

Abstract
Crystallization chaperones have been used to facilitate the crystallization of challenging proteins. Even though the maltose-binding protein (MBP) is one of the most commonly used crystallization chaperones, the design of optimal expression constructs for crystallization of MBP fusion proteins remains a challenge. To increase the success rate of MBP-facilitated crystallization, a series of expression vectors have been designed with either a short flexible linker or a set of rigid helical linkers. Seven death domain superfamily members were tested for crystallization with this set of vectors, six of which had never been crystallized before. All of the seven targets were crystallized, and their structures were determined using at least one of the vectors. Our successful crystallization of all of the targets demonstrates the validity of our approach and expands the arsenal of the crystallization chaperone toolkit, which may be applicable to crystallization of other difficult protein targets, as well as to other crystallization chaperones.
Recombinant expression of fusion proteins containing a target protein and an unrelated expression tags is a common strategy to obtain samples for medical and research applications1. Depending on their function and properties, these tags can be divided into several categories including purification tags, stability tags, and function tags. Many proteins have been tested as expression tags including the glutathione S-transferase (GST), small ubiquitin-like modifier (SUMO), thioredoxin (TRX), disulfide oxidoreductase (DsbA), N utilization substance protein A (NusA), B1 domain of protein G (GB1), oligopeptide derived from pancreatic ribonuclease A (S-tag), and maltose-binding protein (MBP)2.
MBP is one of the most commonly used protein expression tags due to its exceptional performance in enhancing the solubility of the target proteins3. It is the periplasmic portion of the ATP-binding cassette (ABC) maltose/maltodextrin transporter found in many bacterial species4. MBP has a large protein-protein interaction surface on the same side of the maltose ligand-binding pocket, through which it binds to the transmembrane domain of the ABC transporter. It is commonly used as an affinity tag for purification, which takes advantage of its ability to bind amylose. While the mechanism of the solubility-enhancing property of MBP is still not completely understood, its large size, intrinsic solubility, abundance of flexible loops, and high percentage of exposed hydrophilic residues may be contributing factors. Recent studies indicate that MBP does not have chaperone activity originally thought to facilitate the folding of the fused proteins5.
Obtaining diffraction-quality crystals remains the rate-limiting step in solving a novel structure using X-ray crystallography, despite the recent advancements in the areas of molecular cloning, protein production and purification, crystallization screening, data collection using synchrotron radiation, and structure determination programs. Many tools have been developed and employed in the structural biology community to enhance the crystallizability of proteins, including in situ proteolysis6,7,8, lysine methylation9,10,11, surface entropy reduction12,13,14, metal mediated crystallization15, polymer-driven crystallization16, site-specific mutagenesis17, and chaperone-aided crystallization (also see recent reviews refs 18,19). In particular, many high-affinity binding partner molecules have been used to facilitate the crystallization of challenging protein targets such as the G protein-coupled receptors (GPCRs). These high-affinity binders also include antibody derivatives, such as Fab and scFv, and other alternative scaffolds that have a high affinity for the target proteins20. In addition, covalently linked protein tags, either at the termini or within a flexible loop of the target protein, have also been shown to promote the formation of crystallization contacts. Many protein tags have been tested and successfully used as crystallization chaperones. These protein tags include the T4 lysozyme21,22, apocytochrome b562RIL23,24,25, GST26, TRX27, green fluorescent protein (GFP)28,29, SUMO (pdb: 3V7O and 4G50), and MBP (Table S1).
The first report using MBP to facilitate crystallization of unrelated protein/peptide dates back to 1998, when a 26 amino acid-long peptide from a dominant B-cell epitope sequence of a hepatitis B surface antigen was crystallized in the form of an MBP fusion30. The first protein that was crystallized with MBP was the human T cell leukemia virus type 1 GP21 in 199831,32. A few important proteins, otherwise recalcitrant to crystallization, have been fused to MBP to obtain their first structures, such as the GPCR extracellular domains33. Moreover, MBP fusion technique has been used in the structure determination of several protein: protein complexes34,35,36. The synergistic effects of combining fixed-arm linkers and surface entropy reduction mutations in MBP were demonstrated recently13. The advantage of using a short linker instead of a long one was demonstrated by several studies13,30. To explore the method of fusion tag-mediated crystallization, the biggest challenge is still the design of such chimeric expression constructs to maximize the crystallizability of the targets18. Waugh stated in a recent review on crystal structures of MBP fusion proteins that a short helical linker (i.e., NAAA) was a frequently used linker37. Despite that using MBP tag as an expression and crystallization platform is increasingly appreciated38,39, the optimal sequence, length, and structure of the linker between MBP and the target proteins have not been fully investigated, which limits the application of this crystallization strategy.
The death domain superfamily is a structural motif found in many proteins that are involved in physiological, as well as pathological processes. It can be divided into four different subfamilies: caspase recruitment domain (CARD), death domain (DD), death effector domain (DED), and the pyrin domain (PYD)40. These death fold domains often participate in protein-protein interactions leading to the formation of large, oligomeric signaling complexes such as the apoptosome and inflammasome40. Structural information is available for only about a third of the total 99 death domain superfamily members in the human genome, and seven death-fold protein complex structures have been determined through X-ray crystallography, including the PIDDosome41, FAS/FADD-DISC42,43, MyDDosome44, and apoptosome45. Structural characterization of these death domains and their complexes is often challenging because of their low stability and tendency to self-interact and aggregate. Inspired by the successful examples of using MBP as a crystallization chaperone, we designed an expression vector (V28E) coding an E. coli MBP protein13,46 followed by a very short linker with amino acid sequence of valine and aspartate encoded by the Sal I restriction enzyme site (GTCGAC). This vector has limited success in expression and crystallization of several fusion proteins for PYD and CARD domains47,48,49. Here we sought to systematically explore the usage of linkers of various lengths and test their effects on crystallization of death domain superfamily members. We hypothesize that optimization of the linker regions between MBP and the target protein will increase the likelihood of obtaining crystals. To this end, we designed a set of seven expression vectors harboring different helical linker sequences, and selected seven death domains as targets, six of which have never been crystallized before. The design of the linker sequences takes into consideration the rigidity of the helical structure, the various spacing and relative orientations of MBP and target proteins connected by different helical linkers, and the minimal structural perturbation by a helical linker that connects the C-terminal helix from MBP with the N-terminal helix from a death domain superfamily member. Our successful crystallization of all of the targets demonstrates the validity of our approach and expands the arsenal of the crystallization chaperone toolkit.
Methods
Data base mining
A BLAST search using the E. coli MBP sequence was performed with the Protein Data Bank (www.rcsb.org) to collect information on all deposited structures containing an MBP tag. All of the hits were manually inspected for information of the target proteins such as size and isoelectric point (pI).
Design of the MBP expression vectors
The coding sequence for an E. coli MBP protein mutant “E” (D82A/K83A/E172A/N173A/K239A)13,46 was amplified by PCR and ligated into a T7 promoter-based bacterial protein expression vector pET30a (Novagen) using restriction enzyme sites Nde I and Xho I (sequence is shown in Table S2). A Sal I site was introduced following the MBP residue A369. This vector was designated “V28E” that features a short linker of residues VD encoded by the Sal I site. Building on vector V28E, vectors V28E1-E6 were designed to harbor helical links with one residue increment (Table 1). Gene of interest can be ligated into this set of vectors using the same restriction enzymes Not I and Xho I, with the Not I site encoding residues AAA that adopts helical structures. A hexa-histidine tag sequence following the Xho I site was retained from the pET30a backbone to facilitate the purification of the fusion protein through immobilized metal affinity chromatography (IMAC).
Table 1. The MBP fusion linker sequences for the seven expression vectors.
| Vector Name | Linker Sequence | Linker Structure |
|---|---|---|
| V28E | VD | Loop |
| V28E1 | AAA | Helix |
| V28E2 | AAAA | Helix |
| V28E3 | AAAAA | Helix |
| V28E4 | AARAAA | Helix |
| V28E5 | AARAAAA | Helix |
| V28E6 | AARAFAAA | Helix |
Protein expression, purification and crystallization
Expression and purification of the MBP-tagged proteins have been described previously47,48. Briefly, transformed BL21 (DE3) Codon Plus RIPL cells (Stratagene, Santa Clara, CA) were grown at 37 °C and protein expression was induced at 18 °C for at least two hours with 0.3 mM IPTG. Cells were lysed by sonication and the recombinant protein was purified by IMAC columns followed by size-exclusion chromatography. The purified protein was concentrated to 20–50 mg/ml in the presence of 10 mM maltose before setting up crystallization screening.
Coding sequences for seven death domain superfamily members (hNLRP1-CARD, hAIM2-PYD, hCARD8-CARD, zIGBP1-CARD, hNLRP1-PYD, hNLRP12-PYD, and hMNDA-PYD) were ligated into each of the V28E and V28E1-E6 vectors. Several domain boundaries were tested for each of the targets, which resulted in more than 50 expression constructs (Table S2). Crystallization of these fusion proteins was screened using the Mosquito crystallization robot (TTP Labtech, United Kingdom). For a particular target protein, once the structure was determined using one of the constructs, further exploration of crystallization for the same target was terminated. It is therefore possible that some additional crystallization conditions for the target might be missed.
X-ray diffraction data collection and structure determination
X-ray diffraction data were collected at GM/CA beamline at the Advanced Photon Source, Argonne National Laboratory (ANL) (Table S3). Data were processed with the HKL2000 program suite49 and XDS50. Structures were solved using molecular replacement program Phaser51 from the CCP4 program suite52 and an E. coli MBP structure (3DM0) as the search model. The structures were determined through alternative manual model building with Coot53 and refinement with Phenix54. The crystal structures were validated by the Molprobity server55. Figures were prepared using PyMol (The PyMOL Molecular Graphics System, Version 1.5.0.4 Schrödinger, LLC.).
Results
A survey of using MBP as a crystallization chaperone
A survey of the Protein Data Bank (www.rcsb.org) showed that many groups have successfully adopted the MBP fusion strategy in the crystallization of their proteins of interest (Table S1). To date, there are over 50 primary research articles reporting the structures of MBP-tagged fusion proteins and there is still a clear upward trend (Fig. 1a). There are over 100 deposited MBP fusion protein structures in the PDB by the middle of 2016, making MBP one of the most utilized crystallization chaperone (also see Waugh’s recent review ref. 37). The MBP-tagged targets vary in their overall folds, function, and origins (Table S1). The size of the target proteins falls in the range of 12 residues to 431 residues, with the median of 105 residues (Fig. 1b). The isoelectronic points of target proteins are distributed between 4.0 and 11.0, with a medium value of 6.6 (Fig. 1b).
Figure 1. MBP is the most successful crystallization chaperone.
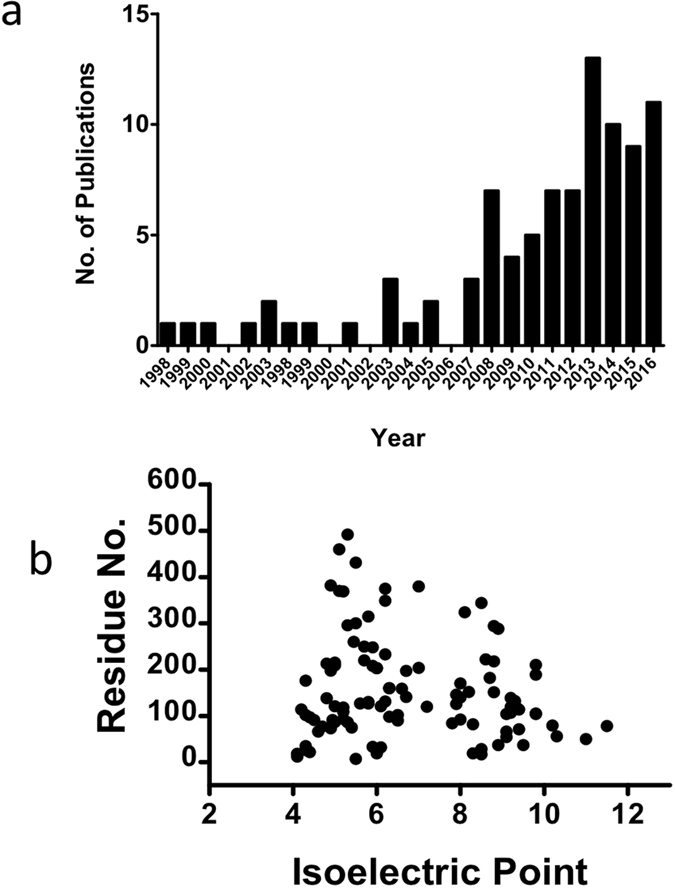
(a) Number of publications reporting MBP fusion protein structures in recent years. (b) Target size distribution and isoelectric point of MBP fusion protein structures. The residue numbers of target proteins were manually verified from the deposited fusion protein structures. Linker and C-terminal artificial sequences such as cloning site and His-tag were not counted.
During our survey of the linker sequences between MBP and the target proteins, we noticed that most linkers are short. This suggests that the relative orientations of MBP and the target proteins may be restricted despite the largely random coil structures of the linker. In agreement with a recent review ref. 37, we have not been able to find consensus linker sequence in these structures, although a linker with the sequence AAA appears to be common.
Crystallization of fusion proteins with a short VD linker
In our study of the death domain superfamily members, obtaining highly purified samples for crystallization has been challenging, as documented for many of these protein-protein interaction domains56. Our initial design of the V28E vector (Fig. 2a) was based on an E. coli MBP protein mutant harboring five mutations that reportedly enhance its crystallizability13,46. The short linker of VD residues followed by the first helical residues of the target proteins may help enhance crystallization. Even though this strategy has resulted in success for some of our targets such as the NLRP1 CARD47, and CARD8 CARD48, and the AIM2 PYD domain47 (Fig. 2b,c), it has failed for other targets such as the zebrafish IGBP1-CARD and human NLRP1-PYD (sequences are listed in Table S2). After extensive testing of the types and concentrations of different commonly used cryo-protectant solutions, we failed to collect useful X-ray diffraction data suitable for structural determination despite the fact that sizable crystals were readily obtained (Fig. 3a,b). The poor diffraction quality of these MBP fusion proteins may be related to the VD linker that introduces internal flexibility of the fusion proteins and micro-scale heterogeneity of the crystals. Indeed, low resolution diffraction is often observed for crystals of large proteins or protein complexes due to structural complexity and internal flexibility50.
Figure 2. Crystallization of MBP fusion proteins with a short VD linker.

(a) The cloning site of the V28E expression vector. Coding sequences for genes of interest can be ligated into the V28E vector using restriction enzyme sites of Sal I and Not I. The other six vectors V28E1-E6 share the overall feature as V28E but use the Not I and Xho I sites for insert ligation. (c) Superposition of the MBP tags in the three fusion protein structures. The three target proteins show different conformation relative to the MBP tag due to the flexibility of VD linker. (b) Different conformations of the linker loops from the three structures in (c).
Figure 3. Structure of the zebrafish IGBP1-CARD crystallized using V28E5.

(a) X-ray diffraction image of zebrafish IGBP1-CARD as MBP-fusion protein expressed with the V28E vector with VD linker. (b) X-ray diffraction image of human NLRP1-PYD as an MBP-fusion protein expressed with the V28E vector containing a VD linker.
As expected, the VD linker region forms a loop bridging the MBP and target proteins in the three solved structures above (Fig. 2b,c). However, conformations of the linker loops among the three structures are drastically different, suggesting that the flexible VD linker may not be optimal for crystallization of some targets such as the IGBP1-CARD and NLRP1-PYD. In addition, target proteins, even those of similar size and fold, may significantly influence the overall structure of the fusion proteins.
Crystallization of fusion proteins containing a series of helical linkers
To design linkers that adopt rigid structures, which can potentially reduce the conformational heterogeneity of the MBP fusion protein, we sought to model the MBP-IPS-1 CARD structure. Here the linker sequence TNSA adopts a continuous helix from the last helix of MBP to the first helix of the IPS-1 CARD which may pose minimal perturbation of the helical structures for either MBP or IPS-1 CARD57 (Fig. 4a). The potential drawback of such a “rigid” linker is that the relative position and orientation of the MBP and the target may be “fixed” such that the chances of packing into ordered crystals may be limited. To explore broader crystallization space, we designed a series of protein expression vectors V28E1-V28E6 containing helical linkers of different lengths (Table 1). The one residue increment of the linker length may change the orientation and location of the target proteins relative to MBP, similar to those of successive residues in an α-helix. Therefore, an increment of one residue in the linker region may allow the target protein to rotate ~100° and translate ~1.5 Å relative to MBP in solution assuming the rigidity of the linker helix (Fig. 4b). As a result, in fusion proteins expressed in vectors V28E1 to V28E6, the orientation of the target protein may change by one and a half turns along the helical axis of the last helix from MBP combined with a translation of ~9 Å. This varied spacing and orientation between MBP and targets may facilitate the presentation of different surface features of the fusion proteins and the sampling of more crystallization space (Table 1).
Figure 4. Design of helical linkers for MBP fusion proteins.

(a) Cartoon representation of MBP-hIPS-1-CARD fusion protein structure (pdb 2VGQ). The last helix of MBP extends and forms a continuous helix with the first helix of CARD. (b) Design of helical linkers. Based on the MBP-hIPS-1-CARD, one residue is inserted in the linker region incrementally from V28E1 to V28E5. The view is shown from the projection of the last helix of MBP. For clarity, only the H3 helix of the CARD is shown in cartoon and in different colors (red for V28E1, orange for V28E2, yellow for V28E3, green for V28E4 and blue for V28E5). The rotation of target protein is ~100° through the insertion of one residue in the linker. (c) X-ray diffraction image of zebrafish IGBP1-CARD as an MBP-fusion protein expressed with the V28E5 vector. By manipulating the linker sequences, X-ray diffraction by the crystals for the IGBP1-CARD fusion protein improved from worse than 6 Å to 1.47 Å. (d) Surface rendering of one MBP tagged IGBP1-CARD structure is shown. The MBP is shown in grey ribbons, the linker residues coded in the V28E5 vector in magenta and the IGBP1-CARD in green. A bound maltotetraose molecule at the MBP is shown as sticks.
Using the set of V28E1-V28E6 vectors, we were able to express, purify and crystallize both the zIGBP1-CARD and the hNLRP1-PYD as MBP-fusion proteins harboring a helical linker. In contrast to crystals of poor diffraction quality using MBP-fusion protein expressed with the V28E vector, X-ray diffraction data of 1.47 Å resolutions were collected for the zIGBP1-CARD crystals using protein expressed with the V28E5 vector (Fig. 4c). This led to structural determination of the first CARD domain from the zebrafish genome58 (Fig. 4d). As shown in Fig. 4d, the V28E5 linker is nearly a perfect α-helix, which joins the last helix of MBP and the first helix of the CARD domain as designed. This is also one of the highest resolution structures for an MBP fusion protein, which may be attributed to the rigidity of the helical linker.
Of note, none of the previously deposited over 100 MBP fusion protein structures were crystallized with more than one linker sequence, probably due to the challenge of the linker design and the lack of systematic approach for linker sequence selection. The set of expression vectors harboring helical linkers (V28E1-V28E6) provide researchers new opportunities to obtain crystals. Indeed, both hNLRP12-PYD and hMNDA-PYD, two targets that failed to crystallize using fusion proteins expressed with the V28E vector, crystallized using fusion proteins expressed with these new vectors (V28E1-V28E6). Remarkably, we were able to crystallize and solve their structures using MBP-fusion proteins expressed with more than one vector. In particular, the hNLRP12-PYD was crystallized as fusion proteins using both V28E4 and V28E6 vectors (Fig. 5a). In the structures, the V28E4 linker adopts a straight α-helix connecting the last helix of MBP to the first helix of PYD, whereas the V28E6 linker is slightly bent. Structural comparison reveals that the PYD rotates 220° from the V28E4 structure to the V28E6 structure (Fig. 5b). Even though the hNLRP12-PYD domains in the two crystal forms adopt different orientations relative to MBP, their structures are essentially identical with a root-mean-square deviation (rmsd) of 0.35 Å (Fig. 5b,c). This suggests that the different linker sequences and crystal lattice packing do not significantly perturb the structures of the target proteins, further validating the linker design strategy.
Figure 5. Structure of the hNLRP12-PYD crystallized in V28E4 and V28E6.

(a) hNLRP12-PYD structures solved using fusion proteins expressed with the V28E4 and V28E6 vectors were superimposed at the MBP region. The MBP tag is colored in grey, the PYD in V28E4 is colored in green, and that in V28E6 is colored in orange. (b) Cylindrical presentation of hNLRP12-PYD. (c) Superposition of the hNLRP12-PYD structures. All of the atoms in PYD were aligned using PyMol and resulted in an rmsd of 0.35 Å.
Similar to the hNLRP12-PYD, the human MNDA PYD domain was crystallized as MBP fusion proteins using three different protein expression vectors V28E3, V28E4, and V28E6. All of these fusion protein crystals diffract very well which led to their structure determination (Fig. 6a). Structural analysis shows that the designed “helical” linkers are slightly distorted probably attributed to crystal packing (Fig. 6b). Nonetheless, the target protein structures from all three different crystals are essentially identical with an rmsd of 0.30–0.40 Å (Fig. 6c), demonstrating again that the MBP fusion strategy enhances the crystallizability without alteration of the structural features of the target proteins.
Figure 6. Superposition of MBP tagged hMNDA-PYD structures.

(a) The MNDA-PYD structures crystallized in V28E3 (magenta), V28E4 (green) and V28E6 (cyan) are superimposed at the MBP region. (b) The V28E6 linker region (blue) forms a slightly distorted helix. (c) hMNDA-PYD structures solved as three fusion proteins are superimposed and are shown in ribbons. All of the atoms in PYD were aligned using PyMol and resulted in rmsds of 0.30–0.40 Å.
Conclusion and Discussion
Despite the abundance of structures for MBP fusion proteins, the design of the fusion protein expression constructs has largely been a trial-and-error process without a clear focus on the sequence or length of the linker region. There has been no report on the rational design of the linker sequences or systematic crystallization screening of MBP fusion proteins, which may have hindered the wider application of this approach in structural biology. In this study, we designed a set of seven vectors for expression of MBP-fusion proteins containing either a short flexible linker or six helical linkers. The six helical linkers were designed based on the following three considerations: first, the α helix is a relatively rigid structure which contrast with the flexible VD linker, therefore may reduce conformational heterogeneity of the fusion proteins; second, helical linkers of various lengths allows the target proteins to be positioned not only at different distances relative to the MBP protein, but also at different orientations because of the 100-degree turn and 1.5 Å translation for each successive residue at a helix. This facilitates the presentation of different surface features of the fusion proteins and the sampling of more crystallization space; third, a helical linker may facilitate the connection between the last helix of MBP and the first helix of a death domain superfamily members with minimal disruption of the helices from MBP or death domain family members. This has proven successful in determining the MBP-IPS-1-CARD structure57.
We tested and successfully crystallized seven targets from the challenging death domain superfamily. Two of them were crystallized in more than one crystal forms containing different linker sequences, demonstrating the power of this linker optimization strategy. We were able to determine the first crystal structures for five of them, including the hAIM2-PYD, hCARD8-CARD, zIGBP1-CARD, hNLRP12-PYD, and hMNDA-PYD. Structures of the hNLRP12-PYD and hMNDA-PYD determined with different linker sequences in different crystal lattices show no significant perturbation of the PYD structures. The structure of the hNLRP1-CARD was determined previously using a selenomethionine-labeled form at 3.1 Å resolution (3KAT). Because essentially identical structures were determined using our MBP-mediated crystallization strategy, and we observed minimal interactions between the MBP tag and the targets in our current studies, our data suggest that fusion protein tags such as MBP are unlikely to interfere with the structure or folding of death domains, and maybe other protein targets as reported previously1,37. Even though we have only tested crystallization of the death domain superfamily members, our MBP fusion strategy may promote crystallization of diverse protein targets, as was demonstrated by previously determined structures of MBP fused with targets of various sizes or folds. Nonetheless, such fusion protein strategy may not be universally applicable for certain difficult protein targets, such as intrinsically disordered proteins that may adopt different conformations depending on different fusion tags they are connected with. Therefore, the fusion protein strategy is one of many different approaches to be tested to promote crystallization.
The linker helix is relatively rigid but can still be influenced by crystal packing forces including the interactions between target protein-target protein, MBP-MBP and MBP-target protein. This may bend/distort the helix to some extent. Indeed, the bending and distortion of the linker helix is observed in some of structures in this study. For example, between the two structures of hNLRP12-PYD fusion proteins, the helix in V28E4 is a straight helix, while that in V28E6 was slightly bent. Structural comparison reveals that the PYD rotates ∼220° from the V28E4 structure to the V28E6 structure (Fig. 5a).
Highly flexible connection between the MBP tag and the target protein may be detrimental to crystallization. To best take advantage of our designed sets of vectors, we recommend removal of all flexible residues at the N-termini of the targets. Our tests demonstrated that the flexible and helical linkers may be complementary to each other in promoting crystallization. Some of our target proteins could only be crystallized with the VD linker while others only with one or more of the rigid helical linkers. Therefore, it is important to test the different linker sequences to enhance the likelihood of crystallization.
The different linker sequences may also facilitate the crystallization of protein complexes. The short linker sequence may allow the MBP tag to stabilize the target protein, but the closely positioned MBP may prevent the target protein from interacting with its partner proteins due to steric hindrance. In such cases, longer and more flexible linker sequences may be tested. The design of the helical linkers with different lengths is based on the α-helical structure that may facilitate the exposure of different surfaces of the target proteins in the context of different fusion proteins. It is therefore possible that the specific protein-protein interaction surfaces may be exposed in one or more of the designed fusion proteins.
The usage of MBP as a crystallization chaperone is perhaps partially due to its high solubility and expansive hydrophilic surface that provides versatile crystal packing interface. There are other proteins such as NusA that may possess similar properties and may be systemically tested as crystallization chaperones. It would be beneficial to test a diverse set of crystallization chaperones for a difficult protein target, because different fusion tags can provide distinct packing interfaces. One caveat of using a helical linker sequence is that the C-terminus of a different protein tag may not end with an extruded helix as in the case of MBP. In such cases, both helical and non-helical linker sequences may need to be explored.
In conclusion, our study provides a strategy for testing MBP and other crystallization chaperones through optimization of the linker sequences. We believe that the use of versatile crystallization chaperones will significantly enhance crystallizability of many challenging protein targets.
Additional Information
Accession codes: Coordinates and structure factors have been deposited in the Protein Data Bank, and the accession codes are presented in Table S3.
How to cite this article: Jin, T. et al. Design of an expression system to enhance MBP-mediated crystallization. Sci. Rep. 7, 40991; doi: 10.1038/srep40991 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
The authors would like to thank Xinhua Ji at the National Cancer Institute for suggestions and discussions, Lars C. Pedersen at the National Institute of Environmental Health Sciences (NIEHS) for providing us the pMAL-MBP wild type and mutant plasmids. X-ray diffraction data were collected at the Advanced Photon Source (APS) GM/CA beamline, which is funded in whole or in part by funds from the National Cancer Institute (Y1-CO-1020) and the National Institute of General Medical Sciences (Y1-GM-1104). Some data was collected at the National Synchrotron Light Source (NSLS) beamline X-25 at the Brookhaven National Laboratory (BNL). Use of the NSLS is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-98CH10886. We’d like to thank the beamline scientists at both synchrotrons for their assistance. T.S.X. is supported by the Case Research Institute and the University Hospitals Case Medical Center. T.J. is supported by the Fundamental Research Funds for the Central Universities and the 100 Talents Program of CAS. Y.L. is supported by the China Postdoctoral Science Foundation (Grant No.: 2015M582007).
Footnotes
Author Contributions T.J. contributed conception, design, data collection and analysis, and manuscript writing; W.C., J.J., J.C., Y.L. K.F., M.H., and P.S. contributed protein purification, data collection, data analysis, and manuscript writing; T.S.X. contributed conception, design, supervision, data interpretation and manuscript writing.
References
- Terpe K. Overview of tag protein fusions: from molecular and biochemical fundamentals to commercial systems. Applied microbiology and biotechnology 60, 523–533, doi: 10.1007/s00253-002-1158-6 (2003). [DOI] [PubMed] [Google Scholar]
- Malhotra A. Tagging for protein expression. Methods in enzymology 463, 239–258, doi: 10.1016/S0076-6879(09)63016-0 (2009). [DOI] [PubMed] [Google Scholar]
- Kapust R. B. & Waugh D. S. Escherichia coli maltose-binding protein is uncommonly effective at promoting the solubility of polypeptides to which it is fused. Protein Sci 8, 1668–1674, doi: 10.1110/ps.8.8.1668 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldham M. L. & Chen J. Crystal structure of the maltose transporter in a pretranslocation intermediate state. Science 332, 1202–1205, doi: 10.1126/science.1200767 (2011). [DOI] [PubMed] [Google Scholar]
- Raran-Kurussi S. & Waugh D. S. The ability to enhance the solubility of its fusion partners is an intrinsic property of maltose-binding protein but their folding is either spontaneous or chaperone-mediated. PLoS One 7, e49589, doi: 10.1371/journal.pone.0049589 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong A. et al. In situ proteolysis for protein crystallization and structure determination. Nature methods 4, 1019–1021, doi: 10.1038/nmeth1118 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abskharon R. N. et al. Combining in-situ proteolysis and microseed matrix screening to promote crystallization of PrPc-nanobody complexes. Protein engineering, design & selection: PEDS 24, 737–741, doi: 10.1093/protein/gzr017 (2011). [DOI] [PubMed] [Google Scholar]
- Gheyi T., Rodgers L., Romero R., Sauder J. M. & Burley S. K. Mass spectrometry guided in situ proteolysis to obtain crystals for X-ray structure determination. Journal of the American Society for Mass Spectrometry 21, 1795–1801, doi: 10.1016/j.jasms.2010.06.015 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sledz P. et al. New surface contacts formed upon reductive lysine methylation: improving the probability of protein crystallization. Protein Sci 19, 1395–1404, doi: 10.1002/pro.420 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter T. S. et al. Lysine methylation as a routine rescue strategy for protein crystallization. Structure 14, 1617–1622, doi: 10.1016/j.str.2006.09.005 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M., Kubota M. & Matsuura Y. Crystallization and improvement of crystal quality for x-ray diffraction of maltooligosyl trehalose synthase by reductive methylation of lysine residues. Acta crystallographica. Section D, Biological crystallography 55, 931–933 (1999). [DOI] [PubMed] [Google Scholar]
- Goldschmidt L., Cooper D. R., Derewenda Z. S. & Eisenberg D. Toward rational protein crystallization: A Web server for the design of crystallizable protein variants. Protein Sci 16, 1569–1576, doi: 10.1110/ps.072914007 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon A. F., Mueller G. A., Zhong X. & Pedersen L. C. A synergistic approach to protein crystallization: combination of a fixed-arm carrier with surface entropy reduction. Protein Sci 19, 901–913, doi: 10.1002/pro.368 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derewenda Z. S. It’s all in the crystals. Acta crystallographica. Section D, Biological crystallography 67, 243–248, doi: 10.1107/S0907444911007797 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laganowsky A. et al. An approach to crystallizing proteins by metal-mediated synthetic symmetrization. Protein Sci 20, 1876–1890, doi: 10.1002/pro.727 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nauli S. et al. Polymer-driven crystallization. Protein Sci 16, 2542–2551, doi: 10.1110/ps.073074207 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggiero A., Smaldone G., Squeglia F. & Berisio R. Enhanced crystallizability by protein engineering approaches: a general overview. Protein and peptide letters 19, 732–742 (2012). [DOI] [PubMed] [Google Scholar]
- Derewenda Z. S. Application of protein engineering to enhance crystallizability and improve crystal properties. Acta crystallographica. Section D, Biological crystallography 66, 604–615, doi: 10.1107/S090744491000644X (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukowska M. A. & Grutter M. G. New concepts and aids to facilitate crystallization. Current opinion in structural biology 23, 409–416, doi: 10.1016/j.sbi.2013.03.003 (2013). [DOI] [PubMed] [Google Scholar]
- Gilbreth R. N. & Koide S. Structural insights for engineering binding proteins based on non-antibody scaffolds. Current opinion in structural biology 22, 413–420, doi: 10.1016/j.sbi.2012.06.001 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Y., Weis W. I. & Kobilka B. K. N-terminal T4 lysozyme fusion facilitates crystallization of a G protein coupled receptor. PLoS One 7, e46039, doi: 10.1371/journal.pone.0046039 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum D. M. et al. GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science 318, 1266–1273, doi: 10.1126/science.1150609 (2007). [DOI] [PubMed] [Google Scholar]
- Chun E. et al. Fusion partner toolchest for the stabilization and crystallization of G protein-coupled receptors. Structure 20, 967–976, doi: 10.1016/j.str.2012.04.010 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C. et al. Structure of the human smoothened receptor bound to an antitumour agent. Nature 497, 338–343, doi: 10.1038/nature12167 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W. et al. Structural basis for allosteric regulation of GPCRs by sodium ions. Science 337, 232–236, doi: 10.1126/science.1219218 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuge M. et al. Use of a fusion protein to obtain crystals suitable for X-ray analysis: crystallization of a GST-fused protein containing the DNA-binding domain of DNA replication-related element-binding factor, DREF. Protein Sci 6, 1783–1786, doi: 10.1002/pro.5560060822 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corsini L., Hothorn M., Scheffzek K., Sattler M. & Stier G. Thioredoxin as a fusion tag for carrier-driven crystallization. Protein Sci 17, 2070–2079, doi: 10.1110/ps.037564.108 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czabotar P. E. et al. Bax crystal structures reveal how BH3 domains activate Bax and nucleate its oligomerization to induce apoptosis. Cell 152, 519–531, doi: 10.1016/j.cell.2012.12.031 (2013). [DOI] [PubMed] [Google Scholar]
- Menetrey J. et al. Processive steps in the reverse direction require uncoupling of the lead head lever arm of myosin VI. Molecular cell 48, 75–86, doi: 10.1016/j.molcel.2012.07.034 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saul F. A., Vulliez-le Normand B., Lema F. & Bentley G. A. Crystal structure of a dominant B-cell epitope from the preS2 region of hepatitis B virus in the form of an inserted peptide segment in maltodextrin-binding protein. Journal of molecular biology 280, 185–192 (1998). [DOI] [PubMed] [Google Scholar]
- Center R. J. et al. Crystallization of a trimeric human T cell leukemia virus type 1 gp21 ectodomain fragment as a chimera with maltose-binding protein. Protein Sci 7, 1612–1619, doi: 10.1002/pro.5560070715 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobe B., Center R. J., Kemp B. E. & Poumbourios P. Crystal structure of human T cell leukemia virus type 1 gp21 ectodomain crystallized as a maltose-binding protein chimera reveals structural evolution of retroviral transmembrane proteins. Proceedings of the National Academy of Sciences of the United States of America 96, 4319–4324 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pioszak A. A. & Xu H. E. Molecular recognition of parathyroid hormone by its G protein-coupled receptor. Proceedings of the National Academy of Sciences of the United States of America 105, 5034–5039, doi: 10.1073/pnas.0801027105 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrick A. N. et al. Structure-function analyses of the human SIX1-EYA2 complex reveal insights into metastasis and BOR syndrome. Nature structural & molecular biology 20, 447–453, doi: 10.1038/nsmb.2505 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan D., Nakatsu T. & Kato H. Crystal structure of peroxisomal targeting signal-2 bound to its receptor complex Pex7p-Pex21p. Nature structural & molecular biology 20, 987–993, doi: 10.1038/nsmb.2618 (2013). [DOI] [PubMed] [Google Scholar]
- Huang D. T. et al. Basis for a ubiquitin-like protein thioester switch toggling E1-E2 affinity. Nature 445, 394–398, doi: 10.1038/nature05490 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waugh D. S. Crystal structures of MBP fusion proteins. Protein Sci 25, 559–571, doi: 10.1002/pro.2863 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokhove M. et al. Easy mammalian expression and crystallography of maltose-binding protein-fused human proteins. J Struct Biol 194, 1–7, doi: 10.1016/j.jsb.2016.01.016 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clifton M. C. et al. A Maltose-Binding Protein Fusion Construct Yields a Robust Crystallography Platform for MCL1. PLoS One 10, e0125010, doi: 10.1371/journal.pone.0125010 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersse K., Verspurten J., Vanden Berghe T. & Vandenabeele P. The death-fold superfamily of homotypic interaction motifs. Trends in biochemical sciences 36, 541–552, doi: 10.1016/j.tibs.2011.06.006 (2011). [DOI] [PubMed] [Google Scholar]
- Park H. H. et al. Death domain assembly mechanism revealed by crystal structure of the oligomeric PIDDosome core complex. Cell 128, 533–546, doi: 10.1016/j.cell.2007.01.019 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott F. L. et al. The Fas-FADD death domain complex structure unravels signalling by receptor clustering. Nature 457, 1019–1022, doi: 10.1038/nature07606 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L. et al. The Fas-FADD death domain complex structure reveals the basis of DISC assembly and disease mutations. Nature structural & molecular biology 17, 1324–1329, doi: 10.1038/nsmb.1920 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S. C., Lo Y. C. & Wu H. Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature 465, 885–890, doi: 10.1038/nature09121 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin H. et al. Structural basis of procaspase-9 recruitment by the apoptotic protease-activating factor 1. Nature 399, 549–557, doi: 10.1038/21124 (1999). [DOI] [PubMed] [Google Scholar]
- Ullah H. et al. Structure of a signal transduction regulator, RACK1, from Arabidopsis thaliana. Protein Sci 17, 1771–1780, doi: 10.1110/ps.035121.108 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin T., Curry J., Smith P., Jiang J. & Xiao T. S. Structure of the NLRP1 caspase recruitment domain suggests potential mechanisms for its association with procaspase-1. Proteins 81, 1266–1270, doi: 10.1002/prot.24287 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin T., Huang M., Smith P., Jiang J. & Xiao T. S. The structure of the CARD8 caspase-recruitment domain suggests its association with the FIIND domain and procaspases through adjacent surfaces. Acta crystallographica. Section F, Structural biology and crystallization communications 69, 482–487, doi: 10.1107/S1744309113010075 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zbyszek Otwinowski W. M. In Methods in enzymology Vol. Macromolecular Crystallography Part A 307–326 (Academic Press, 1997). [Google Scholar]
- Kabsch W. Xds. Acta crystallographica. Section D, Biological crystallography 66, 125–132, doi: 10.1107/S0907444909047337 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. J. et al. Phaser crystallographic software. J Appl Crystallogr 40, 658–674, doi: 10.1107/S0021889807021206 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potterton E., Briggs P., Turkenburg M. & Dodson E. A graphical user interface to the CCP4 program suite. Acta crystallographica. Section D, Biological crystallography 59, 1131–1137, doi: 10.1107/S0907444903008126 (2003). [DOI] [PubMed] [Google Scholar]
- Emsley P. & Cowtan K. Coot: model-building tools for molecular graphics. Acta crystallographica. Section D, Biological crystallography 60, 2126–2132, doi: 10.1107/S0907444904019158 (2004). [DOI] [PubMed] [Google Scholar]
- Adams P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta crystallographica. Section D, Biological crystallography 66, 213–221, doi: 10.1107/S0907444909052925 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta crystallographica. Section D, Biological crystallography 66, 12–21, doi: 10.1107/S0907444909042073 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukits S. F. et al. Solution structure of the tumor necrosis factor receptor-1 death domain. Journal of molecular biology 310, 895–906, doi: 10.1006/jmbi.2001.4790 (2001). [DOI] [PubMed] [Google Scholar]
- Potter J. A., Randall R. E. & Taylor G. L. Crystal structure of human IPS-1/MAVS/VISA/Cardif caspase activation recruitment domain. BMC structural biology 8, 11, doi: 10.1186/1472-6807-8-11 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin T., Huang M., Smith P., Jiang J. & Xiao T. S. Structure of the caspase-recruitment domain from a zebrafish guanylate-binding protein. Acta crystallographica. Section F, Structural biology and crystallization communications 69, 855–860, doi: 10.1107/S1744309113015558 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.