathematical (theoretical) justification of using effect summation as the basis of null interaction is discussed in a review article by members of our group (2). Instead of using only one concentration combination of two agents, the evaluation was performed over a clinically relevant and achievable concentration range. We believe that this approach would enhance the clinical relevance of the results.
It was evident from Fig. 4B that the extent of the synergy resulting from different concentration combinations was different. Since both concentration ranges of the agents investigated were clinically relevant, instead of reporting the extent of interaction for each concentration combination, we seek an objective measure of interaction over the concentration ranges. With multiple concentration combinations, we used the ratio of the VUP of the observed and expected surfaces (VUPobserved/VUPexpected) as a global interaction index for the entire concentration range. For a two-drug combination, the VUP can be conceptualized as the integral killing effect over two concentration ranges: the larger the VUP, the less bactericidal activity. With reference to the expected combined activity of two drugs, additivity was defined as an interactive index of 1 (the observed overall killing of the agent combination is identical to that expected of the sum of all agents in the combination). Furthermore, synergy and antagonism were defined as interaction index values of <1 (the observed overall killing of the agent combination is greater than that expected of the sum of each agent in the combination) and >1 (the observed overall killing of the agent combination is less than that expected of the sum of each agent in the combination), respectively. Double integration could be used to compute VUPexpected because we know the exact concentration-effect relationships of both agents when used alone (from the x-z and y-z planes). In contrast, we do not have any basis for assuming any meaningful structural model for the interaction. Consequently, interpolation (a three-dimensional version of the trapezoidal method) was used to estimate the integral killing (VUPobserved) of the drug combination. With repeated observations of the effect associated with each combination, the variances of the observations were used to compute a 95% confidence interval of the interactive index. Such an objective parameter provided a statistical basis for quantifying and comparing different drug combinations with respect to their bactericidal activity. Furthermore, our approach may be easily modified to study the combined effects of interaction systems involving multiple (more than two) agents. Our results corroborate previous data indicating that meropenem is synergistic with an aminoglycoside when used in combination against P. aeruginosa (19). These data provide further support for exploring the use of this agent in combination therapy to suppress resistance in P. aeruginosa.
The combined effect of antimicrobial agents is a complex biological system to study. Despite our efforts to circumvent obvious limitations of previous models, there are still unresolved issues. The greatest combined effect appeared to be seen with low concentrations of both agents, due to our inability to measure effects which exceed inoculum eradication (e.g., negative bacterial load). The interacting system is thus limited with a maximal effect of inoculum eradication, as mentioned elsewhere (2). The most appropriate way to address this issue is to determine the bacterial load before 24 h of drug exposure, so that the rate (in addition to the extent) of bacterial killing can be determined. The mosaic pattern of interaction (an interaction in which the experimental surface intersects the additive surface at one or several points) was another concern. As shown in Fig. 4B, all the observed data points were on the same side of the additive surface, suggesting that this was not a major issue with our experimental data set. The most appropriate way to address this issue in our opinion is to divide the data points into different regions based on spatial orientation, so that different interactive indices can be computed individually. We wish to point out that none of the existing interaction models addresses either limitation satisfactorily. However, before a more sophisticated model can be formulated, effect summation must first be accepted as a viable method for characterizing combined drug effects, which is the primary objective of this study.
In conclusion, we described a parametric method for characterizing the PDI of antimicrobial agents. Our approach appears robust, and further validation with other drug-drug-pathogen combinations is warranted.
Acknowledgments
This study was supported by AstraZeneca, Wilmington, Del.
REFERENCES
- 1.Bliss, C. I. 1939. The toxicity of poisons applied jointly. Ann. Appl. Biol. 26:585-615. [Google Scholar]
- 2.Boucher, A. N., and V. H. Tam. Submitted for publication.
- 3.Cappelletty, D. M., and M. J. Rybak. 1996. Comparison of methodologies for synergism testing of drug combinations against resistant strains of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 40:677-683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Craig, W. A., and S. C. Ebert. 1990. Killing and regrowth of bacteria in vitro: a review. Scand. J. Infect. Dis. Suppl. 74:63-70. [PubMed] [Google Scholar]
- 5.D'Argenio, D. Z. 1981. Optimal sampling times for pharmacokinetic experiments. J. Pharmacokinet. Biopharm. 9:739-756. [DOI] [PubMed] [Google Scholar]
- 6.Gold, H. S., and R. C. Moellering. 1996. Antimicrobial-drug resistance. N. Engl. J. Med. 335:1444-1453. [DOI] [PubMed] [Google Scholar]
- 7.Greco, W. R., H. S. Park, and Y. M. Rustum. 1990. Application of a new approach for the quantitation of drug synergism to the combination of cis-diamminedichloroplatinum and 1-β-D-arabinofuranosylcytosine. Cancer Res. 50:5318-5327. [PubMed] [Google Scholar]
- 8.Karakoc, B., and A. A. Gerceker. 2001. In-vitro activities of various antibiotics, alone and in combination with amikacin against Pseudomonas aeruginosa. Int. J. Antimicrob. Agents 18:567-570. [DOI] [PubMed] [Google Scholar]
- 9.Khinkis, L. A., L. Levasseur, H. Faessel, and W. R. Greco. 2003. Optimal design for estimating parameters of the 4-parameter Hill model. Nonlinearity Biol. Toxicol. Med. 1:363-377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.King, T. C., D. Schlessinger, and D. J. Krogstad. 1981. The assessment of antimicrobial combinations. Rev. Infect. Dis. 3:627-633. [DOI] [PubMed] [Google Scholar]
- 11.Landman, D., J. M. Quale, D. Mayorga, A. Adedeji, K. Vangala, J. Ravishankar, C. Flores, and S. Brooks. 2002. Citywide clonal outbreak of multiresistant Acinetobacter baumannii and Pseudomonas aeruginosa in Brooklyn, N.Y.: the preantibiotic era has returned. Arch. Intern. Med. 162:1515-1520. [DOI] [PubMed] [Google Scholar]
- 12.Lewis, R. E., D. J. Diekema, S. A. Messer, M. A. Pfaller, and M. E. Klepser. 2002. Comparison of Etest, chequerboard dilution and time-kill studies for the detection of synergy or antagonism between antifungal agents tested against Candida species. J. Antimicrob. Chemother. 49:345-351. [DOI] [PubMed] [Google Scholar]
- 13.Livermore, D. M. 2002. Multiple mechanisms of antimicrobial resistance in Pseudomonas aeruginosa: our worst nightmare? Clin. Infect. Dis. 34:634-640. [DOI] [PubMed] [Google Scholar]
- 14.Meletiadis, J., J. W. Mouton, J. F. Meis, and P. E. Verweij. 2003. In vitro drug interaction modeling of combinations of azoles with terbinafine against clinical Scedosporium prolificans isolates. Antimicrob. Agents Chemother. 47:106-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.National Committee for Clinical Laboratory Standards. 2000. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically. Document M7-A5. National Committee for Clinical Laboratory Standards, Wayne, Pa.
- 16.Neu, H. C. 1992. The crisis in antibiotic resistance. Science 257:1064-1073. [DOI] [PubMed] [Google Scholar]
- 17.Perea, S., G. Gonzalez, A. W. Fothergill, W. R. Kirkpatrick, M. G. Rinaldi, and T. F. Patterson. 2002. In vitro interaction of caspofungin acetate with voriconazole against clinical isolates of Aspergillus spp. Antimicrob. Agents Chemother. 46:3039-3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rochon-Edouard, S., M. Pestel-Caron, J. F. Lemeland, and F. Caron. 2000. In vitro synergistic effects of double and triple combinations of beta-lactams, vancomycin, and netilmicin against methicillin-resistant Staphylococcus aureus strains. Antimicrob. Agents Chemother. 44:3055-3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Song, W., H. J. Woo, J. S. Kim, and K. M. Lee. 2003. In vitro activity of beta-lactams in combination with other antimicrobial agents against resistant strains of Pseudomonas aeruginosa. Int. J. Antimicrob. Agents 21:8-12. [DOI] [PubMed] [Google Scholar]
- 20.Te Dorsthorst, D. T., P. E. Verweij, J. Meletiadis, M. Bergervoet, N. C. Punt, J. F. Meis, and J. W. Mouton. 2002. In vitro interaction of flucytosine combined with amphotericin B or fluconazole against thirty-five yeast isolates determined by both the fractional inhibitory concentration index and the response surface approach. Antimicrob. Agents Chemother. 46:2982-2989. [DOI] [PMC free article] [PubMed] [Google Scholar]