

Abstract
AR-42, a new orally bioavailable, potent, hydroxamate-tethered phenylbutyrate class I/IIB histone deacetylase inhibitor currently is under evaluation in phase 1 and 2 clinical trials and has demonstrated activity in both hematologic and solid tumor malignancies. This report focuses on the preclinical characterization of the pharmacokinetics of AR-42 in mice and rats. A high-performance liquid chromatography–tandem mass spectrometry assay has been developed and applied to the pharmacokinetic study of the more active stereoisomer, S-AR-42, when administered via intravenous and oral routes in rodents, including plasma, bone marrow, and spleen pharmacokinetics (PK) in CD2F1 mice and plasma PK in F344 rats. Oral bioavailability was estimated to be 26 and 100% in mice and rats, respectively. R-AR-42 was also evaluated intravenously in rats and was shown to display different pharmacokinetics with a much shorter terminal half-life compared to that of S-AR-42. Renal clearance was a minor elimination pathway for parental S-AR-42. Oral administration of S-AR-42 to tumor-bearing mice demonstrated high uptake and exposure of the parent drug in the lymphoid tissues, spleen, and bone marrow. This is the first report of the pharmacokinetics of this novel agent, which is now in early phase clinical trials.
Key words: AR-42, histone deacetylase inhibitor, mouse, pharmacokinetics, rat
INTRODUCTION
The dynamic balance of DNA acetylation levels in cells, modulated by histone deacetylase (HDAC) and histone acetyltransferases (HATs), is crucial for regulating chromatin structure and transcriptional dysregulation of genes that are implicated in controlling either cell cycle progression or pathways regulating cell differentiation and/or apoptosis (1–5). Evidence has shown that this epigenetic marking system is associated with inappropriate gene expression in many forms of cancer (3,6,7). Encouraged by the finding that inhibition of HDAC induces cancer cell apoptosis (8,9), inhibitors of HDAC have been developed as a potent and specific strategy for the treatment of solid tumors and hematological malignancies (1,2,10–12). Several classes of HDAC inhibitors have been developed including short-chain fatty acids, such as phenylbutyrate and valproic acid (10); various hydroxamic acid derivatives such, as SAHA and TSA (4,12,13); and cyclic tetrapeptides, such as depsipeptide (2,10,14). These compounds have exhibited in vitro potencies in the mM to nM range (1,10). Recently, a novel class of hydroxamate-tethered phenylbutyrate derivatives has been designed and synthesized with nM HDAC inhibitory activity (13,15–17). One of these compounds, designated R,S-N-hydroxy-4-(3-methyl-2-phenylbutyrylamino)-benzamide (R, S-AR-42, OSU-HDAC-42, NSC 731438) (Fig. 1), exhibited potent cytotoxicity in the NCI 60-cell line screen with a mean GI50 of 0.2 μM (7,11,).
Fig. 1.
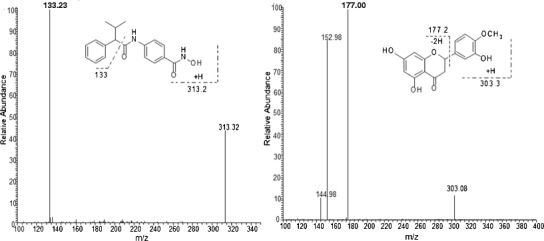
MS/MS spectra of 10 μg/mL AR-42 (left) and internal standard, hesperetin (right). Chemical structures display potential fragmentation of each compound
AR-42 was shown to increase histone H-4 acetylation and p21waf1/CIP1 expression and to decrease pAkt in PC-3 prostate carcinoma cells (7). AR-42 also inhibited the growth of subcutaneous PC-3 xenografts when administered orally to nude mice (50 mg/kg every other day and 25 mg/kg once daily) (11). Immunoblotting demonstrated increased histone H-3 acetylation in tumors from treated mice. More recent data demonstrates that AR-42 appears to have further unique activity among other HDAC inhibitors, including downregulation of CD44, IRF4, and c-Myc within multiple myeloma cells, thus restoring sensitivity to immunomodulatory drug therapy and to a greater extent than panobinostat, a recently FDA-approved HDAC inhibitor (19). Another study compared AR-42 to both vorinostat and romidepsin, two other FDA-approved HDAC inhibitors, in two murine cancer cachexia models (20). The data demonstrated that AR-42 significantly improved the preservation of body weight and prevented tumor burden-induced reductions in lower limb muscle mass and grip strength when compared to even maximally tolerated doses of vorinostat and romidepsin. Critically, AR-42 significantly prolonged survival of cachectic mice, while the other agents did not. Most importantly, AR-42 is well tolerated in the clinic relative to other HDAC inhibitors (21), and it is being actively evaluated in clinical trials for both solid tumors (clinicaltrials.gov ID, NCT02282917) and hematologic malignancies (NCT01129193, NCT01798901, and NCT02569320). Because of higher in vivo potency, the S-isomer, S-N-hydroxy-4-(3-methyl-2-phenylbutyrylamino)-benzamide (S-AR-42) was selected for preclinical and eventual clinical development by the National Cancer Institute. This report describes the preclinical pharmacokinetic characterization of S-AR-42 in mice and rats along with a limited comparison with R-AR-42 pharmacokinetics in rats, which provides some explanation for why the S-AR-42 isomer exhibits greater in vivo effects at reduced doses compared to R-AR-42.
MATERIALS AND METHODS
Materials
Non-formulated S-AR-42 and hesperetin (IS) were supplied by the Drug Synthesis and Chemistry Branch of the National Cancer Institute (Bethesda, MD). R-AR-42 was synthesized as previously described (18). Acetonitrile, methanol, ethyl acetate, and formic acid were of analytical grade and purchased from Fisher Scientific (Pittsburgh, PA). Distilled water was purified using an E-pure water purification system (Barnstead, Dubuque, IA). Phosphate-buffered saline (PBS) was obtained from Invitrogen (Grand Island, NY). CD2F1 mice, F344 rats, mouse plasma, and rat plasma were purchased from Harlan, Inc. (Indianapolis, IN). Ketamine, xylazine, heparin sodium (1000 unit/mL), and saline of medical grade were obtained from The Ohio State University Wexner Medical Center.
Instruments
The quantitative assay for both the S- and R-enantiomers of AR-42 was initially developed on a Perkin-Elmer Sciex API 300 and was later transferred to an Applied Biosystems (AB Sciex) API 3000 triple quadrupole mass spectrometer with partial cross-validation of the method. Both systems were coupled to a Shimadzu HPLC system (Shimadzu, Columbia, MD) equipped with a CBM-20A system controller, SIL-20AC temperature-controlled autosampler, and a CTO-20A column oven.
Preparation of Stock Solutions and Standards
Stock solutions of S-AR-42 and IS, each 1 mg/mL, were prepared in MeOH and kept at −80°C. By serial dilution of S-AR-42 stock solution with blank plasma, working standard solutions at 10, 20, 50, 100, 200, 500, 1000, 2000, 5000, and 10,000 ng/mL were prepared. The working internal standard solution was prepared by 100-fold dilution of IS stock solution using methanol. All these working solutions were prepared on the day of each run and discarded after use.
Sample Preparation Procedure
Ten microliter working standards or working quality control (QC) solutions were spiked to 90 μL blank plasma sample to give final concentrations of 0, 1, 2, 5, 10, 20, 50, 100, 200, 500, and 1000 ng/mL. After vortex mixing for 30 s, 10 μL IS working solution was added, and the samples were vortex mixed again for 30 s. The samples were extracted by ethyl acetate (1000 μL, 30 min) on a mechanical shaker. After centrifugation at 15,900 g for 1 min, the organic layers were separated and transferred to a glass tube and concentrated to dryness with a stream of nitrogen. The residues were reconstituted in 100 μL reconstitution solution (MeOH/water, 60:40 with 0.2% formic acid) and 20 μL was injected into the LC-MS system.
Chromatographic Conditions
Separation was carried out at room temperature using a Thermo Beta Basic C8 column (50 cm × 2.1 mm, 5 μm particle size, Thermo Hypersil-Keystone, Bellefonte, PA), which was coupled to a 2-μm precolumn filter (Thermo Hypersil-Keystone, Bellefonte, PA). The isocratic mobile phase, comprising 60% methanol/40% water/0.2% formic acid, was delivered to the precolumn at a flow rate of 0.2 mL/min and to the ion source at 10 μL/min after a 95:5 (LC:MS) split. The total run time was 6 min.
Mass Spectrometry
The mass spectrometer was operated using electrospray ionization (ESI) with an ionspray voltage of +4200 V. The positive ion multiple-reaction-monitoring mode analysis was performed using nitrogen as the collision gas. Nitrogen curtain gas flow and the ionspray flow were set at 0.6 and 0.9 L/min, respectively. The pressure in the collision cell was set at 0.29 Pa. The orifice voltage and ring voltage were set to +50 and +320 V, respectively. A dwell time of 600 ms and a pause time of 1.2 ms between scans were used to monitor precursor/product ion pairs of S-AR-42 (m/z 313.2–133.2) and IS (m/z 303.2–177.2). The mass spectrometer was tuned daily to its optimum sensitivity and mass accuracy by infusion of a standard calibration solution of polypropylene glycol and a fresh standard solution of S-AR-42 at 5 ng/mL in the HPLC mobile phase as described above.
Assay Validation Experiments
The within-day and between-day accuracy and precision were evaluated by analyzing four to six replicates of blank rat and mouse plasma, mouse urine, and mouse bone marrow sample sets individually spiked with S-AR-42 or R-AR-42 at three concentration levels corresponding to QC-low (5 ng/mL), QC-medium (50 ng/mL), and QC-high (500 ng/mL). The linearity and reproducibility of the calibration curve and QCs were evaluated by analysis of plasma, urine, and bone marrow samples, which were prepared by spiking blank matrix with the working standard and working internal standard solutions.
Recovery and matrix effect were evaluated by measuring the ratio of signals obtained from QC samples prepared and processed as described above (recovery), or samples where AR-42 was added to dried residue after processing (post-spiked samples, matrix effect), to measured signals obtained from QC samples prepared in reconstitution solution.
Stability
For the short-term stability study, S-AR-42 (500 ng/mL) in phosphate-buffered saline (PBS) or mouse plasma were incubated at 4, 24, and 37°C. At the time points of 0, 2, 4, 6, 8, and 24 h following incubation, aliquots of 10 μL each were removed and processed for AR-42 analysis. Long-term stability was evaluated in mouse plasma (10 μg/mL) stored in 10 μL aliquots at −20°C. At various times over a 22-day period, aliquots were diluted 100-fold to 100 ng/mL before processing and analysis.
Plasma Protein Binding
Ultra-filtration was employed to evaluate protein binding of S-AR-42 in mouse plasma. Briefly, triplicate samples of mouse plasma containing S-AR-42 at 0.5, 5, and 10 μg/mL were incubated at 37°C for 1 h. A 0.35 mL aliquot of the plasma sample from each concentration was loaded into a Microcon centrifuge filter device (Regenerated Cellulose 30,000 MWCO, Millipore Corporation, Bedford, MA) for centrifugation at 11,000 g and at 4°C for 50 min. The filters were then carefully removed, and the clear, protein-free solution at the bottom of the vial was obtained. Aliquots of 100 μL each were removed and processed for S-AR-42 analysis.
Protein binding was calculated using the following equation:
Drug Formulation
For preparation of intravenous (i.v.) dosing solutions in mice, S-AR-42 was first dissolved in ethanol followed by addition of twice the volume of PEG 400. Normal saline was then added to produce the final proportion of ethanol, PEG 400, and saline at a ratio of 12:24:64. For oral (p.o.) dosing, the total amount of saline was reduced slightly to achieve a final ratio of ethanol, PEG 400, and saline of 15:30:55. For rats, the ethanol, PEG 400, and saline ratios were 10:20:70 and 15:20:65 for i.v. and p.o. dosing, respectively.
Pharmacokinetic Study in Mice
All animal studies were conducted using protocols approved by The Ohio State University Institutional Animal Care and Use Committee. CD2F1 mice weighing 18–22 g were used for the pharmacokinetic (PK) study. For i.v. administration, approximately 100 μL (adjusted by body weight) dosing solution was administrated via i.v. bolus to reach a dose of 20 mg/kg per mouse (n = 6 per time point). For the p.o. administration study, approximately 200 μL (adjusted by body weight) dosing solution was administrated via oral bolus to reach a dose of 50 mg/kg per mouse (n = 6 per time point). Blood was collected via cardiac puncture at 0.08, 0.25, 0.5, 0.75, 1, 2, 3, 4, 6, 8, 16, 24, 48, and 72 h after dosing. Blood samples were centrifuged at 1000 g for 5 min, and the supernatant of each was collected and kept at −80°C until analysis.
Pharmacokinetic Study in the Rat
Six F344 rats were given i.v. or p.o. administration at 20 or 50 mg/kg, respectively. The plasma samples were collected at each time point via a jugular vein catheter. The time points in this study were 0.08, 0.25, 0.5, 0.75, 1, 2, 3, 4, 6, 8, 16, 24, 48, and 72 h. The quantification method of these samples is the same as described above.
Urinary Excretion
Urine was collected from mice (n = 6) and rats (n = 6) housed in metabolic cages. Urine was collected prior to and at 24 h after S-AR-42 treatment (20 mg/kg S-AR-42 via i.v. bolus administration). Cages were washed using distilled water, which was collected and combined with the respective urine samples prior to storage at −80°C.
AR-42 Uptake and Distribution in Mouse Bone Marrow
To determine bone marrow distribution of AR-42, the human AML MV4-11 cell engrafted immune deficiency mice were used with oral treatment. Specifically, each of the 23–25 g NOD/SCID mice (Jackson Laboratory, Bar Harbor, ME, USA) was engrafted with 0.3 × 106 human AML MV4-11 cells through tail vein on day 0. Engraftments were confirmed within approximately 2 weeks and prior to dosing for PK analysis. AR-42 was dissolved in 0.5% methylcellulose and 1% tween-80 as a 4 mg/mL dosing solution. To conduct oral administration of AR-42 on AML-engrafted mice, 200–250 μL of dosing solution was administered at a dose of 40 mg/kg according to body weight using gavage on day 17 after the engraftment. Plasma, bone marrow, and spleens were harvested from the treated mice at the time schedule of 0 (pre-dose), 0.25, 0.5, 1, 2, 3, 4, 6, 8, 16, 24, 48, and 72 h after dosing (n = 3 mice per time point). All samples were immediately stored at −80°C until analysis. Concentrations of standard, QC, and experimental samples were measured based on measured protein concentration within bone marrow samples and with the assumptions that protein content was 20% of total mass of the bone marrow tissue and that 1 mg tissue equals 1 μL tissue volume.
miR-29b Expression in Bone Marrow
Gene expression of miR-29b in bone marrow and spleen of AML-engrafted NGS mice were measured at four time points (12, 24, 48, and 72 h) using real-time PCR with murine primer/probes for primary-miR-29b-1 as published previously (22). RT-PCR was performed using cDNA reverse-transcribed from 1 μg of total RNA extracted from bone marrow and spleen tissue by TRIzol reagent (Invitrogen) using the manufacturer-recommended protocol. Real-time RT-PCR reactions were performed using TaqMan reagents (Applied Biosystems), and the data were analyzed by version 1.6 software of Sequence Detector. Results that represent the fold change of transcript levels of miR-29b between these two tissue samples were expressed as the mean S.D. from triplicate determinations.
Data Analysis
Plasma concentration-time data were fit using nonlinear least-squares regression using WinNonlin (V 6.0, Pharsight, Mountain View, CA) computer software. Model selection was guided by the Akaike information criteria and standard errors of estimate. Weighting factors of 1/Y2 and 1/Yhat were evaluated.
RESULTS AND DISCUSSION
HPLC-MS/MS Conditions
The electrospray mass spectrum of S-AR-42 indicated the parent [M+H]+ ion at m/z 313.2 was highly abundant. When the parent ion was fragmented, the most highly abundant ion observed was at m/z 133.2 (Fig. 1). The possible structures of observed fragment ions are presented in Fig. 1. The mass spectrum of the IS demonstrated a parent ion [M+H]+ as the base peak at m/z 303.2. The product ion spectrum of the IS revealed m/z 177.2 as the most abundant ion. Thus, the transitions of m/z 313.2 > 133.2 for AR-42 and 303.2 > 177.2 for the IS were selected for monitoring. Chromatograms from blank mouse plasma and mouse plasma with IS (1000 ng/mL) and S-AR-42 (2 ng/mL) are displayed in Fig. 2 and supports the assay selectivity in both mouse and rat plasma using these transitions. Retention times for S-AR-42 and IS were 3.13 and 2.48 min, respectively. Due to moderate tailing at higher AR-42 concentrations, the isocratic run time was kept at 6 min. Peak area ratios of analyte/IS vs. nominal concentrations were used to generate the calibration curves.
Fig. 2.
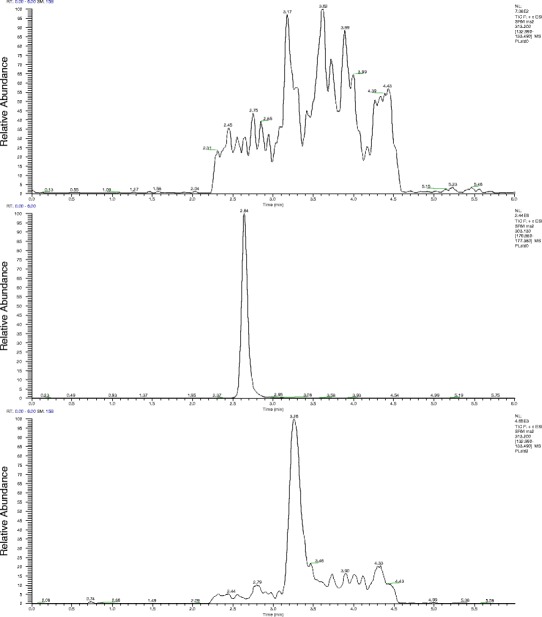
Ion chromatograms of extracts from blank mouse plasma (top) or mouse plasma spiked with 1000 ng/mL hesperetin (IS, middle) and 2 ng/mL S-AR-42 (bottom)
Assay Validation
The initial LC-MS/MS approach indicated negligible interference in the extracts from either mouse or rat plasma (Fig. 2). The limit of detection was 1 ng/mL for S-AR-42 with signal-to-noise ratio >5 using 100 μL mouse or rat plasma and 100 μL reconstituted extract. The lower limit of quantification (LLOQ) for S-AR-42 was set at 2 ng/mL (6 nM), both in mouse plasma and rat plasma. Linearity was demonstrated between the LLOQ to 50 ng/mL (0.16 μM) for low-concentration samples and from 50 to 1000 ng/mL (3.2 μM) for high-concentration samples. Table I displays within-day and between-day validation data for both mouse and rat plasma. Coefficients of variation (CVs) were found to be within acceptable limits according to the FDA criteria (23). Variation of accuracy was less than 10% for all means calculated. Similarly, these data demonstrated adequate reproducibility and sensitivity of the method to characterize plasma pharmacokinetics in mice and rats. Similar validation data was achieved in mouse urine and bone marrow as shown in Table I. Recoveries were determined to be 99–108% for mouse plasma, 83–89% for mouse urine, and 97–111% for rat plasma. Matrix effects were similar at 98–107, 84–94, and 96–110% for mouse plasma, mouse urine, and rat plasma, respectively. These data show high recovery and minimal matrix effect in each of the matrices tested.
Table I.
LC/MS/MS Assay Within-Day and Between-Day Precision and Accurate Data of S-AR-42 in Mouse Plasma, Mouse Urine, Mouse Bone Marrow, and Rat Plasma
| Matrix type | Nominal concentration, μM (ng/mL) | Within-day | Between-day | ||||
|---|---|---|---|---|---|---|---|
| Observed concentration, μM (ng/mL) | CV (%) | Accuracy (%) | Observed concentration, μM (ng/mL) | CV (%) | Accuracy (%) | ||
| Mouse plasma | 0.016 (5) | 0.016 ± 0.002 (5.0 ± 0.5) | 9.5 | 99.4 | 0.016 ± 0.001 (5.1 ± 0.5) | 8.8 | 102.5 |
| 0.16 (50) | 0.15 ± 0.01 (45.6 ± 3.0) | 6.6 | 91.3 | 0.16 ± 0.00 (50.1 ± 0.3) | 0.6 | 100.2 | |
| 1.6 (500) | 1.51 ± 0.08 (471 ± 24) | 5.0 | 94.2 | 1.61 ± 0.02 (503 ± 7) | 1.4 | 100.7 | |
| Mouse urine | 0.016 (5) | 0.016 ± 0.001 (4.8 ± 0.2) | 4.6 | 95.2 | 0.015 ± 0.002 (4.72 ± 0.46) | 9.8 | 94.3 |
| 0.16 (50) | 0.16 ± 0.01 (49.9 ± 6.0) | 5.3 | 98.3 | 0.16 ± 0.02 (48.4 ± 5.1) | 10.6 | 96.9 | |
| 1.6 (500) | 1.46 ± 0.02 (454 ± 7) | 1.5 | 90.9 | 1.47 ± 0.06 (460 ± 20) | 4.4 | 92.0 | |
| Mouse bone marrow | 0.016 (5) | 0.016 ± 0.001 (5.1 ± 0.4) | 8.3 | 102.2 | 0.015 ± 0.001 (4.8 ± 0.4) | 7.6 | 95.3 |
| 0.16 (50) | 0.16 ± 0.01 (49.9 ± 1.8) | 3.5 | 99.9 | 0.17 ± 0.01 (52.2 ± 2.4) | 4.6 | 104.3 | |
| 1.6 (500) | 1.66 ± 0.04 (518 ± 14) | 2.7 | 103.7 | 1.59 ± 0.09 (498 ± 29) | 5.8 | 99.6 | |
| Rat plasma | 0.016 (5) | 0.016 ± 0.002 (5.0 ± 0.7) | 13.2 | 99.2 | 0.016 ± 0.002 (4.9 ± 0.5) | 9.7 | 97.7 |
| 0.16 (50) | 0.16 ± 0.01 (51.3 ± 2.6) | 5.2 | 102.6 | 0.16 ± 0.00 (49.3 ± 0.7) | 1.5 | 98.7 | |
| 1.6 (500) | 1.55 ± 0.05 (483 ± 15) | 3.0 | 96.6 | 1.60 ± 0.01 (502 ± 3) | 0.7 | 100.6 | |
Within-day data are n = 6 (plasma and urine) or n = 4 (bone marrow), and between-day data are n = 18 (plasma and urine) or n = 12 (bone marrow). Data are expressed as mean ± SD
Stability and Protein Binding of S-AR-42 in Mouse Plasma by LC/MS/MS
S-AR-42 was found to be relatively stable in mouse plasma at 4°C with an estimated degradation half-life of 122 h. However, at the higher temperatures tested, plasma S-AR-42 concentrations declined monoexponentially and degraded with half-lives of 11.5 and 5.8 h at 24 and 37°C, respectively. Little or no degradation was observed in PBS buffer, even at 37°C. At −20°C, S-AR-42 was stable in mouse plasma with no apparent decomposition over 22 days. Similar results were observed in rat plasma. S-AR-42 was highly bound to plasma proteins (>96%) in mouse plasma at all concentrations evaluated.
Pharmacokinetics of S-AR-42 in the Mouse Following I.V. and P.O. Bolus Administration
The mean plasma concentration-time profile of S-AR-42 in CD2F1 mice is shown in Fig. 3. The data were fitted to a two-compartment model using WinNonlin with a weighting factor of 1/Y2. The relevant PK parameters were computed as shown in Table II. Following a dosage of S-AR-42 at 20 mg/kg via i.v. bolus administration, plasma concentrations reached ~55 μM then decreased to ~0.02 μM by 72 h after dosing. Distribution and elimination half-lives were 0.39 and 10.1 h, respectively.
Fig. 3.
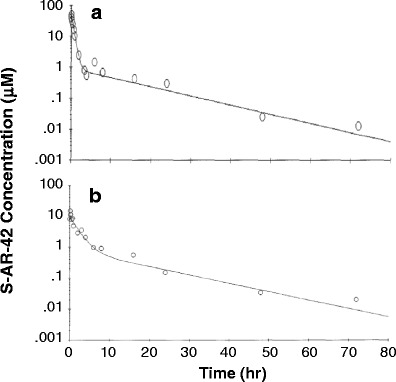
Plasma concentration-time profile of S-AR-42 in the mouse following i.v. (a) and p.o. (b) administration at 20 and 50 mg/kg, respectively. Data points represent means of six replicates at each time point, and lines are fitted data using a two-compartment model
Table II.
Relevant Compartmental Pharmacokinetic Parameters of S-AR-42 in the Mouse After i.v. and p.o. Administration. Parameters Were Estimated by Curve Fitting of all Data (n = 6 Observations per Time Point)
| PK parameter | I.V. administration (20 mg/kg) | P.O. administration (50 mg/kg) |
|---|---|---|
| C 5 min/max (μM) | 55.4 | 14.7 |
| α (h−1) | 1.79 | 0.513 |
| T 1/2 α (h) | 0.387 | 1.35 |
| β (h−1) | 0.068 | 0.062 |
| T 1/2 β (h) | 10.1 | 11.1 |
| Cl (l/h/kg) | 1.47 | 5.35 |
| Vss (l/kg) | 7.16 | |
| AUC0-∞ (μM×h) | 43.7 | 29.9 |
| T max (h) | 0.126 |
The mean plasma concentration-time profile of S-AR-42 after oral administration in CD2F1 mice is also shown in Fig. 3. S-AR-42 was absorbed rapidly following a 50 mg/kg oral administration. Plasma concentrations reached a peak of 14.7 μM at the earliest time point evaluated (5 min, 0.17 h). Relevant PK parameters are displayed in Table II. The initial half-life was 1.35 h, and the terminal half-life was 11.1 h. The area under the curve (AUC) was 29.9 μM×h, which corresponded to an oral bioavailability of 27% as calculated by
The unchanged parent drug was measured in urine for 24 h after i.v. dosing, and the dosage recovery was found to be 1.4% which indicated that renal clearance is a minor pathway for the elimination of parent S-AR-42 in mice.
S-AR-42 was found to suppress intraepithelial neoplasia and blocked disease progression in solid tumor without negative effects on bone marrow (24). More recently, S-AR-42 was demonstrated to have promising effects in AML in combination with decitabine (22), and it is currently under evaluation in an ongoing clinical trial with this combination (clinicaltrials.gov ID NCT01798901). It was therefore important to characterize S-AR-42 uptake in the spleen and bone marrow of AML-diseased mice where the bulk of the leukemic cells are found. Figure 4 shows that following oral dosing of 40 mg/kg three times weekly for 2.5 weeks (50 mg/kg was toxic in diseased mice), S-AR-42 levels achieved were approximately 6 μM (1.86 ng/mg tissue) which persisted for approximately 3–4 h after the final dose then slowly declined until S-AR-42 was undetectable after 48 h. These results demonstrated good penetration of AR-42 in bone marrow correlated with observed miR-29b levels in bone marrow which increased approximately threefold in expression over baseline within 24 h after dosing (Fig. 5). This increased miR-29b expression persisted for up to 48 h before declining to approximately 2.4-fold at 72 h. Similar results were observed in spleen (Fig. 5). These data suggest that repeat oral dosing of AR-42 will provide continuous effects on miR-29b expression in these leukemic cell compartments in vivo.
Fig. 4.
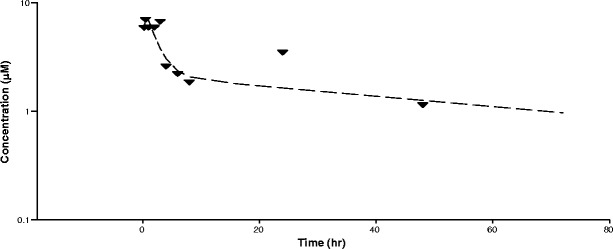
Concentration of AR-42 in mouse bone marrow following the last of eight oral doses at 40 mg/kg. Data represent mean of n = 3 measurements per time point. The line was generated from fitting to a two-compartment model with extravascular input and weighting = 1/year2
Fig. 5.
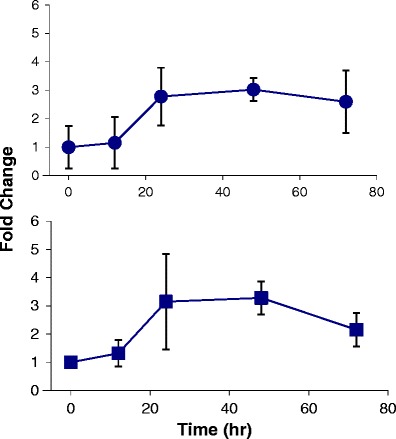
miR-29b expression vs. time profiles for bone marrow (top, circles) and spleen (bottom, squares) in AML-induced NGS mice. Data are fold change in expression relative to the pre-dose measurement (mice not dosed with AR-42). Data are presented as means of n = 3 measurements ± standard deviations
Pharmacokinetic Study of S-AR-42 in the Rat Following I.V. And P.O. Bolus Administration
For comparison of AR-42 in mice vs. rats, we dosed rats (n = 6) with S-AR-42 at the same body mass normalized doses of 20 and 50 mg/kg for i.v. and p.o., respectively. Following an i.v. bolus dose of S-AR-42 at 20 mg/kg, the mean drug plasma concentration reached 35.6 μM then decreased to approximately 0.02 μM by 24 h (Fig. 6). The concentration-time profile of S-AR-42 in rats was also described by a two-compartment model with initial and terminal half-lives of 0.12 and 5.1 h, respectively. Mean AUC and clearance (CL) were calculated as 39.8 μM×h and 1.80 L/h/kg, respectively. Other relevant parameters for i.v. administration are listed in Table III. Twenty-four-hour urinary excretion was estimated to be 5% of the dose.
Fig. 6.
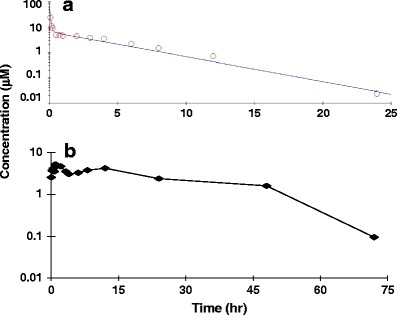
Plasma concentration-time profiles of S-AR-42 in rat #1 following an i.v. bolus administration at 20 mg/kg (a) and p.o. administration at 50 mg/kg (b). Data points are means of n = 6 measurements. The line for the i.v. dosing data was generated from fitting to a two-compartment model
Table III.
Relevant Plasma Compartmental Pharmacokinetic Parameters of S-AR-42 in Rats (n = 6) Following I.V. and P.O. Administration
| PK parameters | Average | SD | CV (%) |
|---|---|---|---|
| I.V. administration (20 mg/kg) | |||
| C 0 (μM) | 35.6 | 24.9 | 70.0 |
| α (h−1) | 7.6 | 4.7 | 62.2 |
| T 1/2 α (h) | 0.12 | 0.07 | 57.1 |
| β (h−1) | 0.22 | 0.09 | 40.5 |
| T 1/2 β (h) | 5.1 | 2.6 | 51.0 |
| MRT (h) | 6.5 | 3.7 | 56.7 |
| Cl (l/h/kg) | 1.8 | 0.6 | 33.0 |
| Vss (l/kg) | 11.0 | 5.5 | 49.5 |
| AUC0-∞ (μM×h) | 39.8 | 14.4 | 36.1 |
| P.O. administration (50 mg/kg) | |||
| C max (μM) | 7.1 | 1.6 | 23.2 |
| T max (h) | 3.3 | 4.5 | 137 |
| λz (h−1) | 0.09 | 0.04 | 37.4 |
| HL-λz (h) | 9.3 | 6.4 | 68.5 |
| Vz/F (L/kg) | 13.7 | 12.1 | 88.2 |
| AUMC (μM×h2) | 3242 | 704 | 21.7 |
| MRT (h) | 19.5 | 5.6 | 28.7 |
| Cl/F (l/h/kg) | 1.0 | 0.2 | 17.7 |
| AUC0-∞ (μM×h) | 171 | 30.9 | 18.1 |
SD standard deviation determined from n = 6 estimates; CV (%) coefficient of variation determined by SD/average*100%
Following a 50 mg/kg p.o. dose of S-AR-42, the drug was found to be rapidly absorbed and the mean maximum plasma concentration (Cmax) of 7.1 μM was reached within 3 h and remained relatively steady until 12 h (Fig. 6). Thereafter, the concentration decreased with time and was still measureable at approximately 0.1 μM up to 72 h. The mean AUC was 170.8 μM×h, which gave a calculated oral bioavailability of ~170% (Table III), suggesting that a portion of the actual i.v. AUC may not have been captured (e.g., under-estimated AUC between 0 and 5 min) or that other factors contributed to an apparent decrease in drug exposure after i.v. dosing. Urinary excretion of unchanged parent drug was approximately 4%, which is consistent with the data obtained from the i.v study.
Pharmacokinetic Study of R-AR-42 in the Rat Following I.V. Bolus Administration
Following a 20 mg/kg i.v. bolus administration of R-AR-42, the plasma concentration-time profile was also described by a two-compartment model with initial and terminal half-lives of 0.3 and 4.1 h, respectively. Plasma R-AR-42 concentration became undetectable after 12 h. The mean initial plasma concentration (C 0) was found to be 66.3 μM and the mean AUC value was 24.8 μM×h, which was lower compared to S-AR-42 (Table III). As our assay is achiral, we cannot detect potential isomeric conversion of AR-42; S-AR-42 has both prolonged plasma exposure of composite AR-42 analyte and more potent in vivo effects suggesting S-R conversion may be minimal.
CONCLUSION
An HPLC-MS/MS method was developed and qualified for the routine determination of AR-42 in mouse and rat plasma. The assay was shown to be specific, accurate, precise, and reproducible. The pharmacokinetics of S-AR-42 showed prolonged plasma exposure compared to the R-isomer following i.v. bolus dose in rats, potentially explaining its greater in vivo effects relative to the R-isomer. The calculated complete oral bioavailability of S-AR-42 in rats (>100%) was higher than in the mouse (26.7%), and renal clearance was found to be a minor pathway for the elimination of parent S-AR-42 in the rat. The drug was well absorbed into mouse bone marrow following oral administration. When combined with AR-42’s distinct HDAC inhibitor pharmacology relative to approved inhibitors, these data demonstrate that S-AR-42 is a promising agent for continued evaluation in the growing number of disease states where HDAC inhibition may be of therapeutic benefit.
Acknowledgments
This study is supported by NCI-N01-CM-52205 (KKC) and NCI-RO1-CA158350 (RG). This work has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
Hao Cheng and Zhiliang Xie contributed equally to this work.
Contributor Information
Mitch A. Phelps, Phone: (614) 832-2547, Email: phelps.32@osu.edu
Kenneth K. Chan, Phone: (614)571-5354, Email: chan.56@osu.edu
References
- 1.Meinke PT, Liberator P. Curr Med Chem. 2001;8:211. doi: 10.2174/0929867013373787. [DOI] [PubMed] [Google Scholar]
- 2.Monneret C. Eur J Med Chem. 2005;40:1. doi: 10.1016/j.ejmech.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 3.Kouraklis G, Theocharis S. Curr Med Chem Anticancer Agents. 2002;2:477. doi: 10.2174/1568011023353921. [DOI] [PubMed] [Google Scholar]
- 4.Hockly E, Richon VM, Woodman B, Smith DL, Zhou X, Rosa E, et al. Proc Natl Acad Sci U S A. 2003;100:2041. doi: 10.1073/pnas.0437870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Richon VM, Zhou X, Rifkind RA, Marks PA. Blood Cells Mol Dis. 2001;27:260. doi: 10.1006/bcmd.2000.0376. [DOI] [PubMed] [Google Scholar]
- 6.Johnstone RW. Nat Rev Drug Discov. 2002;1:287. doi: 10.1038/nrd772. [DOI] [PubMed] [Google Scholar]
- 7.Lu Q, Yang Y-T, Chen C-S, Davis M, Byrd JC, Etherton MR, et al. J Med Chem. 2004;47:467. doi: 10.1021/jm0303655. [DOI] [PubMed] [Google Scholar]
- 8.Marks PA, Richon VM, Rifkind RA. J Natl Cancer Inst. 2000;92:1210. doi: 10.1093/jnci/92.15.1210. [DOI] [PubMed] [Google Scholar]
- 9.Chen C-S, Weng S-C, Tseng P-H, Lin H-P, Chen C-S. J Biol Chem. 2005;280:38879. doi: 10.1074/jbc.M505733200. [DOI] [PubMed] [Google Scholar]
- 10.Jung M. Curr Med Chem. 2001;8:1505. doi: 10.2174/0929867013372058. [DOI] [PubMed] [Google Scholar]
- 11.Kulp SK, Chen C-S, Wang D-S, Chen C-Y, Chen C-S. Clin Cancer Res. 2006;12:5199. doi: 10.1158/1078-0432.CCR-06-0429. [DOI] [PubMed] [Google Scholar]
- 12.Munster PN, Troso-Sandoval T, Rosen N, Rifkind R, Marks PA, Richon VM. Cancer Res. 2001;61:8492. [PubMed] [Google Scholar]
- 13.Suzuki T, Nagano Y, Kouketsu A, Matsuura A, Maruyama S, Kurotaki M, et al. J Med Chem. 2005;48:1019. doi: 10.1021/jm049207j. [DOI] [PubMed] [Google Scholar]
- 14.Roychowdhury S, Baiocchi RA, Vourganti S, Bhatt D, Blaser BW, Freud AG, et al. J Natl Cancer Inst. 2004;96:1447. doi: 10.1093/jnci/djh271. [DOI] [PubMed] [Google Scholar]
- 15.Bouchain G, Delorme D. Curr Med Chem. 2003;10:2359. doi: 10.2174/0929867033456585. [DOI] [PubMed] [Google Scholar]
- 16.Wittich S, Scherf H, Xie C, Brosch G, Loidl P, Gerhaeuser C, et al. J Med Chem. 2002;45:3296. doi: 10.1021/jm0208119. [DOI] [PubMed] [Google Scholar]
- 17.Wang D-F, Wiest O, Helquist P, Lan-Hargest H-Y, Wiech NL. Bioorg Med Chem Lett. 2004;14:707. doi: 10.1016/j.bmcl.2003.11.062. [DOI] [PubMed] [Google Scholar]
- 18.Lu Q, Wang D-S, Chen C-S, Hu Y-D, Chen C-S. J Med Chem. 2005;48:5530. [DOI] [PubMed]
- 19.Canella A, Cordero Nieves H, Sborov DW, Cascione L, Radomska HS, Smith E, et al. Oncotarget. 2015;6:31134. doi: 10.18632/oncotarget.5290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tseng YC, Kulp SK, Lai IL, Hsu EC, He WA, Frankhouser DE, Yan PS, Mo X, Bloomston M, Lesinski GB, Marcucci G, Guttridge DC, Bekaii-Saab T, Chen CS. J Natl Cancer Inst. 2015;107. [DOI] [PMC free article] [PubMed]
- 21.Hofmeister CC, Liu ZF, Bowers MA, Porcu P, Flynn JM, Christian B, Baiocchi RA, Benson DM, Andritsos LA, Greenfield CN, Sell M, Geyer S, Byrd JC, Grever MR. Blood 2012;120.
- 22.Mims A, Walker AR, Huang X, Sun J, Wang H, Santhanam R, et al. Leukemia. 2013;27:871. doi: 10.1038/leu.2012.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guidance for Industry, Bioanalytical Method Validation. Draft Guidance. U.S. Department of Health and Human Services, Food and Drug Administration. September, 2013. Revision 1.
- 24.Sargeant AM, Rengel RC, Kulp SK, Klein RD, Clinton SK, Wang YC, et al. Cancer Res. 2008;68:3999. [DOI] [PubMed]