

1. Disease characteristics
1.1 Name of the disease (synonyms)
16p12.1 microdeletion (hg18/NCBI36). Please note that the updated release of the human reference genome (hg19/GRCh37) annotates this region as 16p12.2.
1.2 OMIM# of the disease
136570.
1.3 Name of the analysed genes or DNA/chromosome segments
Chromosome 16p12.2 (hg19 chr16:g.(?_ 21950000)_(22470000_?)del).
1.4 OMIM# of the gene(s)
UQCRC2 (*191329); EEF2K (*606968); CDR2 (*117340); PDZD9 (no OMIM entry); C16orf52 (no OMIM entry); VWA3A (no OMIM entry); and POLR3E (no OMIM entry).
1.5 Mutational spectrum
The 16p12.2 microdeletion is ~520 kbp in size, located in the short arm of chromosome 16. The genomic rearrangement is mediated by highly identical (>99.5%) segmental duplications (68 kbp) flanking this region. Although the deletion break points are difficult to map owing to the high complexity of the flanking segmental duplications, these are usually located at hg19 chr16:g.(?_ 21950000)_(22470000_?).1, 2 Seven annotated genes or transcripts are located within the unique region flanked by segmental duplications and are as follows: UQCRC2, PDZD9, C16orf52, VWA3A, EEF2K, POLR3E and CDR2. The underlying mechanism for how the heterozygosity of one or more genes in 16p12.2 results in disease is unknown. The seven genes deleted in 16p12.2 have been studied to a variable extent. UQCRC2 is a fundamental protein for the assembly of the mitochondrial respiratory chain complex. CDR2 is an onco-neural antigen and has been associated with cerebellar ataxia.3 POLR3E is a component of the RNA polymerase that synthesizes small RNAs.4 EEF2K is a kinase involved in the regulation of protein synthesis elongation, and has been recently associated with learning and memory, synaptic plasticity and the short-term antidepressant action of ketamine.5, 6, 7, 8 EEF2K-knockout mice have been described to have defects in the reproductive system, and those carrying a homozygous-inactivating mutation leading to 0.5% residual activity showed learning memory deficits and altered brain activity.5 Limited functional information is available on PDZD9, C16orf52 and VWA3A. Even though the basic functionality has been reported for some genes within the region, their exact role towards pathogenicity owing to the 16p12.2 deletion is not known, highlighting the need for in-depth molecular studies of these genes.
1.6 Analytical methods
The presence of the deletion can be detected by genome-wide or targeted approaches that determine copy number in the region. Recent studies using genome-wide technologies such as comparative genomic hybridization and SNP microarrays have enabled discovery of this microdeletion, which otherwise would have been missed by lower resolution assays such as chromosomal banding techniques. The accuracy of estimation of the deletion size depends on the type of microarray used (targeted or whole genome) and the density of probes in the region.
1.7 Analytical validation
Targeted assays such as real-time quantitative PCR, fluorescence in situ hybridization and multiplex ligation-dependent probe amplification can be used for confirming the deletion and for assessment of its transmission within the family.
1.8 Estimated frequency of the disease (Incidence at birth (‘birth prevalence') or population prevalence
If known to be variable between ethnic groups, please report):
The 16p12.2 deletion is associated with incomplete penetrance and variable expressivity, making it difficult to estimate the exact prevalence of the deletion. Girirajan et al.9 reported the deletion in 42 out of 21 127 individuals with developmental delay and intellectual disability who were referred for clinical genetic testing, compared with 8 out of 14 839 control individuals. By comparing the frequency of individuals with 16p12.2 deletion referred for clinical genetic testing with microarrays, the prevalence of this deletion is estimated to be similar (1:15 000 live births) to that of Smith–Magenis syndrome.9, 10 However, this estimate does not take into account mildly-affected and unaffected individuals who carry the 16p12.2 deletion and are not referred to clinics.11
1.9 Diagnostic setting

Comment:
Given the variable expressivity of the microdeletion and the incomplete penetrance, prenatal diagnosis is possible in families carrying the deletion, but the clinical outcomes cannot be predicted. Pre-implantation genetic diagnosis can also be performed, and the same considerations apply.
2. Test characteristics
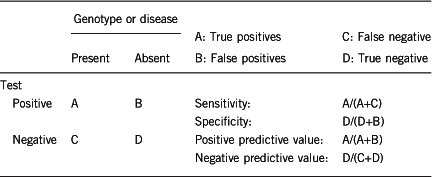
2.1 Analytical sensitivity
(proportion of positive tests if the genotype is present)
Approximately 100% using the analytical methods described.
2.2 Analytical specificity (proportion of negative tests if the genotype is not present)
Approximately 100% using the analytical methods described.
2.3 Clinical sensitivity (proportion of positive tests if the disease is present)
The clinical sensitivity can be dependent on variable factors such as age or family history. In such cases, a general statement should be given, even if a quantification can only be made case by case.
The proportion of positive tests for the deletion, given the phenotype, is not 100% owing to the extensive genetic heterogeneity of neurodevelopmental phenotypes. The 16p12.2 microdeletion does not lead to a recognizable syndrome. The deletion is associated with incomplete penetrance and extensive phenotypic heterogeneity. The phenotypic heterogeneity can potentially be explained by the genetic background of the deletion carrier, including the presence of second-site mutations elsewhere in the genome.9, 12 The frequently described phenotypes in deletion carriers include developmental delay, speech delay, craniofacial and skeletal features, growth retardation, microcephaly, congenital heart defect, epilepsy, psychiatric and behavioural disorders, and hypotonia.9 An increased risk for schizophrenia has also been documented.13
2.4 Clinical specificity (proportion of negative tests if the disease is not present)
The clinical specificity can be dependent on variable factors such as age or family history. In such cases, a general statement should be given, even if a quantification can only be made case by case.
Variable. The incomplete penetrance and the extensive range of phenotypes associated with the deletion preclude a 100% clinical specificity. Deletion carriers may be mildly affected or unaffected.
2.5 Positive clinical predictive value (life-time risk of developing the disease if the test is positive)
This is variable owing to the incomplete penetrance and phenotypic heterogeneity of the deletion. The risk for developing a phenotype is also contingent upon the presence of a positive family history of neurodevelopmental/psychiatric phenotype and the genetic background.
2.6 Negative clinical predictive value (probability of not developing the disease if the test is negative)
Assume an increased risk based on family history for a non-affected person. Allelic and locus heterogeneity may need to be considered.
Index case in that family had been tested:
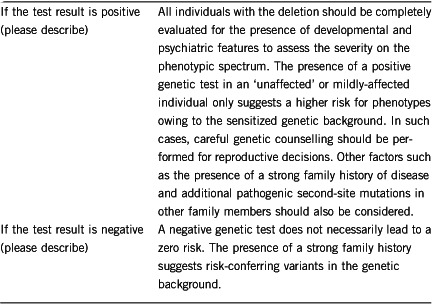
The probability of not developing a disease if the test is negative for the deletion is not 100%. Not finding the deletion in the proband does not preclude the index case from manifesting a phenotype (for example, congenital anomalies or neuropsychiatric disease later in life). Other factors such as locus and allelic heterogeneity, as well as positive family history, can increase the risk of manifestation of the phenotype.
Index case in that family had not been tested:
The probability of not developing a disease if the index case has not been tested for the deletion is not 100%. Other factors such as locus and allelic heterogeneity, as well as positive family history can also increase the risk of manifestation of the phenotype.
3. Clinical utility
3.1 (Differential) diagnostics: The tested person is clinically affected
(To be answered if in 1.9 ‘A' was marked).
3.1.1 Can a diagnosis be made other than through a genetic test?

3.1.2 Describe the burden of alternative diagnostic methods to the patient
Not applicable.
3.1.3 How is the cost effectiveness of alternative diagnostic methods to be judged?
Not applicable.
3.1.4 Will disease management be influenced by the result of a genetic test?

3.2 Predictive Setting: The tested person is clinically unaffected but carries an increased risk based on family history
(To be answered if in 1.9 ‘B' was marked).
3.2.1 Will the result of a genetic test influence lifestyle and prevention?
3.2.2 Which options in view of lifestyle and prevention does a person at-risk have if no genetic test has been done (please describe)?
Not applicable.
3.3 Genetic risk assessment in family members of a diseased person
(To be answered if in 1.9 ‘C' was marked).
3.3.1 Does the result of a genetic test resolve the genetic situation in that family?
Once the deletion has been identified in the index case, genetic testing of the parents should be offered for genetic counselling. Although not reported in the literature, gonadal mosaicism is another possibility. Because of the incomplete penetrance of the deletion, genetic testing should be offered to siblings to evaluate a possibility of gonadal mosaicism. Inheritance of the deletion from a parent would mandate genetic testing in other family members.
3.3.2 Can a genetic test in the index patient save genetic or other tests in family members?
No. Even under the assumption of a de novo occurrence of the deletion in the family, siblings should be offered testing to evaluate a possibility of gonadal mosaicism.
3.3.3 Does a positive genetic test result in the index patient enable a predictive test in a family member?
Yes, only when deletion in the index case is inherited. The identification of an inherited deletion can be followed up by genetic testing of other family members.
3.4 Prenatal diagnosis
(To be answered if in 1.9 ‘D' was marked)
3.4.1 Does a positive genetic test result in the index patient enable a prenatal diagnosis?
Yes, only when the deletion in index case is inherited from a parent. However, the phenotypic outcome cannot be predicted owing to the variable expressivity and incomplete penetrance of the deletion.
4. If applicable, further consequences of testing
Please assume that the result of a genetic test has no immediate medical consequences. Is there any evidence that a genetic test is nevertheless useful for the patient or his/her relatives? (Please describe).
Yes. Identification of the deletion in the patient may have implications for the patient and relatives' reproductive decisions, and may warrant prenatal diagnosis. The deletion is inherited in autosomal dominant manner, and in 95% of cases, it is inherited from a parent. However, the extensive phenotypic variability of 16p12.2 microdeletion precludes accurate prediction of the clinical outcome.
Acknowledgments
This work was supported by EuroGentest2 (Unit 2: ‘Genetic testing as part of health care'), a Coordination Action under FP7 (Grant Agreement Number 261469) and the European Society of Human Genetics.
Footnotes
The authors declare no conflict of interest.
References
- Antonacci F, Kidd JM, Marques-Bonet T et al: A large and complex structural polymorphism at 16p12.1 underlies microdeletion disease risk. Nat Genet 2010; 42: 745–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girirajan S, Eichler EE: Phenotypic variability and genetic susceptibility to genomic disorders. Hum Mol Genet 2010; 19: R176–R187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblum M, Posner J: Paraneoplastic cerebellar degeneration. IA clinical analysis of 55 anti-Yo antibody-positive patients. Neurology 1992; 42: 1931–1931. [DOI] [PubMed] [Google Scholar]
- Dong Z, Bell LR: SIN, a novel Drosophila protein that associates with the RNA binding protein sex-lethal. Gene 1999; 237: 421–428. [DOI] [PubMed] [Google Scholar]
- Gildish I, Manor D, David O et al: Impaired associative taste learning and abnormal brain activation in kinase-defective eEF2K mice. Learn Mem 2012; 19: 116–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCamphill PK, Farah CA, Anadolu MN, Hoque S, Sossin WS: Bidirectional regulation of eEF2 phosphorylation controls synaptic plasticity by decoding neuronal activity patterns. J Neurosci 2015; 35: 4403–4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteggia LM, Gideons E, Kavalali ET: The role of eukaryotic elongation factor 2 kinase in rapid antidepressant action of ketamine. Biol Psychiatry 2013; 73: 1199–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S, Park JM, Kim S et al: Elongation factor 2 and fragile X mental retardation protein control the dynamic translation of Arc/Arg3.1 essential for mGluR-LTD. Neuron 2008; 59: 70–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girirajan S, Rosenfeld JA, Cooper GM et al: A recurrent 16p12.1 microdeletion supports a two-hit model for severe developmental delay. Nat Genet 2010; 42: 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld JA, Coe BP, Eichler EE, Cuckle H, Shaffer LG: Estimates of penetrance for recurrent pathogenic copy-number variations. Genet Med 2012; 15: 478–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girirajan S, Moeschler J, Rosenfeld J: 16p12.2 microdeletion. In: Pagon RA, Adam MP, Ardinger HH et al. (eds): GeneReviews [Internet]. Seattle: University of Washington, pp 1993–2016..
- Girirajan S, Rosenfeld JA, Coe BP et al: Phenotypic heterogeneity of genomic disorders and rare copy-number variants. N Engl J Med 2012; 367: 1321–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees E, Walters JT, Chambert KD et al: CNV analysis in a large schizophrenia sample implicates deletions at 16p12.1 and SLC1A1 and duplications at 1p36.33 and CGNL1. Hum Mol Genet 2014; 23: 1669–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]