

1. DISEASE CHARACTERISTICS
1.1 Name of the disease (synonyms)
There are several subclasses of familial partial lipodystrophy (FPLD): Type 1 FPLD (FPLD1), also known as Köbberling lipodystrophy; Type 2 FPLD (FPLD2), also known as Dunnigan variety; Type 3 FPLD (FPLD3); Type 4 FPLD (FPLD4); Type 5 FPLD (FPLD5); and Type 6 FPLD (FPLD6).
The molecular basis of FPLD1 remains to be identified.1 It is important to underline that FPLD is an expanding group of disorders associating partial lipodystrophy and metabolic complications (see section 1.9), whose classification remains to be clarified. On one hand, FPLD4, FPLD5 and FPLD6 have been described based each on only one to five independent patients. On the other hand, a missense variant was identified in AKT2 in an autosomal dominant form of partial lipodystrophy,2 but the disorder has not entered the classification of FPLD so far.
Moreover, in addition to the diseases known as FPLD in OMIM, partial lipodystrophies can be encountered in other complex syndromes with a Mendelian inheritance, as progeroid or autoinflammatory syndromes. The description of all these rare entities is beyond the scope of this clinical utility gene card, which will focus on so-called FPLD.
1.2 OMIM# of the disease
FPLD2: #151660; FPLD3: #604367; FPLD4: #613877; FPLD5: #615238; FPLD6: #615980.
1.3 Name of the analysed genes or DNA/chromosome segments
FPLD2: LMNA, FPLD3: PPARG, FPLD4: PLIN1, FPLD5: CIDEC, FPLD6: LIPE.
1.4 OMIM# of the gene(s)
LMNA: *150330; PPARG: *601487; PLIN1: *170290; CIDEC: *612120; LIPE: *151750.
1.5 Mutational spectrum
Disease-causing variants in LMNA and PPARG account for >50% of all cases of familial partial lipodystrophies. Molecular defects identified in other genes explain very few cases. There is a specific database listing molecular defects in LMNA http://www.umd.be/LMNA/, but there is no website listing variants in the other genes responsible for FPLD.
LMNA—FPLD2 is an autosomal dominant disorder. More than 85% of molecular defects identified in LMNA (NM_170707) and responsible for FPLD2 affect Arginine 482, which is a hotspot for disease-causing variants: p.(Arg482Trp) (c.1444C>T); p.(Arg482Gly) (c.1444C>G); p.(Arg482Gln) (c.1445G>A); and p.(Arg482Leu) (c.1445G>T).3, 4, 5 The great majority of other LMNA variants implicated in FPLD2 or in atypical lipodystrophies (so-called 'metabolic laminopathies'),6 corresponds to missense changes spread over the different exons of the gene. A few other molecular defects have been reported including a deletion, a duplication, a splice site and a nonsense disease-causing variant (for more details, see http://www.umd.be/LMNA/). A homozygous frameshift variant has been shown to lead to a severe lipodystrophic phenotype associated to cardiac rhythm and conduction disturbances.7 Besides FPLD, LMNA variants have been implicated in a wide spectrum of other laminopathies including Emery–Dreifuss muscular dystrophy, limb-girdle muscular dystrophy type 1B, dilated cardiomyopathy with conduction defect, Charcot–Marie–Tooth neuropathy type 2B1, mandibuloacral dysplasia, Hutchinson–Gilford Progeria syndrome, restrictive dermopathy and other atypical premature aging syndromes, as well as overlapping phenotypes.
PPARG—FPLD3 is an autosomal dominant disorder. Several dozens of disease-causing variants have been described,8 most of them corresponding to missense changes. A few other molecular defects have been reported: nonsense variants, deletions leading to frameshifts, as well as a variant in the promoter. Most of them, affecting the DNA or ligand-binding domains of the transcription factor, are dominant-negative variants. Notably, one compound heterozygote for PPARG variants was shown to present congenital generalized lipodystrophy.9
PLIN1—FPLD4 is an autosomal dominant disorder. Two deletions and one splice site variant, all resulting in frameshifts, have been identified.10, 11 Two of them have been shown to alter physiological functions of perilipin.12
CIDEC—FPLD5 is an autosomal recessive disorder. One nonsense variant disrupting the function of the protein has been reported to date.13
LIPE—FPLD6 is an autosomal recessive disorder. Two deletions resulting in frameshifts have been reported in that gene.14, 15 Functional cellular consequences were reported for one of them.14
1.6 Analytical methods
Sanger sequencing of PCR products corresponding to coding regions and conserved splice sites is performed on a routine basis. Next-generation sequencing, including gene-targeted and whole-exome sequencing approaches, is also used.
1.7 Analytical validation
There are several steps in the analytical validation process:
Sequencing of both DNA strands (forward and reverse) is performed.
When the genetic test is positive, a search of the molecular defects is recommended on a second independent sample from the patient. More generally, identification of the same variant in the affected proband's relatives provides additional confirmation of the result. In recessive forms, when two heterozygous variants or a homozygous variant are found, testing of the patients' parents is recommended to confirm that the defect is biallelic. In dominant forms when a molecular defect is identified, testing of the patient's parents is proposed with genetic counselling.
Newly discovered variants may be searched for in databases listing benign and pathogenic variations, such as dbSNP, HGMD, 1000 Genomes, EVS and ExAC. Pathogenicity of variants can also be tested by functional studies or in silico prediction methods using SIFT (La Jolla, CA, USA), Polyphen-2 (Boston, MA, USA) and Mutation Taster softwares (Berlin, Germany).
Notably, there is to date no external quality assessment dedicated to this specific set of genes proposed by the European Molecular Genetics Quality Network.
1.8 Estimated frequency of the disease (incidence at birth ('birth prevalence') or population prevalence). If known to be variable between ethnic groups, please report)
The population prevalence has been estimated to be about 1 in 100 000.16 FPLD2 due to LMNA variants is the most common variety, but less than 500 patients were reported worldwide. Other clinical entities constitute rarities: about 30 independent families or patients have been reported to harbour disease-causing variants in PPARG, five in PLIN1, one in CIDEC and two in LIPE.17
1.9 Diagnostic setting

Comment: FPLD are characterized by subcutaneous fat loss, mostly in the limbs and extremities resulting in a peripheral muscular appearance. Men are more difficult to diagnose. In certain subclasses, increased subcutaneous fat accumulation has been reported in the face, neck and intra-abdominal region. Lipodystrophic manifestations, which usually appear around puberty, or during adulthood, progressively associate with numerous complications (eg, insulin resistance with acanthosis nigricans, diabetes mellitus, hypertriglyceridaemia, acute pancreatitis, hepatic steatosis, ovarian hyperandrogenism and polycystic ovaries in women). In addition, other symptoms can be associated with specific variants (see Table below).
Main clinical and laboratory characteristics of FPLD subclasses.
| FPLD type (gene involved) | Transmission | Clinical onset of disease | Clinical lipodystrophy | Common signs to all FPLD | Signs of metabolic severity | Possible additional signs |
|---|---|---|---|---|---|---|
| FPLD1 (unknown) | Unknown | Childhood or adulthood Only described in women | Lipoatrophy of buttocks and lower limbs Truncal obesity | FPLD1 to 6: Insulin resistance, diabetes, non-alcoholic fatty liver disease, high triglycerides, low HDL-cholesterol | FPLD1 to 5: Acanthosis nigricans, muscular appearance, prominent veins in limbs, acute pancreatitis, high blood pressure ±hyperandrogenism, polycystic ovaries, atherosclerosis, low leptin and adiponectin levels | — |
| FPLD2 (LMNA) | Autosomal dominant | Around puberty | Lipoatrophy of buttocks, limbs and trunk Accumulation of cervicofacial subcutaneous and/or intra-abdominal fat Subcutaneous lipomas | Skeletal and cardiac muscular dystrophy and/or premature ageing (mainly in patients with non-Arg482 mutations) | ||
| FPLD3 (PPARG) | Autosomal dominant | Adulthood | Lipoatrophy of buttocks and limbs Accumulation of intra-abdominal fat | Very high blood pressure Severe generalized lipoatrophic syndrome with renal insufficiency (one patient described with a biallelic mutation) | ||
| FPLD4 (PLIN1) | Autosomal dominant | Childhood or adulthood (five unrelated families described) | Lipoatrophy of buttocks and limbs | — | ||
| FPLD5 (CIDEC) | Autosomal recessive | Childhood (one case described) | Lipoatrophy of buttocks and limbs | Ketosis-prone diabetes | ||
| FPLD6 (LIPE) | Autosomal recessive | Adulthood (two unrelated families describeds) | Decreased lower-extremity fat Increased visceral fat | — | Muscular dystrophy |
2. TEST CHARACTERISTICS
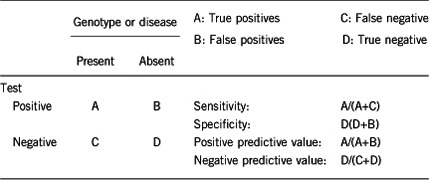
2.1 Analytical sensitivity
(proportion of positive tests if the genotype is present)
Depending on the quality of sequencing methods, the analytical sensitivity is close to 100% for germline variants located in coding regions, and flanking intronic sequences. Single-nucleotide polymorphisms (SNPs) within PCR primer-binding sites can result in preferential amplification of a single allele and constitute a rare cause of missed variant, so that careful checking of primer-binding sites for SNPs is essential. Notably, potential deep intronic variants, variants in promoters, large deletions and duplications would not be detected by Sanger sequencing. The development of next-generation sequencing in routine diagnosis will allow the detection of additional molecular defects including deletions/duplications missed by Sanger sequencing and potentially variants localized in previously uncovered regions.
2.2 Analytical specificity
(proportion of negative tests if the genotype is not present)
The analytical specificity is close to 100%. The analytical validation described above should avoid false positive tests.
2.3 Clinical sensitivity
(proportion of positive tests if the disease is present)
The clinical sensitivity can be dependent on variable factors such as age, sex or family history. In such cases a general statement should be given, even if a quantification can only be made case by case.
The clinical sensitivity is ~50%. Variants in LMNA and PPARG account for ~50% of all cases of partial lipodystrophic syndromes. Very few cases are explained by other known genes. This strongly argues for the involvement of other so far unknown disease-causing genes. In addition, when genetic testing is negative in a patient with symptoms evocative of FPLD, differential diagnoses can be considered (please see 3.1).
2.4 Clinical specificity
(proportion of negative tests if the disease is not present)
The clinical specificity can be dependent on variable factors such as age or family history. In such cases a general statement should be given, even if a quantification can only be made case by case.
A precise quantification is difficult. Since metabolic complications are usually delayed in these diseases and the clinical lipodystrophy can be difficult to diagnose in some patients (for example in young and/or lean patients and men), familial genetic screening can lead to the detection of affected individuals before any clinical expression of the disease.
2.5 Positive clinical predictive value
(life-time risk of developing the disease if the test is positive)
The positive clinical predictive value is high but difficult to quantify precisely as a whole, since it depends on the molecular defect identified. To the best of our knowledge, incomplete penetrance has not been described in typical forms of FPLD2 linked to the genetic alterations of Arg482 in LMNA, although the disease phenotype could be mild in some patients, especially in lean men. Nevertheless, it has been reported in several rare atypical forms.7, 18, 19, 20
2.6 Negative clinical predictive value
(probability of not developing the disease if the test is negative)
Assume an increased risk based on family history for a non-affected person. Allelic and locus heterogeneity may need to be considered.
Index case in that family had been tested:
The negative clinical predictive value is nearly 100%, although a negative test does not exclude the possibility of developing a FPLD due to molecular defects in other genes that were not tested.
Index case in that family had not been tested:
Genetic testing for a clinically unaffected individual is not indicated in this situation. It would only be undertaken if a variant in a gene responsible for FPLD has been identified in the proband.
3. CLINICAL UTILITY
3.1 (Differential) diagnostics: The tested person is clinically affected
(To be answered if in 1.9 'A' was marked)
3.1.1 Can a diagnosis be made other than through a genetic test?

Genetic testing helps to confirm the clinical diagnosis. Indeed, FPLD should be considered in differential diagnosis of patients presenting with familial insulin-resistant diabetes with android fat distribution or rare syndromes of severe insulin resistance with paucity of fat (eg, insulin resistance syndrome due to INSR variants or progeroid syndromes).
3.1.2 Describe the burden of alternative diagnostic methods to the patient
In typical cases, clinical diagnosis is strongly suggested by combining familial history, physical examination, biochemical results and imagery. There are no invasive procedures for the patient.
3.1.3 How is the cost effectiveness of alternative diagnostic methods to be judged?
Clinical investigations, biochemical assays and imaging are actually used to get an accurate clinical evaluation of the patients, which is necessary for their proper management and follow-up. This does not exclude genetic testing and vice versa. Both diagnostic procedures add to the global picture.
3.1.4 Will disease management be influenced by the result of a genetic test?

3.2 Predictive Setting: The tested person is clinically unaffected but carries an increased risk based on family history
(To be answered if in 1.9 'B' was marked).
3.2.1 Will the result of a genetic test influence lifestyle and prevention?
If the test result is 'positive' (please describe):
A regular clinical and biological follow-up of the person is required, including screening for glucose tolerance abnormalities, dyslipidemia, non-alcoholic fatty liver disease and atherosclerosis. Preventive lifestyle and dietary measures can delay the metabolic complications, including acute pancreatitis due to severe hypertriglyceridemia. In addition, the use of metformin should be considered in patients with severe insulin resistance to prevent diabetes. Oral combined contraceptives including ethinyl-estradiol should be contraindicated in women with a positive genetic test. In addition, screening for cardiac rhythm and/or conduction disturbances are recommended when a disease-causing variant is identified in LMNA, allowing primary prevention of severe cardiac complications.
If the test result is negative (please describe):
The individual is not genetically predisposed to FPLD and the risk of developing the disease is reduced to that of the general population.
3.2.2 Which options in view of lifestyle and prevention does a person at-risk have if no genetic test has been done (please describe)?
All at-risk individuals can be followed-up in the same way, whether they have had the genetic test or not.
3.3 Genetic risk assessment in family members of a diseased person
(To be answered if in 1.9 'C' was marked).
3.3.1 Does the result of a genetic test resolve the genetic situation in that family?
Usually yes, by testing the potential family members carrying molecular defect(s).
3.3.2 Can a genetic test in the index patient save genetic or other tests in family members?
Yes.
3.3.3 Does a positive genetic test result in the index patient enable a predictive test in a family member?
Yes.
3.4 Prenatal diagnosis
(To be answered if in 1.9 'D' was marked).
3.4.1 Does a positive genetic test result in the index patient enable a prenatal diagnosis?
Yes. Prenatal diagnosis should be considered case by case. It could be proposed in rare forms of FPLD2 associated with signs of severity such as skeletal and muscular dystrophy and/or premature ageing, in rare autosomal recessive forms of FPLD3, in which severe generalized lipoatrophic syndrome and renal insufficiency have been described, or in other severe situations.
4. IF APPLICABLE, FURTHER CONSEQUENCES OF TESTING
Please assume that the result of a genetic test has no immediate medical consequences. Is there any evidence that a genetic test is nevertheless useful for the patient or his/her relatives? (Please describe).
Acknowledgments
This work was supported by EuroGentest2 (Unit 2: 'Genetic testing as part of health care'), a Coordination Action under FP7 (Grant Agreement Number 261469) and the European Society of Human Genetics. We thank Dr David Savage (Wellcome Trust-Medical Research Council Institute of Metabolic Science, University of Cambridge, UK) for having carefully read and amended the manuscript.
Footnotes
The authors declare no conflict of interest.
References
- Herbst KL, Tannock LR, Deeb SS et al: Köbberling type of familial partial lipodystrophy: an underrecognized syndrome. Diabetes Care 2003; 26: 1819–1824. [DOI] [PubMed] [Google Scholar]
- George S, Rochford JJ, Wolfrum C et al: A family with severe insulin resistance and diabetes due to a mutation in AKT2. Science 2004; 304: 1325–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shackleton S, Lloyd DJ, Jackson SN et al: LMNA, encoding lamin A/C, is mutated in partial lipodystrophy. Nat Genet 2000; 24: 153–156. [DOI] [PubMed] [Google Scholar]
- Speckman RA, Garg A, Du F et al: Mutational and haplotype analyses of families with familial partial lipodystrophy (Dunnigan variety) reveal recurrent missense mutations in the globular C-terminal domain of lamin A/C. Am J Hum Genet 2000; 66: 1192–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigouroux C, Magre J, Vantyghem MC et al: Lamin A/C gene: sex-determined expression of mutations in Dunnigan-type familial partial lipodystrophy and absence of coding mutations in congenital and acquired generalized lipoatrophy. Diabetes 2000; 49: 1958–1962. [DOI] [PubMed] [Google Scholar]
- Decaudain A, Vantyghem MC, Guerci B et al: New metabolic phenotypes in laminopathies: LMNA mutations in patients with severe metabolic syndrome. J Clin Endocrinol Metab 2007; 92: 4835–4844. [DOI] [PubMed] [Google Scholar]
- Le Dour C, Schneebeli S, Bakiri F et al: A homozygous mutation of prelamin-A preventing its farnesylation and maturation leads to a severe lipodystrophic phenotype: new insights into the pathogenicity of nonfarnesylated prelamin-A. J Clin Endocrinol Metab 2011; 96: E856–E862. [DOI] [PubMed] [Google Scholar]
- Miehle K, Porrmann J, Mitter D et al: Novel peroxisome proliferator-activated receptor gamma mutation in a family with familial partial lipodystrophy type 3. Clin Endocrinol (Oxf) 2015; 84: 141–148. [DOI] [PubMed] [Google Scholar]
- Dyment DA, Gibson WT, Huang L, Bassyouni H, Hegele RA, Innes AM: Biallelic mutations at PPARG cause a congenital, generalized lipodystrophy similar to the Berardinelli-Seip syndrome. Eur J Med Genet 2014; 57: 524–526. [DOI] [PubMed] [Google Scholar]
- Gandotra S, Le Dour C, Bottomley W et al: Perilipin deficiency and autosomal dominant partial lipodystrophy. N Engl J Med 2011; 364: 740–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozusko K, Tsang VH, Bottomley W et al: Clinical and molecular characterization of a novel PLIN1 frameshift mutation identified in patients with familial partial lipodystrophy. Diabetes 2015; 64: 299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandotra S, Lim K, Girousse A, Saudek V, O'Rahilly S, Savage DB: Human frame shift mutations affecting the carboxyl terminus of perilipin increase lipolysis by failing to sequester the adipose triglyceride lipase (ATGL) coactivator AB-hydrolase-containing 5 (ABHD5). J Biol Chem 2011; 286: 34998–35006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio-Cabezas O, Puri V, Murano I et al: Partial lipodystrophy and insulin resistant diabetes in a patient with a homozygous nonsense mutation in CIDEC. EMBO Mol Med 2009; 1: 280–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert JS, Yerges-Armstrong LM, Horenstein RB et al: Null mutation in hormone-sensitive lipase gene and risk of type 2 diabetes. N Engl J Med 2014; 370: 2307–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farhan SM, Robinson JF, McIntyre AD et al: A novel LIPE nonsense mutation found using exome sequencing in siblings with late-onset familial partial lipodystrophy. Can J Cardiol 2014; 30: 1649–1654. [DOI] [PubMed] [Google Scholar]
- Hegele RA, Pollex RL: Genetic and physiological insights into the metabolic syndrome. Am J Physiol Regul Integr Comp Physiol 2005; 289: R663–R669. [DOI] [PubMed] [Google Scholar]
- Garg A: Clinical review#: Lipodystrophies: genetic and acquired body fat disorders. J Clin Endocrinol Metab 2011; 96: 3313–3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muschke P, Kolsch U, Jakubiczka S, Wieland I, Brune T, Wieacker P: The heterozygous LMNA mutation p.R471G causes a variable phenotype with features of two types of familial partial lipodystrophy. Am J Med Genet A 2007; 143 A: 2810–2814. [DOI] [PubMed] [Google Scholar]
- Rankin J, Auer-Grumbach M, Bagg W et al: Extreme phenotypic diversity and nonpenetrance in families with the LMNA gene mutation R644C. Am J Med Genet A 2008; 146 A: 1530–1542. [DOI] [PubMed] [Google Scholar]
- Andre P, Schneebeli S, Vigouroux C, Lascols O, Schaaf M, Chevalier P: Metabolic and cardiac phenotype characterization in 37 atypical Dunnigan patients with nonfarnesylated mutated prelamin A. Am Heart J 2015; 169: 587–593. [DOI] [PubMed] [Google Scholar]