

Abstract
The insulin receptor substrate (IRS) proteins are adaptor molecules that integrate signals generated by receptors that are implicated in human breast cancer. We investigated the specific contribution of IRS-2 to mammary tumor progression using transgenic mice that express the polyoma virus middle T antigen (PyV-MT) in the mammary gland and IRS-2-null (IRS-2−/−) mice. PyV-MT-induced tumor initiation and growth were similar in wild-type (WT) and IRS-2−/− mice. However, the latency and incidence of metastasis were significantly decreased in the absence of IRS-2 expression. The contribution of IRS-2 to metastasis is intrinsic to the tumor cells, because IRS-2−/− mammary tumor cells did not metastasize when grown orthotopically in the mammary fat pads of WT mice. WT and IRS-2−/− tumors contained similar numbers of mitotic cells, but IRS-2−/− tumors had a higher incidence of apoptosis than did WT tumors. In vitro, IRS-2−/− mammary tumor cells were less invasive and more apoptotic in response to growth factor deprivation than their WT counterparts. In contrast, IRS-1−/− tumor cells, which express only IRS-2, were highly invasive and were resistant to apoptotic stimuli. Collectively, our findings reveal an important contribution of IRS-2 to breast cancer metastasis.
The differentiation and function of mammary epithelial cells are dependent upon the combined action of growth factor/hormone receptors and adhesion receptors, which act in concert to control the signals required for normal cell function (10, 24). In breast cancer, many of these same receptors contribute to tumor progression, and it is clear that growth factor stimulation and matrix composition determine the fate of these malignant cells (4, 41). Insulin-like growth factor (IGF) signaling is implicated in normal mammary gland development and function as well as in breast cancer (34). IGF-1 and IGF-2 are expressed in the normal mammary gland, and they promote mitogenesis and survival of normal mammary epithelial cells, as well as breast carcinoma cells, in vitro (23, 34). Furthermore, IGF-1 or IGF-2 overexpression in transgenic mice promotes mammary tumorigenesis (14). The IGFs signal through the IGF-1 receptor (IGF-1R), which is frequently overexpressed in human breast cancer and is a prognostic indicator of tumor recurrence and reduced patient survival (32, 49). Importantly, increased levels of IGF-1R signaling can interfere with Herceptin action and cause resistance to this breast cancer therapy (1, 28).
The IGF-1R mediates its functions through activation of an intrinsic tyrosine kinase in its cytoplasmic domain (reviewed in reference 34). Upon activation, the receptor is autophosphorylated and recruits intracellular substrates, including the insulin receptor substrate (IRS) proteins, Shc and SH2b (34). Although the importance of IGF-1R-dependent signaling cascades for breast carcinoma progression has been demonstrated by dominant-negative approaches, the contributions of individual downstream effectors to specific IGF-1R functions have not been elucidated in vivo (9). To understand how the IGF-1R contributes to tumor progression, it is necessary to establish which intracellular signaling pathways are essential for its function. The IRS proteins, including IRS-1, -2, -3, and -4, are adaptor molecules that mediate their function downstream of the IGF-1R by organizing signaling complexes at sites of receptor activation (52). The IRS proteins contain multiple phosphotyrosine binding motifs through which they recruit additional downstream effectors. IRS-1 and IRS-2 are expressed ubiquitously, including in mammary epithelial cells, whereas IRS-3 and IRS-4 are restricted in their localization and are found predominantly in adipose tissue and brain, respectively (20, 21, 23).
Cell-based and mouse experiments suggest that IRS-1 and IRS-2 play distinct physiological roles. IRS-1-null mice are born small and are insulin resistant, but they never develop diabetes, owing to robust proliferation and survival of pancreatic β cells (2). In contrast, IRS-2-null mice are born with a small brain but a normal body size, and they develop early-onset diabetes because of a combination of peripheral insulin resistance and β-cell failure (38, 53, 54). Although some of the differences in the null phenotypes may be related to differential tissue expression, studies on the metabolic functions of the IRS proteins suggest that IRS-1 and IRS-2 cannot substitute functionally for one another (5, 11). With regard to breast cancer, IRS-1 is regulated by estrogen, and this isoform is the predominant IRS family member that is activated by IGF-1 signaling in estrogen receptor-positive (ER+) breast carcinoma cell lines to promote growth and survival (18, 22, 30). IRS-1 levels decrease in response to tamoxifen and ICI 182,780, which might explain, at least in part, the inhibition of breast carcinoma growth by these antiestrogens (12, 35). In primary human breast cancers, high levels of IRS-1 can be predictive of a greater incidence of recurrence and a decreased patient survival rate (22, 33).
In contrast to IRS-1, few studies have addressed the role of IRS-2 in breast cancer. While IRS-1 expression is regulated by estrogen, IRS-2 expression is regulated by progesterone and cyclic AMP-mediated activation of CREB (8, 50). In ER− breast carcinoma cell lines, which lack or have decreased IRS-1 expression but retain IRS-2 expression, IGF-1R is still required for metastasis but no longer promotes mitogenesis (9). Recently, it was shown that IRS-2 is an essential intermediate in the activation of phosphatidylinositol 3-kinase (PI3K) and promotion of breast carcinoma invasion by the α6β4-integrin receptor (40). IRS-2 also mediates IGF-1-dependent breast carcinoma cell migration (19, 55). To investigate rigorously the contribution of IRS-2 to the development and progression of breast cancer, we used a mouse model of mammary tumorigenesis and IRS-2-null mice to determine the effect of IRS-2 expression on tumor initiation, growth, and progression to metastasis. The data obtained support a specific role for IRS-2 in mammary tumor metastasis.
MATERIALS AND METHODS
Mice.
IRS-2−/+ mice were generated and described previously (54). Female C57Bl/6 IRS-2+/− mice were crossed with male FVB mice that were transgenic (+/−) for the polyoma virus middle T (PyV-MT) antigen under the control of the mouse mammary tumor virus (MMTV) promoter (Jackson Labs). Female IRS-2+/− offspring were bred to male IRS-2+/− offspring that were also transgenic for the PyV-MT antigen (PyV-MT+/−). Female offspring from this cross that were PyV-MT+/− and were wild type for the IRS-2 allele (WT) or homozygous null for the IRS-2 allele (IRS-2−/−) were saved for analysis. Genotyping was performed by PCR using oligonucleotides specific for the IRS-2 gene (5′-CTTGGCTACCATGTTGTTATTGTC-3′ and 5′-AGCTCTGGAGGTTTACTTTCCTAG-3′), the neomycin gene (5′-GCTACCCGTGATATTGCTGAAGAG-3′), and as described previously for the PyV-MT transgene (46).
Beginning at 50 days of age, the mice were palpated every 10 days to detect the onset of mammary tumor development. At the 100- or 110-day endpoint, the tumors were dissected and measured using calipers. Total tumor volume was determined using the following formula: volume = (4/3)(π)(1/2 × smaller diameter)2 (1/2 × larger diameter). Portions of the tumors and the lungs were either snap frozen or fixed in 10% buffered formalin.
To examine the ability of IRS-2−/− tumor cells to grow and metastasize in a WT host, IRS-2−/− mammary tumor cells (106) were resuspended in Matrigel (50 μl) and injected into the left number 3 thoracic mammary gland of female nu/nu mice. After 12 weeks, the tumors and lungs were dissected and portions of each were either snap frozen or fixed in 10% buffered formalin.
Histology and IHC.
Formalin-fixed tissue was embedded in paraffin and sections were either stained with hemolysin and eosin (H&E) or with antibodies for immunohistochemistry (IHC) analysis. For IHC, tissue sections were deparaffinized and rehydrated before quenching endogenous peroxidase activity in 3% H2O2 for 5 min. To detect myoepithelial cells, tissue sections were blocked in M.O.M. Mouse IgG Blocking Reagent (Vector Laboratories, Burlingame, Calif.) and then incubated with a 1:500 dilution of a smooth-muscle actin-specific monoclonal antibody (clone 1A4; Sigma, St. Louis, Mo.). The primary antibody was amplified with M.O.M. Biotinylated Anti-Mouse IgG Reagent and was detected using the Vectastain Elite ABC reagent (Vector Laboratories) with 3,3′-diaminobenzidine (DAB) as the substrate. To detect endothelial cells, tissue sections were digested with N-p-tosyl-l-phenylalanyl chloromethyl ketone-trypsin (0.05%) for 28 min before incubating overnight at 4°C with rat anti-mouse CD31 (2.5 μg/ml; Pharmingen). The primary antibody was amplified with biotinylated goat-anti-rat immunoglobulin G (IgG) (Vector Laboratories) and was detected using the Vectastain Elite ABC reagent with DAB as the substrate. To detect apoptotic cells, tissue sections were stained using the ApopTag Plus Peroxidase In Situ Apoptosis Detection kit according to the manufacturer's instructions (Chemicon, Temecula, Calif.).
Whole-mount preparation.
The number 4 inguinal mammary gland was removed from PyV-MT-expressing mice and was spread onto a glass slide. The mammary glands were fixed for 2 to 4 h in Carnoy's fixative and then were incubated sequentially for 15 min each in 70, 50, and 25% ethanol (EtOH). After a 5-min wash in H2O, the glands were stained overnight in carmine alum stain. The mammary glands were then incubated sequentially for 15 min each in 75, 95, and 100% EtOH before being cleared in xylene. The cleared mammary glands were mounted with permount.
Real-time quantitative PCR (RQ-PCR).
Total RNA was isolated from lung tissue by using the RNeasy extraction kit (QIAGEN). PyV-MT mRNA was amplified with the following primers and probes: forward primer, 5′-AGCCCGATGACAGCATATCC-3′; reverse primer, 5′-GGTCTTGGTCGCTTTCTGGA-3′; Taqman probe, 5′-CGGACCCCCCCAGAACTCCTGT-3′. GAPDH mRNA was amplified as a control (Applied Biosystems).
Tumor cell isolation and characterization.
Mammary tumor cell lines were established from the PyV-MT-derived tumors. The mammary tumors were removed, placed in Dulbecco's modified Eagle medium (DMEM) containing penicillin/streptomycin (P/S) and 2 mg of collagenase/ml, and minced into small pieces with a razor blade. The minced tumor tissue was incubated with shaking at 37°C for 3 h, at which time the cell suspension was centrifuged and washed five times in phosphate-buffered saline (PBS) containing 5% fetal calf serum (FCS). The final cell pellet was resuspended in DMEM/F12 containing 2% FCS and P/S, and the cells were plated (2 × 106 cells/100-mm-diameter dish) into collagen I-coated plates. The isolated cells were subcloned by limiting dilution to obtain pure populations of mammary tumor cells. To confirm the epithelial origin of the isolated subclones, the cells were screened by immunoblotting for their expression of the epithelium-specific β4-integrin subunit (29).
Immunoprecipitation and immunoblotting.
Frozen tumors were homogenized at 4°C in a 50 mM Tris buffer, pH 8, containing 150 mM NaCl, 1% NP-40, 0.5% deoxycholic acid, 0.1% sodium dodecyl sulfate, 1 mM sodium orthovanadate, and protease inhibitors (Complete Mini; Roche Applied Science, Indianapolis, Ind.). Tissue culture cells were solubilized at 4°C for 10 min in a 20 mM Tris buffer, pH 7.4, containing 0.14 M NaCl, 1% NP-40, 10% glycerol, 1 mM sodium orthovanadate, 2 mM phenylmethylsulfonyl fluoride, and 5-μg/ml concentrations of aprotinin, pepstatin, and leupeptin. Nuclei were removed by centrifugation at 12,000 × g for 10 min. Aliquots of tissue or cell extracts containing equivalent amounts of protein were incubated for 3 h at 4°C with antibodies and protein-A Sepharose (Amersham Biosciences) with constant agitation. The beads were washed three times in the extraction buffer. Laemmli sample buffer was added to the samples, which were then incubated at 100°C for 4 min. Immunoprecipitates as well as aliquots of total cell extracts were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to nitrocellulose filters.
For immunoblotting, the filters were blocked for 1 h with a 50 mM Tris buffer, pH 7.5, containing 0.15 M NaCl, 0.05% Tween-20 (TBST), and 5% (wt/vol) Carnation dry milk. The filters were incubated overnight at 4°C in the same buffer containing primary antibodies. After three 10-min washes in TBST, the filters were incubated for 1 h in blocking buffer containing horseradish peroxidase-conjugated secondary antibodies. After three 10-min washes in TBST, proteins were detected by enhanced chemiluminescence (Pierce, Rockford, Ill.). For RC-20 phosphotyrosine immunoblots, the filters were blocked for 1 h with a 10 mM Tris buffer, pH 7.5, containing 0.5 M NaCl, 0.1% Tween-20 (RC-20 buffer), and 2% (wt/vol) Carnation dry milk. The filters were washed briefly in RC-20 buffer and then were incubated overnight at 4°C in RC-20 buffer containing 3% (wt/vol) bovine serum albumin (BSA) and a 1:500 dilution of the RC-20 antibody. After being washed, the filters were incubated for 1 h in blocking buffer containing horseradish peroxidase-conjugated streptavidin and the proteins were detected by enhanced chemiluminescence.
The following antibodies were used for immunoprecipitation or immunoblotting: IRS-1 and IRS-2 (Upstate Biotechnology Inc., Lake Placid, N.Y.), phosphotyrosine (RC-20; BD Biosciences, San Diego, Calif.), p85 (generous gift from Alex Toker), β4-integrin subunit (generous gift from Art Mercurio), estrogen receptor-α (ERα; Santa Cruz Biotechnology, Santa Cruz, Calif.), progesterone receptor (PR; Santa Cruz Biotechnology), cyclin D1 (Biosource, Camarillo, Calif.), and IGF-1R β-subunit (Santa Cruz Biotechnology).
Invasion assay.
Matrigel invasion assays were performed as described previously (42, 43) using 6.5-mm-diameter Transwell chambers (8 μm pore size; Costar). Matrigel, purified from the Englebreth-Holm-Swarm tumor, was diluted in cold distilled water, added to the transwells (7.5 μg/well), and dried in a sterile hood. The Matrigel was then reconstituted with medium for an hour at 37°C before the addition of cells. Cells (0.25 × 105) were resuspended in serum-free DMEM containing 0.1% BSA, and cells were added to each well. Conditioned NIH-3T3 medium was added to the bottom wells of the chambers. After 3 h, the cells that had not invaded were removed from the upper face of the filters with cotton swabs and the cells that had invaded to the lower surface of the filters were fixed in methanol and then stained with a 0.2% solution of crystal violet in 2% ethanol. Invasion was quantitated by visual counting. The mean of five individual fields in the center of the filter where invasion was the highest was obtained for each well.
Apoptosis assays.
Cells (7.5 × 105) were plated in 100-mm-diameter tissue culture dishes in complete growth medium and were allowed to adhere for 5 h. The dishes were washed one time to remove nonadherent cells and then were incubated in either complete growth medium or DMEM containing 0.1% BSA for 24 h. For analysis of DNA content, adherent cells were collected by trypsinization and were combined with nonadherent cells from the culture medium. After centrifugation, the cell pellet was washed once in cold PBS and the cells were fixed in 100% EtOH and stored overnight at −20°C. The fixed cells were washed once in PBS and then were resuspended in PBS containing 0.1% Triton X-100, 0.1 mM EDTA, 0.05 mg of RNase A (50U/mg)/ml, and 50 μg of propidium iodide/ml. The cells were analyzed by flow cytometry using a Becton Dickinson FACSCalibur after an hour of incubation at room temperature.
To assay for total caspase activity, cells were plated on glass coverslips in 24-well plates and were treated as described above. After serum starvation for 24 h, the cells were incubated with 10 μM FITC-VAD-FMK (Promega) for 20 min in the dark at 37°C. The cells were washed two times in PBS and were fixed for 30 min in 10% buffered formalin. After washing in PBS, the cells were incubated for 5 min in Hoechst stain and were then mounted onto glass slides.
RESULTS
IRS-1 and IRS-2 are phosphorylated in MMTV-PyV-MT antigen-derived tumors.
To examine the contribution of IRS-2 to mammary tumor initiation, growth, and metastasis, we evaluated the ability of the PyV-MT oncogene to promote mammary tumorigenesis and progression in the absence of IRS-2 expression (13). To validate the use of this model to study IRS-2 function, we assessed the expression of IRS-2 and other IRS family members in PyV-MT-derived mammary tumors. IRS-1 and IRS-2 are expressed in normal mammary epithelial cells, and expression persists in the mammary tumors (23) (Fig. 1). We did not detect expression of IRS-3 or IRS-4 (data not shown). In addition, both IRS-1 and IRS-2 are constitutively tyrosine phosphorylated and associate with the downstream effector PI3K (Fig. 1), indicating that IRS-dependent signaling pathways are active in the PyV-MT mammary tumors.
FIG. 1.
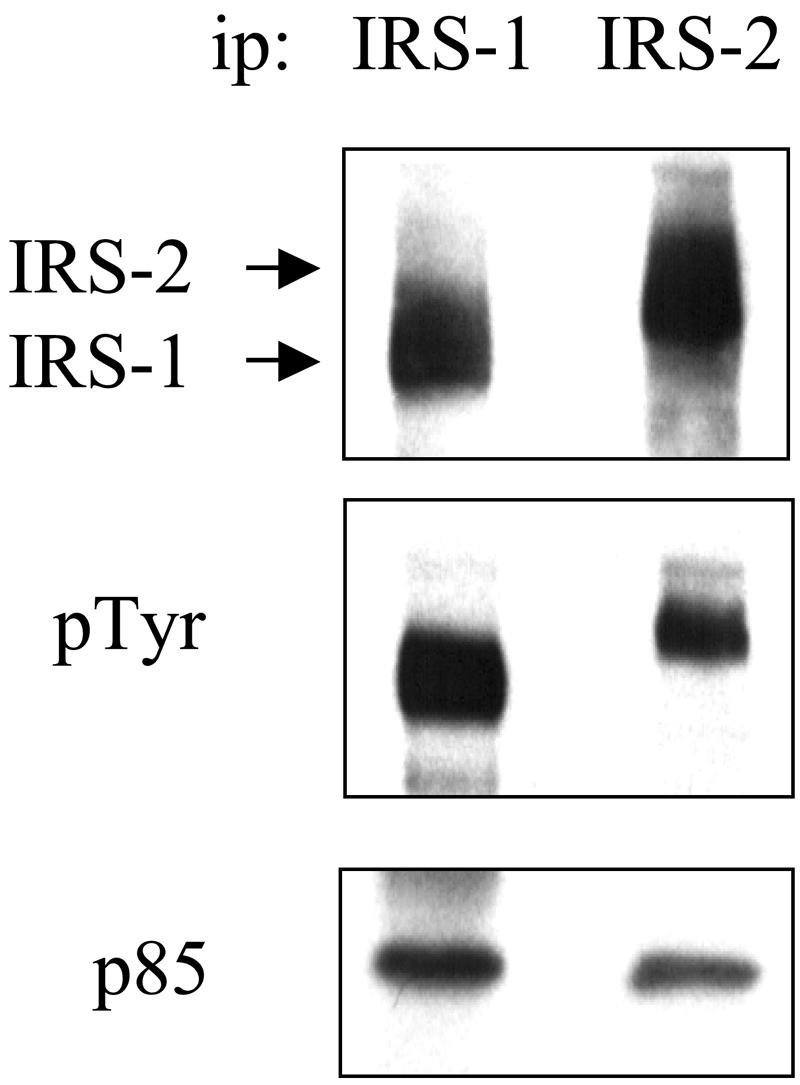
IRS-1 and IRS-2 expression and activation in PyV-MT mammary tumors. Aliquots of PyV-MT mammary tumor extracts that contained equivalent amounts of total protein were immunoprecipitated (ip) with IRS-1- or IRS-2-specific antibodies and then were immunoblotted with RC-20 (middle panel). The immunoblots were subsequently stripped and reprobed with IRS-1- or IRS-2-specific (top panel) and p85-specific (lower panel) polyclonal antisera.
Expression of IRS-2 is not required for PyV-MT-mediated tumor initiation and growth.
To assess the contribution of IRS-2 to PyV-MT-dependent tumorigenesis, female mice that were WT for the IRS-2 allele or homozygous null for the IRS-2 allele (IRS-2−/−) and that expressed the PyV-MT transgene (PyV-MT+/−) were analyzed for mammary tumor development. Tumors were first detected at a similar age in the WT and IRS-2−/− mice (Fig. 2A and Table 1). Moreover, the total tumor burden of the IRS-2−/− mice did not differ significantly from that of their WT littermates at either 100 or 110 days of age (Fig. 2B). Taken together, these results demonstrate that IRS-2 expression is not required for PyV-MT-dependent mammary tumor initiation or primary tumor growth.
FIG. 2.

Analysis of IRS-2 involvement in PyV-MT mammary tumor development. Female IRS-2+/− mice were bred with male IRS-2+/− mice that were also transgenic (+/−) for the PyV-MT antigen. (A) Female WT and IRS-2−/−, PyV-MT+/− mice were monitored for the age at which mammary tumors were first palpable. (B) Female WT and IRS-2−/− PyV-MT+/− mice were analyzed for their total tumor burden at 100 or 110 days of age. Shown is the mean tumor volume (± standard errors of the means) from each of the time points. The number of mice analyzed for each time point is indicated. WT at 100 days versus IRS-2 at 100 days, P = 0.69; WT at 110 days versus IRS-2 at 110 days, P = 0.68.
TABLE 1.
Onset of palpable mammary tumors in WT and IRS-2-null PyV-MT mice
| Genotype | Age (days) | Mean (± SEM) onset of mammary tumors (days) |
|---|---|---|
| WT | 100 | 76a (± 2) |
| WT | 110 | 71b (± 1) |
| IRS-2−/− | 100 | 78 (± 7) |
| IRS-2−/− | 110 | 75 (± 3) |
WT at 100 days versus IRS-2−/− at 100 days, P = 0.73.
WT at 110 days versus IRS-2−/− at 110 days, P = 0.09.
Pathology of IRS-2-null tumors.
Whole-mount analysis of the IRS-2−/− mammary glands revealed that mammary tumors developed in a multifocal manner, similar to that observed for the WT PyV-MT mice (13) (Fig. 3A and B). The histological grade of the IRS-2−/− and WT tumors was assessed using the Nottingham grading system. Both the WT and IRS-2−/− tumors were judged to be high grade, with areas of solid tumor nests lacking significant glandular structure and containing large, pleitrophic nuclei with little cytoplasm (Fig. 3C and D). Overall, a smaller percentage of the total tumor mass was scored as high grade in the IRS-2−/− tumors than in the WT tumors. The remaining tumor mass was judged to be low grade, with more homogeneous, small nuclei and a glandular morphology (Fig. 3E). In these regions of the tumors, myoepithelial cells were still present surrounding the glands as detected by staining with antibodies that recognize smooth-muscle actin (Fig. 3F). Tumor angiogenesis was assessed by staining for the endothelial cell-specific marker CD31 (39). Both the WT and IRS-2−/− tumors were well vascularized, and marked differences in tumor angiogenesis were not evident (Fig. 3G and H).
FIG. 3.

Whole-mount analysis and histological characteristics of WT (A, C, G) and IRS-2 null (B, D-F, H) PyV-MT mammary tumors. (A and B) The number 4 inguinal mammary glands from WT and IRS-2−/−, PyV-MT+/− mice were fixed and stained with carmine alum stain. Bar, 1 mm. (C and D) Solid nodular high-grade tumors that lack glandular structure and contain minimal stroma. (E) Glandular, low-grade tumor region from an IRS-2−/− mouse. Myoepithelial cells are still present surrounding the dilated glands, as demonstrated by the positive smooth-muscle actin staining (F). (C-F) Bar, 100 μm. (G and H) Mammary tumors from WT (G) and IRS-2−/− (H) mice stained with CD31 to detect tumor angiogenesis. Bar, 50 μm.
The changes that occur when PyV-MT mammary tumors progress from hyperplasia to frank carcinoma have been described at the molecular level (26). To determine if mammary tumor progression is inhibited or delayed in the absence of IRS-2 expression, we evaluated the expression of specific markers that are known to be increased or decreased at the protein level in late-stage, invasive tumors. ER and PR are expressed in normal mammary epithelial cells, and expression of both of these hormone receptors diminishes or is lost as tumors become malignant (7, 26, 45). In contrast, cyclin D1 expression persists throughout progression and is expressed at higher levels in late-stage carcinomas than in the hyperplastic and adenoma-mammary intraepithelial neoplasia stages, which reflects an increased mitotic activity in these aggressive tumors (26, 47). As shown in Fig. 4A, ERα, PR-B, and cyclin D1 were expressed at similar levels in WT and IRS-2−/− tumors, indicating that IRS-2 expression and function are not required for PyV-MT mammary tumor progression to malignancy. The mitotic indices of the WT and IRS-2−/− tumors were also comparable in the tumor sections, which is in keeping with the equivalent cyclin D1 expression levels (data not shown). We also examined the expression of the IGF-1R, a major upstream activator of the IRS proteins, to determine if this signaling pathway was disrupted in the absence of IRS-2 expression (34). Although variability existed in the expression of the IGF-1R, the levels did not differ between the two tumor groups. The tumors from IRS-2−/− mice lacked IRS-2 expression, and IRS-1 expression did not increase to compensate for this loss. (Fig. 4A).
FIG. 4.

Biochemical analysis of representative WT and IRS-2−/−, PyV-MT mammary tumors. Aliquots of mammary tumor extracts that contained equivalent amounts of total protein were immunoblotted with IRS-1-, IRS-2-, β4-, p85-, cyclin D1-, ERα-, and PR-specific antibodies. Aliquots of mammary tumor extracts that contained equivalent amounts of total protein were immunoprecipitated and then immunoblotted with antisera specific for the IGF-1R β subunit. WT, mice that are homozygous positive for the IRS-2 gene; IRS-2−/−, mice that are homozygous null for the IRS-2 gene.
Expression of IRS-2 is required for mammary tumor metastasis.
To evaluate the importance of IRS-2 function for mammary tumor metastasis, the lungs from each of the tumor-bearing mice were removed and five independent sections from each lung were examined microscopically for metastatic lesions. In keeping with previously published reports, lung metastasis was observed with a high frequency in the WT PyV-MT mice (13). The incidence of metastasis increased over time, and 70% of the WT mice had detectable metastatic lesions in the lungs by 110 days (Fig. 5A). In contrast, lung metastasis was observed in only 13% of the IRS-2−/− mice at 110 days. Furthermore, metastatic lesions were first detected in the WT mice at 90 days of age (data not shown), whereas metastasis was not observed in the IRS-2−/− mice until 110 days.
FIG. 5.

Analysis of IRS-2 involvement in PyV-MT mammary tumor metastasis. (A) Lungs from female WT and IRS-2−/−, PyV-MT+/− mice were sectioned and screened microscopically for the presence of metastatic lesions. Five representative sections from each lung were analyzed. Shown is the percentage of the mice that scored positively for metastasis at each time point. (B) PyV-MT mRNA was amplified and quantitated from the lungs of 110-day-old WT (1 to 9) and IRS-2−/− (10 to 16) PyV-MT-expressing mice by RQ-PCR. Number 17, control RNA from the lungs of a mouse lacking PyV-MT expression. WT, mice that are homozygous positive for the IRS-2 gene; IRS-2−/−, mice that are homozygous null for the IRS-2 gene.
To quantitate metastasis in the lungs of the WT and IRS-2−/− mice, RQ-PCR was performed with primers that recognize PyV-MT mRNA. PyV-MT is not expressed in the lungs of normal mice (Fig. 5B, mouse no. 17), and a positive signal is indicative of the presence of mammary tumor cells. Although the relative level of PyV-MT mRNA detected in the lungs of the WT mice was variable, a positive signal was detected in the lungs from all but one of the WT mice. In contrast, a positive signal for PyV-MT mRNA was detected in the lungs from only one of the IRS-2−/− mice, and the relative level of expression was low. Importantly, the IRS-2−/− mouse that was positive for PyV-MT mRNA by RQ-PCR was also positive for metastasis by visual examination of the lung sections.
Analysis of PyV-MT-derived cell lines.
To investigate the mechanism by which IRS-2 promotes metastasis, tumor cell lines were established from PyV-MT-derived mammary tumors. WT tumor cells expressed both IRS-1 and IRS-2, whereas tumor cells isolated from the IRS-2−/− mice expressed only IRS-1 (Fig. 6A). In addition, cell lines were isolated from IRS-1−/− PyV-MT-derived mammary tumors to examine the behavior of tumor cells that express only IRS-2 (Fig. 6A). All of the cell lines expressed the β4-integrin subunit, confirming their epithelial origin (29). In addition, the expression and activation of the IGF-1R, a major upstream regulator of the IRS proteins, was similar in all of the cell lines.
FIG. 6.

Characterization of WT and IRS-2-null mammary tumor cell lines. (A) Aliquots of cell extracts from WT, IRS-2−/−, and IRS-1−/− mammary tumor cells that contained equivalent amounts of total protein were immunoblotted with antibodies specific for IRS-1, IRS-2, the β4-integrin subunit, and the p85 regulatory subunit of PI3K as a loading control. PyV-MT mammary tumor cells were stimulated for 10 min with IGF-1 (100 ng/ml), and aliquots of cell extracts that contained equivalent amounts of total protein were immunoprecipitated with antisera specific for the IGF-1R β subunit. The immunoprecipitates were immunoblotted with phosphotyrosine-specific antibodies (pIGF-1Rβ). The immunoblots were subsequently stripped and reprobed with IGF-1R β-subunit-specific antisera (IGF-1R). Two independent subclones for each tumor genotype are shown. (B) H&E-stained section from an IRS-2−/− tumor grown orthotopically in the mammary fat pad of a WT mouse. The tumors are solid, with little glandular structure, and are high grade. (C) CD31 staining of an IRS-2−/− tumor grown orthotopically in the mammary fat pad of a WT mouse. WT, mice that are homozygous positive for the IRS-2 gene; IRS-2−/−, mice that are homozygous null for the IRS-2 gene; IRS-1−/−, mice that are homozygous null for the IRS-1 gene.
The ability of tumors to grow and metastasize is influenced by the stromal microenvironment. To determine if the requirement for IRS-2 expression for mammary tumor metastasis is an intrinsic property of the tumor cells and not the result of systemic or stromal changes in the IRS-2−/− mice, the ability of IRS-2−/− mammary tumor cells to develop tumors and metastasize in a WT host was examined. When IRS-2−/− mammary tumor cells were injected into the mammary fat pad of female nu/nu mice, 100% of the mice developed tumors. The tumors were allowed to grow for 12 weeks, at which time 75% of the tumors were >1 cm in diameter. Upon histological examination, the tumor mass was found to contain predominantly solid tumor nests, with little glandular structure (Fig. 6B). As was observed for the IRS-2−/− transgenic tumors, the IRS-2−/− orthotopic tumors were also well vascularized (Fig. 6C). However, no lung metastasis was observed, which supports the hypothesis that mammary tumor cells require IRS-2 expression intrinsically for metastasis (data not shown).
To assess the contribution of IRS-2 to mammary tumor cell invasion, the ability of the tumor-derived cell lines to invade through Matrigel was examined. Although IRS-2−/− tumor cells were capable of invasion, their rate was reduced approximately fivefold compared to that of the WT tumor cells (Fig. 7A). In contrast, the invasive rate of the IRS-1−/− tumor cells increased markedly. To determine if IRS-2 is important for metastatic tumor cell survival, the ability of the PyV-MT-derived tumor cells to survive in response to serum deprivation was assessed. A greater increase in cell death was observed for the IRS-2−/− tumor cells than for the WT or IRS-1−/− tumor cells when they were deprived of serum (Fig. 7B). To confirm that the cell death we observed in the IRS-2−/− cells after withdrawal of serum resulted from apoptosis, caspase activation was monitored with the CaspACE FITC-VAD-FMK in situ marker. Apoptotic cells become fluorescent when FITC-VAD-FMK binds to activated caspases. FITC-VAD-FMK-positive cells were detected in the IRS-2−/− tumor cells, but not in the WT tumor cells, after serum deprivation (Fig. 7C). None of the tumor cells showed evidence of caspase activation in the presence of 10% FCS.
FIG. 7.

Analysis of WT and IRS-2-null mammary tumor cell function. (A) Mammary tumor cells were assayed for their ability to invade Matrigel. The data shown are from two individual subclones of each cell type and are the mean values (± standard deviations) of three (IRS-1−/−) or four (WT and IRS-2−/−) independent assays done in triplicate. (B) Mammary tumor cells were maintained in culture medium containing either 10% FCS or 0.1% BSA for 24 h. Adherent and nonadherent cells were collected and stained with propidium iodide. The DNA content of the cells was analyzed by flow cytometry. The data shown represent the percentage of sub-G1 cells in the presence of 0.1% BSA and 10% FCS and are the mean (± standard deviations) of three (WT) or four (IRS-1−/− and IRS-2−/−) independent experiments. (C) WT and IRS-2−/− mammary tumor cells were maintained in culture medium containing either 10% FCS or 0.1% BSA for 24 h. Adherent cells were incubated in the presence of FITC-VAD-FMK to detect apoptotic cells. Representative images of the cells maintained in 0.1% BSA are shown. The Hoechst nuclear stain verifies the apoptotic morphology of the fluorescein isothiocyanate-labeled cells. WT, mice that are homozygous positive for the IRS-2 gene; IRS-2−/−, mice that are homozygous null for the IRS-2 gene; IRS-1−/−, mice that are homozygous null for the IRS-1 gene.
To determine if an increased apoptotic rate was also present in the IRS-2−/− tumors, ApopTag staining was performed on tumor sections. Although apoptotic cells were present in both WT and IRS-2−/− tumors, the percentage of apoptotic cells was increased in the IRS-2−/− tumors (Fig. 8). These results provide additional evidence of the involvement of IRS-2 in mammary tumor cell survival signaling pathways.
FIG. 8.

Terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling analysis of WT and IRS-2−/− mammary tumors. Left panel, representative images from WT and IRS-2−/− mammary tumors stained with Apoptag reagent. Right panel, percentage of ApopTag-positive nuclei in the tumor sections. The percentage was determined using the following formula: number of ApopTag positive nuclei/total nuclei. n = 20 fields (four random fields from five tumors) for the WT mice, and n = 28 fields (four random fields from seven tumors) for the IRS-2−/− mice.
DISCUSSION
Our results establish an important role for IRS-2 in mammary tumor metastasis. Using the PyV-MT mouse model of mammary tumorigenesis, we showed that mice lacking expression of the IRS-2 adaptor protein develop mammary tumors with a latency and growth rate similar to those of their WT littermates and that IRS-2 is not required for tumor angiogenesis. Importantly, however, the ability of PyV-MT-derived mammary tumors to metastasize to the lungs is significantly impaired in the absence of IRS-2 expression. This deficiency in metastasis is also observed when IRS-2−/− mammary tumor cells are grown orthotopically in the mammary fat pads of WT mice, supporting an intrinsic function for IRS-2 in the mammary tumor cells. We have also established a mechanism for IRS-2 function in mammary tumor metastasis. Specifically, IRS-2 promotes mammary tumor cell invasion and resistance to stress-induced apoptosis. Taken together, our findings highlight an important contribution of IRS-2 to breast cancer and identify a novel metastasis-associated signaling pathway.
Our study is the first to establish a specific role for IRS-2 in tumor metastasis and to identify the mechanistic basis for this function. Hallmarks of the metastatic phenotype are the ability to invade from the primary tumor site into the stromal microenvironment and to survive in foreign environments (15). Our data reveal that IRS-2 contributes to both of these essential metastatic steps. Mammary tumor cells that lack IRS-2 expression are less invasive, whereas IRS-1−/− cells that express only IRS-2 are more invasive, than WT cells that express both isoforms. These findings are in agreement with previous studies demonstrating a role for IRS-2 in the ability of the α6β4 integrin to promote invasion (40), as well as with more recent studies that have associated increased motility with IRS-2 function in variants of MDA-MB-231 and MCF-7 breast carcinoma cells (19, 55). Our results also support that IRS-2 contributes to tumor metastasis through the regulation of cell survival signaling pathways. A higher incidence of apoptosis was observed in IRS-2−/− tumors than in WT tumors. Furthermore, IRS-2−/− tumor cells undergo apoptosis at a higher rate in the absence of growth factor stimulation than tumor cells that express IRS-2. In contrast, loss of IRS-1 expression did not increase apoptosis. Rather, cells expressing only IRS-2 (IRS-1−/−) were more resistant to stress-induced apoptosis. Presumably, tumor cells lacking IRS-2 are more sensitive to apoptotic stimuli and are less likely to survive outside of their normal microenvironment, where intrinsic survival mechanisms are required. A hypothesis that emerges from the present data is that IRS-2 functions in an autocrine signaling pathway that is essential for invasion and survival when exogenous stimuli are not present, such as during metastasis to foreign tissues.
Our data on the importance of IRS-2 for mammary tumor metastasis and the contrasting behavior of the IRS-1−/− and IRS-2−/− mammary tumor cells indicate that IRS-1 and IRS-2 are not functionally redundant in breast cancer. IRS-2-null tumors express IRS-1, but this isoform is not sufficient to support the metastasis of mammary tumor cells to the lungs. Moreover, IRS-2, but not IRS-1, promotes tumor cell invasion and survival in vitro. These results suggest that IRS-2 is capable of activating a unique signaling pathway that is essential for tumor progression and that this pathway is not activated sufficiently, or at all, through IRS-1. The importance of IRS-2 for mammary tumor cell invasion and survival suggests an involvement of a PI3K-dependent signaling pathway in the function of this adaptor protein (6). IRS-2 was previously implicated in the activation of PI3K by the α6β4 integrin, and the importance of this signaling pathway for promoting breast carcinoma cell invasion and survival has previously been demonstrated (3, 40, 43). Although IRS-1 and IRS-2 each contain multiple PI3K binding motifs and can recruit and activate PI3K, the distinct intracellular compartmentalization of IRS-1 and IRS-2 could influence the functional outcomes of their activation of downstream signaling pathways (52). This mechanism for differential IRS function has been described for IRS-2-dependent insulin-stimulated Glut4 translocation and glucose uptake in brown adipocytes (11). In addition to PI3K, other downstream effectors might play a role in the metastasis-promoting functions of IRS-2, because this adaptor protein contains potential binding motifs for many intracellular signaling molecules, including the tyrosine phosphatase SHP-2, the adapters Nck, Grb-2, and Crk, and the tyrosine kinase fyn (52). Many of these effectors function in signaling pathways that could regulate metastasis.
Although few studies have examined IRS expression in human breast cancer, one study did observe a decrease in IRS-1 expression during tumor progression, with IRS-1 expressed at low levels in poorly differentiated breast cancers (37). It is intriguing to speculate that IRS-2 is the predominant IRS isoform expressed in these later stages of tumor progression. The fact that IRS-1−/− mammary tumor cells that express only IRS-2 are highly invasive and are resistant to apoptotic stress suggests that these tumors would be more aggressive. With regard to this possibility, IRS-2 expression is maintained in ER− breast carcinoma cells, while IRS-1 expression is decreased or lost (40). Loss of ER expression is considered a poor prognostic marker, and ER− tumors tend to be more aggressive than tumors that retain ER expression (44).
Our findings that IRS-1 and IRS-2 play distinct roles in mammary tumor progression raise the question of whether these adaptor proteins contribute to the progression of other types of cancers. IGF-1 signaling has been implicated in the behavior of many other carcinomas, including prostate, colon, thyroid, and pancreatic (17, 25, 27, 36, 51). It is likely that the IRS proteins function downstream of the IGF-1R in these cancers as well and contribute to tumor development and progression. Strong evidence to support a role for the IRS proteins in progression comes from recent studies on the tumor-suppressor tuberous schlerosis complex (TSC1-2) (31). This complex consists of two gene products, tuberin and hamartin, which negatively regulate the activity of the growth-regulatory kinase S6K1 (31, 48). S6K1 regulates insulin and IGF-1-dependent activation of PI3K by suppressing IRS gene expression and also by phosphorylating the IRS proteins and targeting them for degradation in a negative feedback loop (16). In tumors that arise from TSC1-2 mutations, which are termed harmartomas, S6K activity is elevated and IRS expression is downregulated. These tumors can grow quite large, but they have a low malignancy potential, as was observed in IRS-2−/− mammary tumors (31).
In summary, we have identified a novel role for the IRS-2 adaptor protein in mammary tumor metastasis. Although IRS-2 is not required for tumor initiation or primary tumor growth, signaling through this adaptor protein is important for promoting tumor cell invasion and survival, hallmarks of the metastatic phenotype. Our data reveal that the relative expression levels of IRS-2 in tumors could significantly affect disease progression for breast cancer patients. Therefore, pathways that regulate IRS-2 expression, as well as downstream pathways activated through IRS-2, represent potential novel therapeutic targets.
Acknowledgments
We thank Olivier Kocher for his help with analysis of the tumor pathology.
This work was supported by NIH grants CA90583 (L.M.S.) and DK38712 (M.F.W).
REFERENCES
- 1.Albanell, J., and J. Baselga. 2001. Unraveling resistance to trastuzumab (herceptin): insulin-like growth factor-I receptor, a new suspect. J. Natl. Cancer Inst. 93:1830-1832. [DOI] [PubMed] [Google Scholar]
- 2.Araki, E., M. A. Lipes, M. E. Patti, J. C. Bruning, B. Haag III, R. S. Johnson, and C. R. Kahn. 1994. Alternative pathway of insulin signalling in mice with targeted disruption of the IRS-1 gene. Nature 372:186-190. [DOI] [PubMed] [Google Scholar]
- 3.Bachelder, R. E., A. Marchetti, R. Falcioni, S. Soddu, and A. M. Mercurio. 1999. Activation of p53 function in carcinoma cells by the α6β4 integrin. J. Biol. Chem. 274:20733-20737. [DOI] [PubMed] [Google Scholar]
- 4.Boudreau, N., and M. J. Bissell. 1998. Extracellular matrix signaling: integration of form and function in normal and malignant cells. Curr. Opin. Cell Biol. 10:640-646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bruning, J. C., J. Winnay, B. Cheatham, and C. R. Kahn. 1997. Differential signaling by insulin receptor substrate 1 (IRS-1) and IRS-2 in IRS-1-deficient cells. Mol. Cell. Biol. 17:1513-1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cantley, L. C. 2002. The phosphoinositide 3-kinase pathway. Science 296:1655-1657. [DOI] [PubMed] [Google Scholar]
- 7.Conneely, O. M., B. M. Jericevic, and J. P. Lydon. 2003. Progesterone receptors in mammary gland development and tumorigenesis. J. Mammary Gland Biol. Neoplasia 8:205-214. [DOI] [PubMed] [Google Scholar]
- 8.Cui, X., Z. Lazard, P. Zhang, T. A. Hopp, and A. V. Lee. 2003. Progesterone crosstalks with insulin-like growth factor signaling in breast cancer cells via induction of insulin receptor substrate-2. Oncogene 22:6937-6941. [DOI] [PubMed] [Google Scholar]
- 9.Dunn, S. E., M. Ehrlich, N. J. Sharp, K. Reiss, G. Solomon, R. Hawkins, R. Baserga, and J. C. Barrett. 1998. A dominant negative mutant of the insulin-like growth factor-I receptor inhibits the adhesion, invasion, and metastasis of breast cancer. Cancer Res. 58:3353-3361. [PubMed] [Google Scholar]
- 10.Farrelly, N., Y.-J. Lee, J. Oliver, C. Dive, and C. H. Streuli. 1999. Extracellular matrix regulates apoptosis in mammary epithelium through a control on insulin signaling. J. Cell Biol. 144:1337-1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fasshauer, M., J. Klein, K. Ueki, K. M. Kriauciunas, M. Benito, M. F. White, and C. R. Kahn. 2000. Essential role of insulin receptor substrate-2 in insulin stimulation of Glut4 translocation and glucose uptake in brown adipocytes. J. Biol. Chem. 275:25494-25501. [DOI] [PubMed] [Google Scholar]
- 12.Guvakova, M. A., and E. Surmacz. 1997. Tamoxifen interferes with the insulin-like growth factor I receptor (IGF-IR) signaling pathway in breast cancer cells. Cancer Res. 57:2606-2610. [PubMed] [Google Scholar]
- 13.Guy, C. T., R. D. Cardiff, and W. J. Muller. 1992. Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol. Cell. Biol. 12:954-961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hadsell, D. L., and S. G. Bonnette. 2000. IGF and insulin action in the mammary gland: lessons from transgenic and knockout models. J. Mammary Gland Biol. Neoplasia 5:19-30. [DOI] [PubMed] [Google Scholar]
- 15.Hanahan, D., and R. A. Weinberg. 2000. The hallmarks of cancer. Cell 100:57-70. [DOI] [PubMed] [Google Scholar]
- 16.Harrington, L. S., G. M. Findlay, A. Gray, T. Tolkacheva, S. Wigfield, H. Rebholz, J. Barnett, N. R. Leslie, S. Cheng, P. R. Shepherd, I. Gout, C. P. Downes, and R. F. Lamb. 2004. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J. Cell Biol. 166:213-223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hellawell, G. O., G. D. Turner, D. R. Davies, R. Poulsom, S. F. Brewster, and V. M. Macaulay. 2002. Expression of the type 1 insulin-like growth factor receptor is up-regulated in primary prostate cancer and commonly persists in metastatic disease. Cancer Res. 62:2942-2950. [PubMed] [Google Scholar]
- 18.Jackson, J. G., M. F. White, and D. Yee. 1998. Insulin receptor substrate-1 is the predominant signaling molecule activated by insulin-like growth factor-I, insulin, and interleukin-4 in estrogen receptor-positive human breast cancer cells. J. Biol. Chem. 273:9994-10003. [DOI] [PubMed] [Google Scholar]
- 19.Jackson, J. G., X. Zhang, T. Yoneda, and D. Yee. 2001. Regulation of breast cancer cell motility by insulin receptor substrate-2 (IRS-2) in metastatic variants of human breast cancer cell lines. Oncogene 20:7318-7325. [DOI] [PubMed] [Google Scholar]
- 20.Lavan, B. E., V. R. Fantin, E. T. Chang, W. S. Lane, S. R. Keller, and G. E. Lienhard. 1997. A novel 160-kDa phosphotyrosine protein in insulin-treated embryonic kidney cells is a new member of the insulin receptor substrate family. J. Biol. Chem. 272:21403-21407. [DOI] [PubMed] [Google Scholar]
- 21.Lavan, B. E., W. S. Lane, and G. E. Lienhard. 1997. The 60-kDa phosphotyrosine protein in insulin-treated adipocytes is a new member of the insulin receptor substrate family. J. Biol. Chem. 272:11439-11443. [DOI] [PubMed] [Google Scholar]
- 22.Lee, A. V., J. G. Jackson, J. L. Gooch, S. G. Hilsenbeck, E. Coronado-Heinsohn, C. K. Osborne, and D. Yee. 1999. Enhancement of insulin-like growth factor signaling in human breast cancer: estrogen regulation of insulin receptor substrate-1 expression in vitro and in vivo. Mol. Endocrinol. 13:787-796. [DOI] [PubMed] [Google Scholar]
- 23.Lee, A. V., P. Zhang, M. Ivanova, S. Bonnette, S. Oesterreich, J. M. Rosen, S. Grimm, R. C. Hovey, B. K. Vonderhaar, C. R. Kahn, D. Torres, J. George, S. Mohsin, D. C. Allred, and D. L. Hadsell. 2003. Developmental and hormonal signals dramatically alter the localization and abundance of insulin receptor substrate proteins in the mammary gland. Endocrinology 144:2683-2694. [DOI] [PubMed] [Google Scholar]
- 24.Lee, Y. J., and C. H. Streuli. 1999. Extracellular matrix selectively modulates the response of mammary epithelial cells to different soluble signaling ligands. J. Biol. Chem. 274:22401-22408. [DOI] [PubMed] [Google Scholar]
- 25.LeRoith, D., and C. T. Roberts, Jr. 2003. The insulin-like growth factor system and cancer. Cancer Lett. 195:127-137. [DOI] [PubMed] [Google Scholar]
- 26.Lin, E. Y., J. G. Jones, P. Li, L. Zhu, K. D. Whitney, W. J. Muller, and J. W. Pollard. 2003. Progression to malignancy in the polyoma middle T oncoprotein mouse breast cancer model provides a reliable model for human diseases. Am. J. Pathol. 163:2113-2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin, Y., A. Tamakoshi, S. Kikuchi, K. Yagyu, Y. Obata, T. Ishibashi, T. Kawamura, Y. Inaba, M. Kurosawa, Y. Motohashi, and Y. Ohno. 2004. Serum insulin-like growth factor-I, insulin-like growth factor binding protein-3, and the risk of pancreatic cancer death. Int. J. Cancer 110:584-588. [DOI] [PubMed] [Google Scholar]
- 28.Lu, Y., X. Zi, Y. Zhao, D. Mascarenhas, and M. Pollak. 2001. Insulin-like growth factor-I receptor signaling and resistance to trastuzumab (herceptin). J. Natl. Cancer Inst. 93:1852-1857. [DOI] [PubMed] [Google Scholar]
- 29.Mercurio, A. M., I. Rabinovitz, and L. M. Shaw. 2001. The alpha 6 beta 4 integrin and epithelial cell migration. Curr. Opin. Cell Biol. 13:541-545. [DOI] [PubMed] [Google Scholar]
- 30.Nolan, M. K., L. Jankowska, M. Prisco, S. Xu, M. A. Guvakova, and E. Surmacz. 1997. Differential roles of IRS-1 and SHC signaling pathways in breast cancer cells. Int. J. Cancer. 72:828-834. [DOI] [PubMed] [Google Scholar]
- 31.Pan, D., J. Dong, Y. Zhang, and X. Gao. 2004. Tuberous sclerosis complex: from Drosophila to human disease. Trends Cell Biol. 14:78-85. [DOI] [PubMed] [Google Scholar]
- 32.Resnik, J. L., D. B. Reichart, K. Huey, N. J. Webster, and B. L. Seely. 1998. Elevated insulin-like growth factor I receptor autophosphorylation and kinase activity in human breast cancer. Cancer Res. 58:1159-1164. [PubMed] [Google Scholar]
- 33.Rocha, R. L., S. G. Hilsenbeck, J. G. Jackson, C. L. VanDenBerg, C. Weng, A. V. Lee, and D. Yee. 1997. Insulin-like growth factor binding protein-3 and insulin receptor substrate-1 in breast cancer: correlation with clinical parameters and disease-free survival. Clin. Cancer Res. 3:103-109. [PubMed] [Google Scholar]
- 34.Sachdev, D., and D. Yee. 2001. The IGF system and breast cancer. Endocr. Relat. Cancer 8:197-209. [DOI] [PubMed] [Google Scholar]
- 35.Salerno, M., D. Sisci, L. Mauro, M. A. Guvakova, S. Ando, and E. Surmacz. 1999. Insulin receptor substrate 1 is a target for the pure antiestrogen ICI 182,780 in breast cancer cells. Int. J. Cancer. 81:299-304. [DOI] [PubMed] [Google Scholar]
- 36.Sandhu, M. S., D. B. Dunger, and E. L. Giovannucci. 2002. Insulin, insulin-like growth factor-I (IGF-I), IGF binding proteins, their biologic interactions, and colorectal cancer. J. Natl. Cancer Inst. 94:972-980. [DOI] [PubMed] [Google Scholar]
- 37.Schnarr, B., K. Strunz, J. Ohsam, A. Benner, J. Wacker, and D. Mayer. 2000. Down-regulation of insulin-like growth factor-I receptor and insulin receptor substrate-1 expression in advanced human breast cancer. Int. J. Cancer 89:506-513. [DOI] [PubMed] [Google Scholar]
- 38.Schubert, M., D. P. Brazil, D. J. Burks, J. A. Kushner, J. Ye, C. L. Flint, J. Farhang-Fallah, P. Dikkes, X. M. Warot, C. Rio, G. Corfas, and M. F. White. 2003. Insulin receptor substrate-2 deficiency impairs brain growth and promotes tau phosphorylation. J. Neurosci. 23:7084-7092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Senger, D. R., K. P. Claffey, J. E. Benes, C. A. Perruzzi, A. P. Sergiou, and M. Detmar. 1997. Angiogenesis promoted by vascular endothelial growth factor: regulation through α1β1 and α2β1 integrins. Proc. Natl. Acad. Sci. USA 94:13612-13617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shaw, L. M. 2001. Identification of insulin receptor substrate 1 (IRS-1) and IRS-2 as signaling intermediates in the α6β4 integrin-dependent activation of phosphoinositide 3-OH kinase and promotion of invasion. Mol. Cell. Biol. 21:5082-5093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shaw, L. M. 1999. Integrin function in breast carcinoma progression. J. Mammary Gland Biol. Neoplasia 4:367-376. [DOI] [PubMed] [Google Scholar]
- 42.Shaw, L. M., C. Chao, U. M. Wewer, and A. M. Mercurio. 1996. Function of the integrin alpha 6 beta 1 in metastatic breast carcinoma cells assessed by expression of a dominant-negative receptor. Cancer Res. 56:959-963. [PubMed] [Google Scholar]
- 43.Shaw, L. M., I. Rabinovitz, H. H. F. Wang, A. Toker, and A. M. Mercurio. 1997. Activation of phosphoinositide 3-OH kinase by the alpha-6-beta-4 integrin promotes carcinoma invasion. Cell 91:949-960. [DOI] [PubMed] [Google Scholar]
- 44.Sheikh, M. S., M. Garcia, P. Pujol, J. A. Fontana, and H. Rochefort. 1994. Why are estrogen-receptor-negative breast cancers more aggressive than the estrogen-receptor-positive breast cancers? Invasion Metastasis 14:329-336. [PubMed] [Google Scholar]
- 45.Sommer, S., and S. A. Fuqua. 2001. Estrogen receptor and breast cancer. Semin. Cancer Biol. 11:339-352. [DOI] [PubMed] [Google Scholar]
- 46.Spicer, A. P., G. J. Rowse, T. K. Lidner, and S. J. Gendler. 1995. Delayed mammary tumor progression in Muc-1 null mice. J. Biol. Chem. 270:30093-30101. [DOI] [PubMed] [Google Scholar]
- 47.Sutherland, R. L., and E. A. Musgrove. 2004. Cyclins and breast cancer. J. Mammary Gland Biol. Neoplasia 9:95-104. [DOI] [PubMed] [Google Scholar]
- 48.Tee, A. R., D. C. Fingar, B. D. Manning, D. J. Kwiatkowski, L. C. Cantley, and J. Blenis. 2002. Tuberous sclerosis complex-1 and -2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc. Natl. Acad. Sci. USA 99:13571-13576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Turner, B. C., B. G. Haffty, L. Narayanan, J. Yuan, P. A. Havre, A. A. Gumbs, L. Kaplan, J. L. Burgaud, D. Carter, R. Baserga, and P. M. Glazer. 1997. Insulin-like growth factor-I receptor overexpression mediates cellular radioresistance and local breast cancer recurrence after lumpectomy and radiation. Cancer Res. 57:3079-3083. [PubMed] [Google Scholar]
- 50.Vassen, L., W. Wegrzyn, and L. Klein-Hitpass. 1999. Human insulin receptor substrate-2 (IRS-2) is a primary progesterone response gene. Mol Endocrinol. 13:485-494. [DOI] [PubMed] [Google Scholar]
- 51.Vella, V., L. Sciacca, G. Pandini, R. Mineo, S. Squatrito, R. Vigneri, and A. Belfiore. 2001. The IGF system in thyroid cancer: new concepts. Mol. Pathol. 54:121-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.White, M. F. 2002. IRS proteins and the common path to diabetes. Am. J. Physiol. Endocrinol. Metab. 283:E413-E422. [DOI] [PubMed] [Google Scholar]
- 53.Withers, D. J., D. J. Burks, H. H. Towery, S. L. Altamuro, C. L. Flint, and M. F. White. 1999. Irs-2 coordinates Igf-1 receptor-mediated beta-cell development and peripheral insulin signalling. Nat. Genet. 23:32-40. [DOI] [PubMed] [Google Scholar]
- 54.Withers, D. J., J. S. Gutierrez, H. Towery, D. J. Burks, J. M. Ren, S. Previs, Y. Zhang, D. Bernal, S. Pons, G. I. Shulman, S. Bonner-Weir, and M. F. White. 1998. Disruption of IRS-2 causes type 2 diabetes in mice. Nature 391:900-904. [DOI] [PubMed] [Google Scholar]
- 55.Zhang, X., S. Kamaraju, F. Hakuno, T. Kabuta, S. Takahashi, D. Sachdev, and D. Yee. 2004. Motility response to insulin-like growth factor-I (IGF-I) in MCF-7 cells is associated with IRS-2 activation and integrin expression. Breast Cancer Res. Treat. 83:161-170. [DOI] [PubMed] [Google Scholar]