

Abstract
Much progress has been made in understanding the pathophysiology and treatment of atypical hemolytic uremic syndrome (aHUS). Plasma therapy is the mainstay of treatment for aHUS. The availability of the first effective anti-complement therapeutic agent, eculizumab, has dramatically changed the outlook of this disease. However, its use in clinical practice raises important questions, such as who should receive the drug, when to start such therapy, and is it safe to stop treatment once the disease is controlled. We describe here for the 1st time in India, use of eculizumab in a 12-year-old boy with aHUS. We also describe in this report challenges faced in procuring the drug, and an ideal, evidence-based method of treating aHUS in children.
Keywords: Eculizumab, hemolytic uremic syndrome, plasma exchange, soliris
Introduction
Atypical hemolytic uremic syndrome (aHUS) is a rare, and life threatening, disease. In most cases, it is caused by chronic, uncontrolled activation of the complement system. The first line of treatment for aHUS was plasmapheresis until September 2011, when the Food and Drug Administration approved eculizumab for the treatment of pediatric and adult patients with aHUS. Eculizumab has been available in the United States since 2007 for the treatment of paroxysmal nocturnal hemoglobinuria. This additional indication makes eculizumab the first drug to be approved for the treatment of aHUS.[1,2,3,4] We describe here for the first time in India, use of eculizumab in a 12-year-old boy with aHUS, and the challenges faced.
Case Report
A 12-year-old Caucasian boy was admitted in September 2014, with complaints of pain abdomen for 4 days, petechial spots over face for 3 days, cola-colored urine and decreased urine output for 1 day. On evaluation, the child had hypertension (blood pressure: 140/90), anemia (hemoglobin = 9 g/dl), severe thrombocytopenia (platelet count 20,000/mm3), acute kidney injury (blood urea 183 mg/dl; serum creatinine 4 mg/dl), lactate dehydrogenase 10,000 IU/ml, and schistocytes on peripheral smear suggestive of hemolysis. Malarial antigen, dengue antigen, widal, leptospiral serology, and all cultures were negative. There was no history of diarrhea, dysentery, or fever prior to this illness.
A diagnosis of aHUS was made and plasma exchange (PE) initiated within 24 h of admission. Following three daily PEs, his hematuria, platelet count, serum creatinine, and urine output improved [Figure 1]. Initially, the PE was done daily for 6 days, followed by alternate days for 2 weeks, then thrice a week for 2 weeks, according to the European Pediatric Study Group guidelines for HUS.[1] The child was discharged on day 7 of admission, and the rest of the PEs were done on an out-patient basis. A complement factor assay was sent to a collaborative laboratory in France which was found to be normal [Table 1]. Since it was an unexplained aHUS, the need for long-term PEs or eculizumab and a complete genetic sequencing workup was discussed with the family. The treatment could only be stopped once the complete genetic work-up was negative. Since the child was an American citizen, the American Embassy, Insurance agency and Alexion Pharmaceuticals, Germany were contacted. The drug is not available in India and a special acquiring permission from the drug controller of India followed by custom duty exemption was taken. The child was already immunized for meningococci according to Centers for Disease Control and Prevention schedule.
Figure 1.
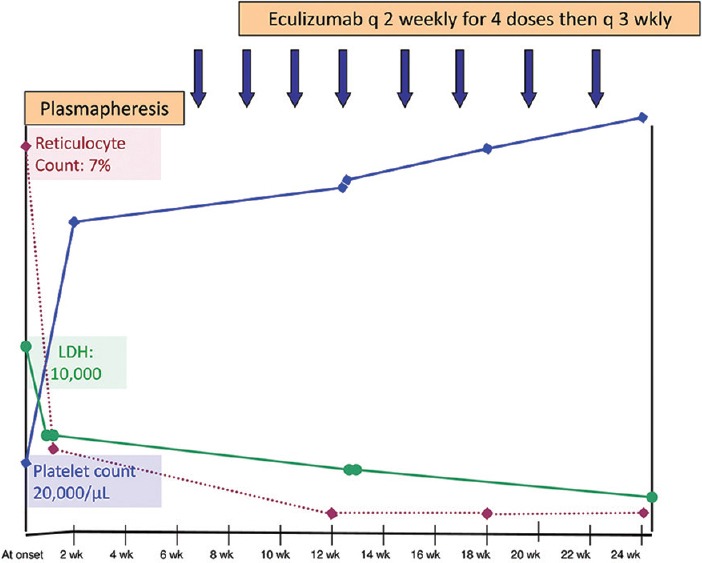
Trend of hemolytic parameters with plasma exchanges and eculizumab
Table 1.
Results of the complement exploration performed in the family members

Subsequently, the child was started on eculizumab (Soliris) 1200 mg q 2 weekly, along with penicillin prophylaxis for meningococci. After three doses, the child developed leukopenia (total leukocyte count 3500/mm3 and absolute neutrophil count 800/mm3). The child was put on cotrimoxazole and acyclovir prophylaxis, and the dose interval was increased to q 3 weekly. The child also developed hallucinations after three doses of eculizumab, which could be attributed to Intensive Care Unit psychosis/drug, which improved on adding risperidone and increasing the drug interval. The child was continued on q 3 weekly eculizumab, with normal renal functions, and a close monitoring for CH50 and hemolysis. A trough level of eculizumab at 3-week dose interval was 135.8 mcg/mL (normal trough used in paroxysmal nocturnal hemoglobinuria 100 mcg/mL).
The gene sequencing results were available in February 2015 and it was normal [Table 1]. It was discussed with the family that the possibility of finding a major genetic defect has been ruled out, though there is still a possibility of relapse. Hence, eculizumab was stopped in April 2015, in view of the absence of any symptoms and normal complement factors and gene sequencing.
Currently the child is 9 months off eculizumab and is doing well, with a normal renal function, no proteinuria, and a normal urine examination with no relapses. The child is under regular follow-up.
Discussion
Despite poor quality evidence, the mainstay of treatment for aHUS was plasma therapy till 2009. The outcome for children with aHUS is poor, and because of the rarity of these disorders, clinical experience is scanty. The guideline for the investigation and initial therapy in aHUS published by the European Pediatric Study Group for HUS in 2009, based on anecdotal case reports, retrospective series and expert consensus, advocated early, frequent, and high volume PEs.[1,2,3] The recently published audit of this guideline indicated considerable morbidity associated with plasma therapy in children.[2] Recently, eculizumab has been proposed as the first-line treatment for aHUS, to avoid PE and the complications of central venous double lumen catheters. When possible, eculizumab treatment should be initiated within 24–48 h of onset or admission. If eculizumab is not (or not immediately) available, PE (or plasma infusions if PE is not possible) should be started as recommended in the 2009 guideline.[1,4] The ideal way of treating aHUS has been highlighted in Figure 2. We switched our patient to maintenance eculizumab after the drug was procured from Germany after getting all the necessary legal permissions.
Figure 2.

Current evidence-based management of atypical hemolytic uremic syndrome (Adapted from Loirat et al.[4])
Eculizumab is a monoclonal antibody that binds to the complement protein C5, inhibiting its cleavage to C5a and C5b. This prevents circulation of the pro-inflammatory C5a peptide and generation of the cytotoxic terminal complement complex C5b-9 (referred to as the membrane attack complex [MAC]).[4,5]
Eculizumab (Soliris®, Alexion) is available through a restricted access program. It is supplied in 300 mg single-use vials. The most common adverse effects reported by the 37 adults and adolescents receiving eculizumab in the two prospective aHUS studies were hypertension (in 35% of patients), headache (30%), anemia (24%), leukopenia (16%), diarrhea (32%), vomiting (22%), and nausea (19%). Our patient has significant leukopenia requiring increasing the dose interval and adding cotrimoxazole and acyclovir prophylaxis. The reduction in MAC formation produced by eculizumab eliminates the body's defense mechanisms against Neisseria meningitidis under normal conditions. Patients should be immunized with meningococcal vaccine at least 2 weeks prior to administering eculizumab unless the risks of delaying therapy outweigh the risk of a meningococcal infection.[5]
The risk of relapse in patients with aHUS is influenced by the genetic background, and the interval between flares is extremely difficult to predict. The issue of the optimal duration of eculizumab treatment has not yet been properly addressed. The European Medicines Agency has approved life-long eculizumab treatment for patients with aHUS.[6] However, the high cost of the drug and the uncertainties surrounding the natural history of aHUS in patients for whom eculizumab prevented the progression to end-stage renal disease, raise the question of whether life-long treatment is warranted for all patients with aHUS. Given that the natural history of aHUS differs depending on the underlying genetic abnormalities, treatments could be tailored on the basis of an individual's complement genetics. Life-long treatment may be appropriate in patients with aHUS who have mutations associated with poor outcomes (e.g. CFH or C3/CFB gain-of-function mutations).[6,7] We also carefully planned to stop eculizumab in our patient, since he had no major genetic mutations, with a normal complement panel.
There is a need to completely evaluate all patients with aHUS for complement abnormalities and antibodies to factor H. These patients should get early PE/eculizumab. The nonavailability of the drug in India raises ethical concerns. HUS being a common cause of acute kidney injury in children in India should get timely and appropriate management with lifesaving eculizumab or PEs.[8] To improve the availability of the drug in Indian market, and subsequently decrease the cost of drug, collaboration among medical experts and health authorities must occur in order to implement a feasible plan of action.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgements
We thank Dr. Carla Nester, University of Iowa, USA and Dr. Craig B Langman, MD, Head, Kidney Diseases, Feinberg School of Medicine, Chicago IL, USA for the help in the management of the patient.
References
- 1.Ariceta G, Besbas N, Johnson S, Karpman D, Landau D, Licht C, et al. Guideline for the investigation and initial therapy of diarrhea-negative hemolytic uremic syndrome. Pediatr Nephrol. 2009;24:687–96. doi: 10.1007/s00467-008-0964-1. [DOI] [PubMed] [Google Scholar]
- 2.Johnson S, Stojanovic J, Ariceta G, Bitzan M, Besbas N, Frieling M, et al. An audit analysis of a guideline for the investigation and initial therapy of diarrhea negative (atypical) hemolytic uremic syndrome. Pediatr Nephrol. 2014;29:1967–78. doi: 10.1007/s00467-014-2817-4. [DOI] [PubMed] [Google Scholar]
- 3.Legendre CM, Licht C, Muus P, Greenbaum LA, Babu S, Bedrosian C, et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med. 2013;368:2169–81. doi: 10.1056/NEJMoa1208981. [DOI] [PubMed] [Google Scholar]
- 4.Loirat C, Fakhouri F, Ariceta G, Besbas N, Bitzan M, Bjerre A, et al. An international consensus approach to the management of atypical hemolytic uremic syndrome in children. Pediatr Nephrol. 2016;31:15–39. doi: 10.1007/s00467-015-3076-8. [DOI] [PubMed] [Google Scholar]
- 5.Soliris® Prescribing Information. Alexion Pharmaceuticals, Inc.; October. 2015. [Last accessed on 2015 Oct 23]. Available from: http://www.soliris.net/indications/index.php .
- 6.Noris M, Caprioli J, Bresin E, Mossali C, Pianetti G, Gamba S, et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol. 2010;5:1844–59. doi: 10.2215/CJN.02210310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ardissino G, Testa S, Possenti I, Tel F, Paglialonga F, Salardi S, et al. Discontinuation of eculizumab maintenance treatment for atypical hemolytic uremic syndrome: A report of 10 cases. Am J Kidney Dis. 2014;64:633–7. doi: 10.1053/j.ajkd.2014.01.434. [DOI] [PubMed] [Google Scholar]
- 8.Sinha A, Gulati A, Saini S, Blanc C, Gupta A, Gurjar BS, et al. Prompt plasma exchanges and immunosuppressive treatment improves the outcomes of anti-factor H autoantibody-associated hemolytic uremic syndrome in children. Kidney Int. 2014;85:1151–60. doi: 10.1038/ki.2013.373. [DOI] [PubMed] [Google Scholar]