

Significance
Human cytochromes P450 (CYPs) play a leading role in detoxication by metabolizing drugs and other foreign compounds. CYP3A4 is the most important CYP because it oxidizes the majority of administered therapeutics and is implicated in drug–drug interactions, drug toxicity, and other adverse effects. To date, little is known about how CYP3A4 adjusts and reshapes the active site to accommodate and regioselectively oxidize a wide variety of compounds. The CYP3A4–midazolam cocrystal structure reveals a profound structural reorganization triggered by the substrate, which was anticipated but never before observed, which helps us better understand and explain experimental results and, by representing a conformational snapshot, could be used for computer modeling and molecular dynamics simulations to improve the outcomes for drug metabolism predictions.
Keywords: CYP3A4, drug metabolism, midazolam, substrate binding, crystal structure
Abstract
Human cytochrome P450 3A4 (CYP3A4) is a major hepatic and intestinal enzyme that oxidizes more than 60% of administered therapeutics. Knowledge of how CYP3A4 adjusts and reshapes the active site to regioselectively oxidize chemically diverse compounds is critical for better understanding structure–function relations in this important enzyme, improving the outcomes for drug metabolism predictions, and developing pharmaceuticals that have a decreased ability to undergo metabolism and cause detrimental drug–drug interactions. However, there is very limited structural information on CYP3A4–substrate interactions available to date. Despite the vast variety of drugs undergoing metabolism, only the sedative midazolam (MDZ) serves as a marker substrate for the in vivo activity assessment because it is preferentially and regioselectively oxidized by CYP3A4. We solved the 2.7 Å crystal structure of the CYP3A4–MDZ complex, where the drug is well defined and oriented suitably for hydroxylation of the C1 atom, the major site of metabolism. This binding mode requires H-bonding to Ser119 and a dramatic conformational switch in the F–G fragment, which transmits to the adjacent D, E, H, and I helices, resulting in a collapse of the active site cavity and MDZ immobilization. In addition to providing insights on the substrate-triggered active site reshaping (an induced fit), the crystal structure explains the accumulated experimental results, identifies possible effector binding sites, and suggests why MDZ is predominantly metabolized by the CYP3A enzyme subfamily.
Human cytochromes P450 (CYPs) play a dominant role in the metabolism of drugs and other foreign chemicals, providing one of the primary means for phase I xenobiotic detoxication. Among the 57 human CYPs, CYP3A4 is the most abundant liver and intestinal enzyme, and it oxidizes the vast majority of pharmaceuticals (1). CYP3A4 has very broad substrate specificity and can metabolize molecules widely differing in size and chemical structure through alkyl carbon and aromatic ring hydroxylation, O- and N-dealkylation, and epoxidation (2). Furthermore, CYP3A4 has the ability to simultaneously accommodate multiple molecules in the active site, which could lead to cooperative binding, atypical (non-Michaelis–Menten) kinetic behavior, and drug–drug interactions (3).
Despite its central role in drug metabolism, there is limited understanding of how CYP3A4 adapts to and regioselectively oxidizes a wide range of diverse compounds. The X-ray structure of ligand-free CYP3A4 was determined more than a decade ago (4, 5), but since then, cocrystal structures with only three substrates were reported: erythromycin, progesterone, and bromoergocryptine (6–8). Among these, only bromoergocryptine is bound in a productive mode, whereas erythromycin is positioned unsuitably for N-demethylation, and progesterone is docked outside the active site pocket. Therefore, more structural information is needed for better understanding the structural plasticity of CYP3A4, interpretation of complex kinetic data, and improvement of the outcomes for drug metabolism predictions.
Here we describe the cocrystal structure of CYP3A4 with the drug midazolam (MDZ) bound in a productive mode. MDZ (Fig. 1) is the benzodiazepine most frequently used for procedural sedation (9) and is a marker substrate for the CYP3A family of enzymes that includes CYP3A4 (10). MDZ is hydroxylated by CYP3A4 predominantly at the C1 position, whereas the 4-OH product accumulates at high substrate concentrations (up to 50% of total product) and inhibits the 1-OH metabolic pathway (11–14). The reaction rate and product distribution strongly depend on the assay conditions and can be modulated by α-naphthoflavone (ANF), testosterone, and other small molecules (12–18). Metabolism of MDZ is further complicated by the drug’s ability to act as a NADPH and time-dependent inhibitor of CYP3A4, possibly by covalent modification of the protein (19). The crystal structure of the CYP3A4–MDZ complex helps us better understand and explain these and other experimental findings by providing direct insights into the protein–substrate interactions and substrate-induced reshaping of the active site.
Fig. 1.
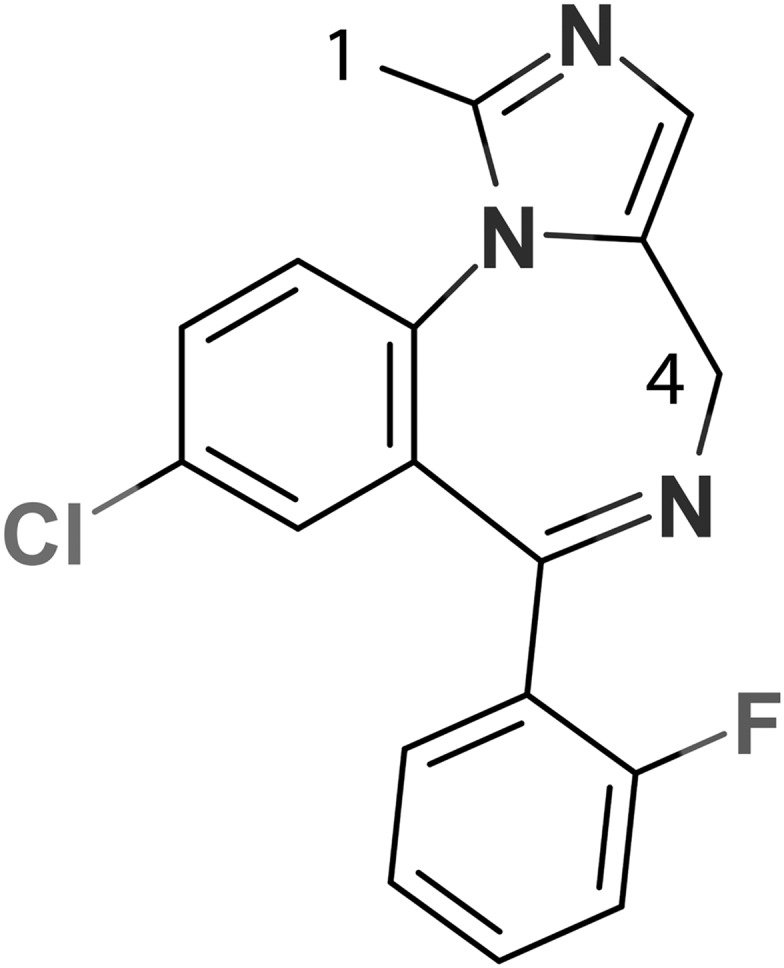
Structure of MDZ with the indicated primary (C1) and secondary (C4) hydroxylation sites.
Results and Discussion
High Ionic Strength Promotes Formation of the CYP3A4–MDZ Complex.
Among structurally diverse compounds oxidized by CYP3A4, MDZ is the most specific substrate and the best in vivo probe for the CYP3A4 activity prediction (10). Because of this unique property and the very limited structural information on the CYP3A4–substrate interactions available so far, it was of great interest to determine the crystal structure of the MDZ-bound CYP3A4. However, our initial attempts were unsuccessful because, similar to most other substrates tested, MDZ dissociated from CYP3A4 during crystallization. Because high ionic strength promotes association of small molecules to CYP3A4 (8), we tested whether an increase in the phosphate concentration could facilitate the CYP3A4–MDZ complex formation. Phosphate was chosen because it stimulates MDZ hydroxylation activity of CYP3A4 by increasing kcat for both the C1 and C4 hydroxylation reactions (12). As shown in Fig. 2A, MDZ is a classic type I ligand that induces a low-to-high spin shift in the heme iron. When the phosphate concentration is elevated from 0.1 to 0.6 M, the amplitude of the absorbance change remains the same, but the titration curve saturates at a threefold lower MDZ concentration, leading to a proportional decrease in the dissociation constant (Kd), from 4.8 ± 0.4 to 1.8 ± 0.2 μM, respectively. Thus, the phosphate ions could stimulate metabolism of MDZ, in part, by increasing its binding affinity for CYP3A4.
Fig. 2.

(A and B) Spectral changes induced by MDZ in the wild type and S119A CYP3A4, respectively. Arrows indicate direction of absorbance changes. (Left insets) Difference spectra between the ligand-free and MDZ-bound forms. (Right insets) Titration plots and hyperbolic fittings from which the Kd values were derived. A low-to-high spin shift in CYP3A4 S119A induced by testosterone is shown in Fig. S2.
Crystal Structure of the CYP3A4–MDZ Complex.
Phosphate ions promoted CYP3A4-MDZ cocrystallization as well. The brown color of crystalline CYP3A4 indicated that the protein is substrate-bound and in a high-spin form. The crystal structure was refined to 2.7 Å with R and Rfree of 22.4% and 29.2%, respectively (Table S1 and Fig. S1). Three molecules present in the asymmetric unit were highly similar and had MDZ bound to the active site in a similar orientation (Fig. 3 A and B). The better-defined molecule A was used for structural and comparative analyses discussed here.
Table S1.
Data collection and refinement statistics
| Ligand | MDZ |
| PDB ID | 5TE8 |
| Data statistics | |
| Space group | P212121 |
| Unit cell parameters | a = 64 Å, b = 118 Å, |
| Molecules per asymmetric unit | c = 206 Å; α, β, γ = 90° |
| Resolution range, Å | 3 |
| Total reflections | 77.52–2.70 (2.80–2.70) |
| Unique reflections | 244,816 |
| Redundancy | 43,549 |
| Completeness | 5.6 (5.9) |
| Average I/σI | 99.4 (99.3) |
| Rpim | 5.8 (1.5) |
| CC 1/2 | 0.115 (0.556) |
| Refinement statistics | 0.976 (0.717) |
| R/Rfree† | 22.4/29.2 |
| Number of atoms | |
| Protein | 11,524 |
| Solvent | 111 |
| r.m.s. deviations | |
| Bond lengths, Å | 0.007 |
| Bond angles, ° | 1.334 |
| Ramachandran outliers, % | 0.1 |
| Average B-factors, Å2 | |
| Molecule A | 45.9 |
| Ligand A | 32.5 |
| Molecule B | 54.2 |
| Ligand B | 33.4 |
| Molecule C | 74.4 |
| Ligand C | 57.6 |
Values in parentheses are for the highest resolution shell.
Rfree was calculated from a subset of 5% of the data that were excluded during refinement.
Fig. S1.

(A) Brown color of crystalline MDZ-bound CYP3A4 indicates that the heme iron is in a high-spin form. (B) The CYP3A4–MDZ complex crystallizes in the P212121 space group, with three molecules per asymmetric unit. MDZ is shown in magenta and space-filling representation. (C) Average B-factors (for the main and side chain atoms) plotted vs. residue number of each peptide chain. Average B-factors for MDZ are listed in Table S1.
Fig. 3.

Crystal structure of the CYP3A4–MDZ complex. (A) Superposition of three molecules present in the asymmetric unit shows their similar fold (root-mean-square deviation between the ordered Cα-atoms is 0.6–0.7 Å). (B) Close-up view at the active site of molecule A. MDZ is bound above the heme plane with the C1–Fe distance of 4.4 Å and forms a hydrogen bond with Ser119 via the imidazole ring nitrogen. The chlorophenyl ring is within the Van der Waals distance from the Ile369–Ala370 fragment, whereas the fluorophenyl ring is flanked by the Leu216, Pro218, and Leu482 side chains. These interactions immobilize MDZ and orient suitably for hydroxylation of the C1-atom, the primary oxidation site. Simulated annealing Fo–Fc composite omit map for MDZ (in green mesh) is contoured at 3σ. (C) Superposition of the ligand-free (Protein Data Bank ID 1TQN; in light/dark green and black) and MDZ-bound (in gray, magenta and red) structures. Structural elements undergoing reorganization are labeled. MDZ is in orange and space-filling representation. A 12-Å shift of Leu216 (shown in cyan) is indicated by an arrow. (D) Slice through the superimposed 1TQN (cyan and magenta) and MDZ-bound structures (yellow and blue) with semitransparent surfaces to show that the substrate-induced conformational switch leads to the active site collapse.
The diazepine/imidazole moiety of MDZ is near parallel to and 3.4–3.7 Å above the heme. In addition to hydrophobic and π–π stacking interactions with the cofactor, the heterocyclic ring establishes hydrophobic and Van der Waals interactions with the Arg105, Ile289, Ala305, Ala307, and Thr309 side chains, whereas the imidazole nitrogen is H-bonded to Ser119. The fluorophenyl group, in contrast, is sandwiched between the Leu216/Pro218 and Leu482 side chains. This arrangement and H-bonding to Ser119 limit rotational, lateral, and vertical movements of MDZ and fix it in position suitable for the C1 atom hydroxylation, with the C1–Fe distance of 4.4 Å.
Having a large and malleable active site, CYP3A4 is expected to undergo conformational changes to accommodate and regiospecifically oxidize chemically diverse compounds. However, until now, the active site reshaping has never been observed. In the previously determined structures of CYP3A4, the ligands mainly affected the F–F′ loop conformation and the relative orientation of the F, F′, G′, and G helices, but these changes were fairly modest (see Fig. 3.10 in ref. 20). The CYP3A4-MDZ cocrystal structure provides a glimpse into how a small substrate molecule could trigger large transformations of the active site, which can be considered an induced fit. To bring MDZ closer to the heme and properly orient, conformational changes in several regions take place (Fig. 3C). The most drastic reorganization occurs in the F–G helix–loop fragment that comprises the upper wall of the active site cavity. This highly flexible fragment is considerably longer in CYP3A4 (residues 202–263) and contains additional F′- and G′-helical regions and connecting loops. A 12-Å movement of Leu216 toward/with the fluorophenyl ring of MDZ leads to a complete and partial unwinding of the F′ and G helices, respectively; a one-turn extension of the F helix; and a 20° rotation of the G′ helix. The G′ helix shift, in turn, forces the B–C loop to move 5 Å away from the active site, whereas the C-terminal loop shifts inward by 3 Å to fill the space emptied by the 211–216 residues, bringing Leu482 closer to MDZ. Together, these conformational perturbations lead to a collapse of the upper wall of the active site cavity (∼20% volume decrease; Fig. 3D) and engulfment and immobilization of MDZ. Furthermore, changes in the F–G fragment are transmitted to the D, E, and H helices and the N-terminal end of the I helix, which shift up or down by 0.6–1.6 Å and involve residues as far as 36 Å away from the catalytic center.
Ser119 Is Critical for MDZ Binding.
Because MDZ establishes only a single H bond with the Ser119 residue, we tested whether the S119A replacement affects the binding affinity for CYP3A4. The purified S119A mutant exists partially in a high-spin form, but MDZ does not cause any further increase in high-spin content. Instead, an increase in MDZ concentration leads to a high-to-low spin conversion (Fig. 2B), resulting in formation of the resting (water-coordinated) form. In the presence of 0.6 M phosphate, absorbance changes were similar but more pronounced because the high-spin content in CYP3A4 S119A is higher under these conditions (∼15% vs. ∼8% in 0.1 M phosphate). Based on spectral perturbations induced by MDZ, it was possible to derive the Kd values, which were ionic-strength-independent (6.4 ± 0.5 and 6.2 ± 0.6 μM at 0.1 and 0.6 M phosphate, respectively) and 1.3- to 3.4-fold higher than the respective values for the wild type. Because the high-spin transition in CYP3A4 S119A could be induced by testosterone (Fig. S2), we conclude that the H-bond to Ser119 is crucial for achieving the MDZ binding mode observed in the crystal structure.
Fig. S2.

A low-to-high spin conversion in CYP3A4 S119A (0.9 μM) induced by a 40- and 80-fold excess of testosterone.
Further evidence for the importance of Ser119 in MDZ association and metabolism was provided by Khan et al. (21), who showed both that the S119A mutation switches the preferential oxidation site from the C1 to C4 atom via a sevenfold increase in Km for the 1-OH MDZ formation and a fourfold increase of Vmax for the 4-OH metabolite production, and that the majority of spectral changes during the CYP3A4–MDZ complex formation are caused by the substrate oriented suitably for the C1 hydroxylation. Our spectral data agree with these results and indicate that Ala at position 119 disfavors the crystallographic binding mode and promotes an alternative orientation in which MDZ cannot displace the distal water ligand and, as the product analysis showed (21), preferentially undergoes hydroxylation at the C4 site. Together, the experimental and structural data, as well as the finding that substitution of Ser119 profoundly changes regioselectivity and total activity of steroid hydroxylation (22), identify Ser119 as the key active site residue that defines the substrate binding affinity, orientation, and oxidation sites.
Structural Explanation of Other Mutagenesis Results.
In addition to Ser119, Khan et al. investigated the role of 14 other residues in MDZ metabolism (21). The mutation sites in their study were chosen on the basis of the homology model because the X-ray structure of CYP3A4 was not available at that time. Four of the mutated residues (Ala305, Thr309, Ile369, and Ala370, shown in green in Fig. 4) lie within the Van der Waals distance from the diazepine/imidazole portion of MDZ. Changes in these residues’ size could directly affect the binding affinity and orientation of MDZ and, hence, alter catalytic rate and product distribution. As expected, the latter parameters were affected by all investigated mutations (A305G/V, T309A/F, I369V/W, and A370V/F) to a different extent (21). The T309A, A370F, and I369W substitutions had the highest effect and decreased the overall activity by several-fold. Furthermore, the T309F mutation completely switched preferentiality in hydroxylation to the C1 site, and A305V, I369W, and A370V to C4. The latter three mutations disfavor the observed 1-OH orientation because of clashes with the MDZ chlorophenyl ring. Polar Thr309, in contrast, is strategically positioned near the dioxygen-binding site (Fig. 4B) and, similar to homologous residues in other P450s, could be involved in the proton delivery network. As suggested by Khan et al. (21), a marked decrease in the catalytic turnover caused by the T309A/F mutations could, in part, result from the altered proton shuttle. In silico modeling shows that Phe in position 309 would allow the 1-OH orientation if MDZ turns away by 10–15° around the H-bonded imidazole nitrogen. That the 4-OH metabolite cannot be formed by the T309F mutant suggests an existence of only one binding site for this orientation, located in the vicinity of Thr309.
Fig. 4.

(A and B) Side view and top view, respectively, at the active site of the CYP3A4–MDZ complex, where residues mutated by Khan et al. (21) are highlighted. Residues located within the Van der Waals distance or more remotely from MDZ (in orange) are depicted in green and cyan, respectively. Some secondary structure elements were deleted to provide a better view.
Other mutated residues are situated further away (shown in cyan in Fig. 4) but could influence MDZ binding and metabolism indirectly. Phe108, Ile120, and Phe304, for instance, are part of the newly formed hydrophobic cluster near the fluorophenyl-flanking Leu216 (Fig. 5A). By changing the side chain volume and interresidue contacts, the F108A/W, I120A/W, and F304A/W mutations could affect the mobility and positioning of Leu216 and, as a consequence, orientation of MDZ. The importance of hydrophobic interactions at this site is evident from the notable mutational effect on the metabolic activity and product distribution (21). The Ala mutants’ activities were more or less similar to those of the wild-type CYP3A4, but Trp at either position led to a two- to threefold increase in the reaction rate and a three- to fourfold higher preference for the C1 hydroxylation. The bulkier indole ring could strengthen hydrophobic contacts holding the cluster together, thereby helping to maintain the Leu216 orientation optimal for the 1-OH association mode. In other words, the F108W, I120W, and F304W mutations most likely stabilize the 1-OH binding mode, rather than prevent the 4-OH orientation. Because Pro107 precedes Phe108, the P107A/W mutations could increase this fragment’s flexibility and assist the aforementioned cluster formation to some extent. Indeed, both mutations elevate the catalytic rate by 30–50% and promote the C1 hydroxylation (21).
Fig. 5.

(A) Reorganization in the hydrophobic cluster (in cyan) near the MDZ-flanking Leu216 (in blue) assists the 1-OH MDZ orientation. Corresponding residues from the ligand-free 1TQN structure are shown in black. (B) Location of CYP3A4 SNVs whose functional effects on the MDZ metabolism were investigated. The I and E helices are in beige and pink, respectively; the F–G fragment is in green. (C and D) Residues comprising the peripheral progesterone binding site (in different colors) and the F–G fragment (in black) in the ligand-free and MDZ-bound CYP3A4, respectively.
It is also of interest to discuss the mutational effect of two residues from the I helix, Ile301 and Tyr307. An 80% decrease in the 1-OH product formation caused by the I301W substitution can be explained by steric hindrance and/or interference with the H-bond formation to the nearby Ser119. That, similar to the T309F mutation, I301W completely inhibits formation of the 4-OH metabolite is another indication that the binding site for this orientation is near the I helix and largely overlaps with the crystallographic binding mode. The side group of Tyr307, in contrast, is directed parallel to the I helix and forms hydrophobic interactions with Leu211. In the ligand-free CYP3A4, this and the neighboring Leu210 comprise the F′–F loop, which becomes part of the extended F helix upon formation of the CYP3A4–MDZ complex. Therefore, substitutions in residues 210, 211, and 307 could influence MDZ orientation by altering hydrophobic interactions and conformational dynamics in the F–G fragment. This prediction is in line with the experimental findings showing that replacement of Leu210 or Leu211 with Trp has a moderately negative effect on the activity (30–45% decrease) and virtually no influence on the product distribution (21). In contrast, introduction of a smaller and less hydrophobic Ala instead of Leu210 and Tyr307 would alter the active site architecture and, as experimentally found, the oxidation site: L210A and 307A shift preference toward the C4-atom hydroxylation, and Y307W to C1 (21).
Structural Effect of Allelic Variants on MDZ Metabolism.
The L373P substitution, which was also investigated by Khan et al. (21), occurs naturally and represents a single-nucleotide variation (SNV) in human CYP3A4 (allele CYP3A4.12) (23). Leu373 is 6.8 Å away from the C4 atom and comprises the β strand that lines the wall of the heme-binding pocket (Figs. 4 and 5B). If, similar to the Leu side chain, the phenyl ring is positioned below the heme plane, the L373F mutation could only change the environment and redox properties of the heme, but not the substrate association mode. However, the L373F variant displayed a 10-fold lower preference for the C1 hydroxylation relative to the wild type (21). This could happen if the phenyl ring protrudes into the active site and, through steric clashing, disfavors the 1-OH binding mode.
Among numerous coding SNVs altering the functional activity of CYP3A4 (24), the MDZ hydroxylase reaction has been investigated in two more variants: T185S and L293P (alleles CYP3A4.16 and CYP3A4.18, respectively) (21, 25, 26). Neither of these remote mutations is in position to directly affect the CYP3A4-MDZ interaction (Fig. 5B). Leu293 is at the N-terminal edge of the I helix and forms hydrophobic interactions with Phe113 from the B–C-connecting loop. Although both regions undergo MDZ-induced positional shifts, the L293P mutation should not significantly affect conformational dynamics in CYP3A4 because of its remote and surface location. Indeed, the L293P variant exhibits only a mild (∼15%) decrease in the 1-OH product formation (the 4-OH metabolic pathway was not investigated) (26). In contrast, the E helix Thr185 lies right underneath Phe203 from the F helix (Fig. 5B). It is possible that the conformational changes induced by MDZ are transmitted to the E helix through the Thr185–Phe203 interaction. In this case, even slight changes in the volume of residue 185 could affect conformational dynamics and modulate the substrate binding and oxidation. This possibility could explain why the T185S mutation alters both 1-OH and 4-OH metabolic pathways via a 30–50% change in the Vmax and/or Km values (25).
Insights into the Effector and Cooperative Binding Mechanism.
Increased production of the 4-OH metabolite and inhibition of the 1-OH pathway observed at high MDZ concentrations (11–14) is an example of negative homotropic substrate cooperativity in CYP3A4. The underlying mechanism for this phenomenon, as well as the effector role of ANF, testosterone, or carbamazepine, were investigated by NMR spectroscopy in combination with molecular modeling techniques, using the ligand-free 1TQN structure as a working model to deduce MDZ orientations consistent with the experimental results (14, 17). Both studies were conducted at high substrate:CYP3A4 ratios (260:1 or 1,000:1), when the C1 hydroxylation is no longer favored and the 1-OH/4-OH product ratio approaches unity. It was shown that under these conditions, the substrate cannot closely approach the heme, as the distances between the C1 and C4 protons of MDZ and the heme iron were equal (7.6–8.0 and 7.8 Å, respectively), and 2–3 Å longer than allowed for hydrogen extraction. The NMR data suggested also that in the absence of effectors, MDZ freely rotates/slides along the heme plane with no favorable orientation (17) or forms stacked dimers (14). The movements of MDZ were restricted by ANF and testosterone, which bound near the heme and promoted the C1 and C4 hydroxylation, respectively (17). Carbamazepine, in turn, was proposed to form stacked heterodimers with MDZ to orient it favorably for the C4 hydroxylation (14).
Taking into account both the NMR and structural data, one can conclude that overcrowding of the active site disallows conformational changes observed in the crystal structure, and that the 1-OH stimulation by ANF at low MDZ concentrations (12) must proceed via a different mechanism because there is no space within the catalytic cavity for the simultaneous binding of the effector and MDZ in the crystallographic mode. As previously suggested, ANF could promote metabolism by enhancing the CYP3A4–redox partner interaction (12) and/or oxygen activation events (27), or modulate conformational dynamics in CYP3A4 by docking to the surface site. One potential effector-binding area identified in the progesterone-bound structure of CYP3A4 is above the phenylalanine cluster (5, 8). However, this peripheral site disintegrates upon MDZ binding (Fig. 5C), and hence, its occupation by ANF would inhibit, rather than promote, the substrate-dependent conformational switch.
Because MDZ triggers structural changes both within and far away from the active site, ANF could modulate the drug metabolism by associating with the affected areas (e.g., near the F–G fragment or the D, E, H, or I helices) and changing conformational dynamics. There are several surface clefts in the MDZ-bound CYP3A4 where ANF and other effector molecules could potentially bind (Fig. 6A and Fig. S3A). Two pockets are adjacent to the G′–G fragment and the hydrophobic cluster near the MDZ-flanking Leu216 (Fig. 6B), the areas undergoing structural rearrangement critically important for the proper MDZ orientation. Both pockets preexist in ligand-free CYP3A4, although with a distinct depth and shape (Fig. S3B). It is possible that occupation of one or both of these sites by ANF could affect the F–G fragment reorganization and the Leu216–cluster formation, which, in turn, would modulate the MDZ binding and metabolism.
Fig. 6.

Possible ANF binding sites in the MDZ-bound CYP3A4. (A) Surface clefts on the proximal face with manually docked ANF molecules (in purple). The F–G fragment is in green; MDZ in orange. Potential ANF binding sites on the distal side are shown in Fig. S3. (B) Side view at the pockets, occupation of which by ANF could modulate conformational dynamics in the F–G fragment (in gray and cyan) and the Leu216-cluster formation. Selected residues comprising the potential ANF binding sites are shown in yellow sticks.
Fig. S3.

(A) Distal face of CYP3A4 with two potential α-naphthoflavone (ANF) binding sites. Cavities were displayed using the detection cutoff of 20 solvent radii, and ANF (in purple) was docked manually. It is generally accepted that the central groove on the P450 distal face serves as a redox partner docking site. By occupying this site, ANF and other effectors could modulate redox partner binding and, as a consequence, an electron flow to the heme. Two other hydrophobic pockets, occupation of which by ANF could affect MDZ metabolism, are indicated by a dashed rectangle and discussed in the main text. (B) Side view on the highlighted area near the F–G fragment in the ligand-free CYP3A4 (1TQN structure). Surface clefts preexist, but one (on the right) is deeper and the other is shallower, relative to those in the CYP3A4–MDZ complex (see Fig. 6B). ANF could bind to both sites regardless of whether the drug is bound or not, but in a different orientation. ANF molecules manually docked to the ligand-free and MDZ-bound CYP3A4 are displayed in orange and purple, respectively.
Also, it is evident from the X-ray structure that the C4-atom of MDZ cannot approach the catalytic center without expansion of the active site. That formation of the 4-OH metabolite is increased at high MDZ concentrations (11–14) also implies that overcrowding and widening of the active site is a prerequisite for this orientation. The latter requirement could explain why the 1-OH but not 4-OH metabolic pathway leads to CYP3A4 inactivation (21). An NADPH and time-dependent loss of the CYP3A4 activity was attributed to covalent protein modification (19), although no direct evidence for protein–metabolite adduct formation was presented. According to the X-ray structure, the protruding C1-atom of MDZ is only 3.3–3.5 Å away from the Leu216 and Ala305 side chains. Thus, there is a possibility that the free radical intermediate, formed after the C1 hydrogen extraction, could attack and covalently attach to either of these residues without significant movement of MDZ. In contrast, because of a central position in the heterocyclic ring and the active site widening, a transient radical formed at the C4 atom might be too far from any residue for the covalent adduct formation.
Summing up, the CYP3A4–MDZ complex structure provides unique insights into the substrate-induced readjustment and reshaping of the active site. Structural reorganization involves the entire F–G segment and triggers long-range residue movements that transmit to the areas far away from the active site. Based on the structural and previously reported experimental results, it was possible to conclude that MDZ has two highly overlapping binding sites near the I helix, and depending on the substrate concentration, a collapse or widening of the active site is required for the 1- and 4-OH metabolite formation. The extended and highly flexible F–G fragment, Ser119, Leu216, Pro218, and Leu482 were identified as the key elements that assist the substrate to associate in a productive mode. These elements are not present in other drug-metabolizing CYPs (Figs. S4–S6), which could explain why MDZ is predominantly oxidized by the CYP3A enzyme subfamily. The X-ray structure also helps to better understand functional effect of SNVs and other genetic variations and, by representing a conformational snapshot, could be used for computer modeling and molecular dynamics simulations to improve the outcomes for the in vivo drug metabolism predictions.
Fig. S4.

Structure-based sequence alignment of human drug-metabolizing CYPs. Only fragments containing residues critical for MDZ binding (depicted in bold red) are shown. The B′–C peptide residues occupying the CYP3A4 Ser119 position (shown in Fig. S5) are highlighted in black/gray.
Fig. S6.

Amino acid sequence alignment of the CYP3A enzyme subfamily preformed with Clustal Omega. Residues critical for MDZ binding are highlighted in bold red.
Fig. S5.

Structural overlay of human drug-metabolizing CYPs: CYP3A4 (1WOE; black), CYP1A2 (2HI4; cyan), CYP2B6 (molecule A of 4RQL; pink), CYP2C9 (molecule A of 1OG2; yellow), and CYP2D6 (molecule A of 4WNU; green). MDZ (in magenta) is shown in the crystallographic binding mode. Neither of the B′–C loop residues aligning with Ser119 of CYP3A4 can H-bond and accommodate MDZ in a similar mode. The length and topology of the F–G and C-terminal loops significantly varies among CYPs, due to which it is not possible to accurately pinpoint residues corresponding to MDZ-flanking leucines 216 and 482. Nevertheless, the outlined sequence and structural dissimilarities, and preservation of all key elements in CYP3A5 and CYP3A7 (Fig. S6) could explain why MDZ is predominantly oxidized by the CYP3A enzyme subfamily.
Materials and Methods
Protein Purification, Crystallization, and Structure Determination.
The wild-type and S119A mutant of human Δ3–22 CYP3A4 were expressed and purified as previously described (28). Crystallization and determination of the X-ray structure of the CYP3A4–MDZ complex are described in SI Materials and Methods. Data collection and refinement statistics are summarized in Table S1. The atomic coordinates and structure factors were deposited to the Protein Data Bank (ID code: 5TE8).
Equilibrium Titrations.
Titration of 1 μM WT and S119A CYP3A4 with MDZ (DMSO solution) was carried out in 0.1 or 0.6 M phosphate buffer at pH 7.4. The Kd values were estimated from hyperbolic fits to the plots of the maximal absorbance change vs. MDZ concentration using IgorPro (WaveMetrics).
SI Materials and Methods
The CYP3A4–MDZ complex was crystallized by a sitting drop vapor diffusion method. CYP3A4 (130 mg/mL in 0.6 mM phosphate buffer at pH 7.4, 1 mM DTT) was mixed with 50 mM MDZ (in DMSO), with a final molar protein:ligand ratio of 1:2. After 20 min incubation, the mixture was centrifuged to remove the precipitate. The crystallization drop consisted of 0.4 μL MDZ-bound protein and 0.6 μL solution #7 from the Hampton Research PegIon2 kit [0.1 M sodium malonate at pH 7.0, 12% (wt/vol) polyethylene glycol 3350]. Brown plate-like crystals grew within several days and, after harvesting, were flash frozen in liquid nitrogen, using Paratone-N oil as a cryoprotectant. The X-ray diffraction data were collected at the Advanced Light Source beamline 8.2.2. The structure was solved by molecular replacement with the PHASER software (29) and 1TQN as a search model. The initial model was refined with the Crystallographic Object-Oriented Toolkit (COOT) (30) and the macromolecular refinement program REFMAC (29).
Acknowledgments
This work was supported by National Institutes of Health Grants GM57353 (to T.L.P.) and ES025767 (to I.F.S.) and involves research carried out at the Advanced Light Source supported by the director, Office of Science, Office of Basic Energy Sciences, of the US Department of Energy under Contract No. DE-AC02-05CH11231.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 5TE8).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1616198114/-/DCSupplemental.
References
- 1.Guengerich FP. Cytochrome P-450 3A4: Regulation and role in drug metabolism. Annu Rev Pharmacol Toxicol. 1999;39:1–17. doi: 10.1146/annurev.pharmtox.39.1.1. [DOI] [PubMed] [Google Scholar]
- 2.Rendic S, Di Carlo FJ. Human cytochrome P450 enzymes: A status report summarizing their reactions, substrates, inducers, and inhibitors. Drug Metab Rev. 1997;29(1-2):413–580. doi: 10.3109/03602539709037591. [DOI] [PubMed] [Google Scholar]
- 3.Atkins WM. Non-Michaelis-Menten kinetics in cytochrome P450-catalyzed reactions. Annu Rev Pharmacol Toxicol. 2005;45:291–310. doi: 10.1146/annurev.pharmtox.45.120403.100004. [DOI] [PubMed] [Google Scholar]
- 4.Yano JK, et al. The structure of human microsomal cytochrome P450 3A4 determined by X-ray crystallography to 2.05-A resolution. J Biol Chem. 2004;279(37):38091–38094. doi: 10.1074/jbc.C400293200. [DOI] [PubMed] [Google Scholar]
- 5.Williams PA, et al. Crystal structures of human cytochrome P450 3A4 bound to metyrapone and progesterone. Science. 2004;305(5684):683–686. doi: 10.1126/science.1099736. [DOI] [PubMed] [Google Scholar]
- 6.Ekroos M, Sjögren T. Structural basis for ligand promiscuity in cytochrome P450 3A4. Proc Natl Acad Sci USA. 2006;103(37):13682–13687. doi: 10.1073/pnas.0603236103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sevrioukova IF, Poulos TL. Structural and mechanistic insights into the interaction of cytochrome P4503A4 with bromoergocryptine, a type I ligand. J Biol Chem. 2012;287(5):3510–3517. doi: 10.1074/jbc.M111.317081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sevrioukova IF, Poulos TL. Anion-dependent stimulation of CYP3A4 monooxygenase. Biochemistry. 2015;54(26):4083–4096. doi: 10.1021/acs.biochem.5b00510. [DOI] [PubMed] [Google Scholar]
- 9.Tobias JD, Leder M. Procedural sedation: A review of sedative agents, monitoring, and management of complications. Saudi J Anaesth. 2011;5(4):395–410. doi: 10.4103/1658-354X.87270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thummel KE, Wilkinson GR. In vitro and in vivo drug interactions involving human CYP3A. Annu Rev Pharmacol Toxicol. 1998;38:389–430. doi: 10.1146/annurev.pharmtox.38.1.389. [DOI] [PubMed] [Google Scholar]
- 11.Gorski JC, Hall SD, Jones DR, VandenBranden M, Wrighton SA. Regioselective biotransformation of midazolam by members of the human cytochrome P450 3A (CYP3A) subfamily. Biochem Pharmacol. 1994;47(9):1643–1653. doi: 10.1016/0006-2952(94)90543-6. [DOI] [PubMed] [Google Scholar]
- 12.Mäenpää J, Hall SD, Ring BJ, Strom SC, Wrighton SA. Human cytochrome P450 3A (CYP3A) mediated midazolam metabolism: The effect of assay conditions and regioselective stimulation by alpha-naphthoflavone, terfenadine and testosterone. Pharmacogenetics. 1998;8(2):137–155. [PubMed] [Google Scholar]
- 13.Ghosal A, Satoh H, Thomas PE, Bush E, Moore D. Inhibition and kinetics of cytochrome P4503A activity in microsomes from rat, human, and cdna-expressed human cytochrome P450. Drug Metab Dispos. 1996;24(9):940–947. [PubMed] [Google Scholar]
- 14.Roberts AG, et al. The structural basis for homotropic and heterotropic cooperativity of midazolam metabolism by human cytochrome P450 3A4. Biochemistry. 2011;50(50):10804–10818. doi: 10.1021/bi200924t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang RW, Newton DJ, Liu N, Atkins WM, Lu AY. Human cytochrome P-450 3A4: In vitro drug-drug interaction patterns are substrate-dependent. Drug Metab Dispos. 2000;28(3):360–366. [PubMed] [Google Scholar]
- 16.Emoto C, Murayama N, Yamazaki H. Effects of enzyme sources on midazolam 1′-hydroxylation activity catalyzed by recombinant cytochrome P450 3A4 in combination with NADPH-cytochrome P450 reductase. Drug Metab Lett. 2008;2(3):190–192. doi: 10.2174/187231208785425737. [DOI] [PubMed] [Google Scholar]
- 17.Cameron MD, et al. Cooperative binding of midazolam with testosterone and alpha-naphthoflavone within the CYP3A4 active site: A NMR T1 paramagnetic relaxation study. Biochemistry. 2005;44(43):14143–14151. doi: 10.1021/bi051689t. [DOI] [PubMed] [Google Scholar]
- 18.Hosea NA, Miller GP, Guengerich FP. Elucidation of distinct ligand binding sites for cytochrome P450 3A4. Biochemistry. 2000;39(20):5929–5939. doi: 10.1021/bi992765t. [DOI] [PubMed] [Google Scholar]
- 19.Schrag ML, Wienkers LC. Covalent alteration of the CYP3A4 active site: Evidence for multiple substrate binding domains. Arch Biochem Biophys. 2001;391(1):49–55. doi: 10.1006/abbi.2001.2401. [DOI] [PubMed] [Google Scholar]
- 20.Sevrioukova IF, Poulos TL. Current approaches for investigating and predicting cytochrome P450 3A4-ligand interactions. Adv Exp Med Biol. 2015;851:83–105. doi: 10.1007/978-3-319-16009-2_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khan KK, He YQ, Domanski TL, Halpert JR. Midazolam oxidation by cytochrome P450 3A4 and active-site mutants: An evaluation of multiple binding sites and of the metabolic pathway that leads to enzyme inactivation. Mol Pharmacol. 2002;61(3):495–506. doi: 10.1124/mol.61.3.495. [DOI] [PubMed] [Google Scholar]
- 22.Roussel F, Khan KK, Halpert JR. The importance of SRS-1 residues in catalytic specificity of human cytochrome P450 3A4. Arch Biochem Biophys. 2000;374(2):269–278. doi: 10.1006/abbi.1999.1599. [DOI] [PubMed] [Google Scholar]
- 23.Eiselt R, et al. Identification and functional characterization of eight CYP3A4 protein variants. Pharmacogenetics. 2001;11(5):447–458. doi: 10.1097/00008571-200107000-00008. [DOI] [PubMed] [Google Scholar]
- 24.Werk AN, Cascorbi I. Functional gene variants of CYP3A4. Clin Pharmacol Ther. 2014;96(3):340–348. doi: 10.1038/clpt.2014.129. [DOI] [PubMed] [Google Scholar]
- 25.Maekawa K, et al. Functional characterization of CYP3A4.16: Catalytic activities toward midazolam and carbamazepine. Xenobiotica. 2009;39(2):140–147. doi: 10.1080/00498250802617746. [DOI] [PubMed] [Google Scholar]
- 26.Kang YS, et al. The CYP3A4*18 genotype in the cytochrome P450 3A4 gene, a rapid metabolizer of sex steroids, is associated with low bone mineral density. Clin Pharmacol Ther. 2009;85(3):312–318. doi: 10.1038/clpt.2008.215. [DOI] [PubMed] [Google Scholar]
- 27.Ueng YF, Kuwabara T, Chun YJ, Guengerich FP. Cooperativity in oxidations catalyzed by cytochrome P450 3A4. Biochemistry. 1997;36(2):370–381. doi: 10.1021/bi962359z. [DOI] [PubMed] [Google Scholar]
- 28.Sevrioukova IF, Poulos TL. Pyridine-substituted desoxyritonavir is a more potent inhibitor of cytochrome P450 3A4 than ritonavir. J Med Chem. 2013;56(9):3733–3741. doi: 10.1021/jm400288z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Winn MD, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67(Pt 4):235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 4):486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]