

Abstract
The objective of this work was to establish the quantitative relationship between Lanreotide Autogel® (LAN) on serum chromogranin A (CgA) and progression-free survival (PFS) in patients with nonfunctioning gastroenteropancreatic neuroendocrine tumors (GEP-NETs) through an integrated pharmacokinetic/pharmacodynamic (PK/PD) model. In CLARINET, a phase III, randomized, double-blind, placebo-controlled study, 204 patients received deep subcutaneous injections of LAN 120 mg (n = 101) or placebo (n = 103) every 4 weeks for 96 weeks. Data for 810 LAN and 1298 CgA serum samples (n = 632 placebo and n = 666 LAN) were used to develop a parametric time-to-event model to relate CgA levels and PFS (76 patients experienced disease progression: n = 49 placebo and n = 27 LAN). LAN serum profiles were described by a one-compartment disposition model. Absorption was characterized by two parallel pathways following first- and zero-order kinetics. As PFS data were considered informative dropouts, CgA and PFS responses were modeled jointly. The LAN-induced decrease in CgA levels was described by an inhibitory E MAX model. Patient age and target lesions at baseline were associated with an increment in baseline CgA. Weibull model distribution showed that decreases in CgA from baseline reduced the hazard of disease progression significantly (P < 0.001). Covariates of tumor location in the pancreas and tumor hepatic tumor load were associated with worse prognosis (P < 0.001). We established a semimechanistic PK/PD model to better understand the effect of LAN on a surrogate endpoint (serum CgA) and ultimately the clinical endpoint (PFS) in treatment-naive patients with nonfunctioning GEP-NETs.
Electronic supplementary material
The online version of this article (doi:10.1208/s12248-016-9884-3) contains supplementary material, which is available to authorized users.
KEY WORDS: chromogranin A, lanreotide, neuroendocrine tumors, population PK/PD, time-to-event analysis
INTRODUCTION
Endocrine tumors are rare, with an incidence approaching five cases/100,000/year (1). They are typically slow-growing tumors (2–5) that arise from endocrine cells located in the gastrointestinal system or the pancreas; most patients have distant metastases at diagnosis (1). The ideal initial treatment is surgical removal of the tumor, but as many patients have inoperable tumors, medical therapy is required.
Somatostatin analogs (SSAs) are the main treatment for gastroenteropancreatic neuroendocrine tumors (GEP-NETs). The efficacy of Lanreotide Autogel (LAN) (known as Depot in the USA) in patients with GEP-NETs has been demonstrated in a randomized, double-blind, placebo-controlled, multicenter phase III clinical trial (6). LAN has been approved recently for the treatment of GEP-NETs in the European Union and the USA (7,8).
According to the European Society for Medical Oncoloty (ESMO) Clinical Practice Guidelines for GEP-NETs, treatment efficacy should be assessed both by imaging procedures (i.e., computed tomography [CT] scans or magnetic resonance imaging [MRI]) and biochemical markers (9). GEP-NETs secrete endocrine markers such as chromogranin A (CgA), the plasma levels of which are elevated in patients with GEP-NETs, and CgA has been reported to be a sensitive tumor marker for disease monitoring: not only does it reflect tumor load, but it is also an indicator of tumor growth (3,10–13).
Whereas prognostic factors are defined to predict disease outcome in the absence of therapy, predictive factors provide information on the potential benefit from treatment (14,15). To date, the most significant prognostic factors identified for GEP-NETs include the size of the primary tumor (1,2) with worse prognosis for pancreatic tumors (9,11,16), presence of metastasis (1,2,5,9), proliferative index (2,17), high hepatic tumor load (3,11,18,19), and CgA expression (3,11,13). It has been suggested that CgA levels are a predictive factor for outcome.
To our knowledge, there is currently no quantitative model to describe the effects of somatostatin analogs in the treatment of GEP-NETs. We now establish an integrated pharmacokinetic/pharmacodynamic (PK/PD) model for biomarker and clinical endpoint effects of LAN, using longitudinal CgA and progression-free survival (PFS) data from the phase III clinical trial CLARINET (6). This model can also be used to evaluate the outcome of alternative study designs (dose level, dosing interval) in patients with GEP-NETs. As a result of this modeling exercise, the prognostic and predictive factors of PFS in these patients have been identified.
METHODS
Study Population
CLARINET is a phase III, randomized, double-blind, comparative, placebo-controlled, parallel group, multicenter study (6). A total of 204 treatment-naive patients with nonfunctioning GEP-NETs located in the pancreas, midgut (small intestine and appendix), hindgut (large intestine, rectum, anal canal, and anus), or of unknown origin were enrolled (33% with hepatic tumor load >25%; 103 treatment, 101 placebo). Patients in the treated group received an extended release aqueous gel formulation of 120 mg LAN every 28 days for 2 years. Table I summarizes the demographic and disease characteristics of the patients included in the analysis.
Table I.
Baseline Demographic and Disease Characteristics of Patients
| Characteristics | Lanreotide arm (n = 101) | Placebo arm (n = 103) |
|---|---|---|
| Age (years) [median (range)] | 64 (30–83) | 63 (31–92) |
| Male (n) | 53 (52.5%) | 54 (52.4%) |
| Weight (kg) [median (range)] | 77 (46–128) | 75 (40–133) |
| CgA level (ng/mL) [median (range)] | 157.6 (14.1–32,920) |
187.7 (17.4–36,110) |
| Tumor origin (n) | ||
| Pancreas | 42 (41.6%) | 49 (47.6%) |
| Midgut | 33 (32.7%) | 39 (37.9%) |
| Hindgut | 11 (10.9%) | 3 (2.9%) |
| Unknown or other | 15 (14.8%) | 12 (11.7%) |
| Hepatic tumor load (n, %) | ||
| 0% | 18 (17.5%) | 16 (15.8%) |
| 0 to <10% | 40 (38.8%) | 33 (32.7%) |
| 10 to <25% | 17 (16.5%) | 13 (12.9%) |
| 25 to <50% | 12 (11.7%) | 23 (22.8%) |
| ≥50% | 16 (15.5%) | 16 (15.8%) |
| Target lesions (n) | ||
| <2 | 33% | 36% |
| 3 or 4 | 23% | 22% |
| 5 or 6 | 37% | 34% |
| >6 | 7% | 10% |
| Progressive status (n, %) | 4 (4.0%) | 4 (3.9%) |
All patients provided written informed consent consistent with the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use–Good Clinical Practice and local legislation. The study was performed in accordance with the Declaration of Helsinki and was approved by the institutional review board of the ethics committee at each study site.
Assessment of LAN, CgA, and Tumor Progression
Serum LAN was measured at (i) baseline; (ii) between the first and second administrations (weeks 1–4)—either, two blood samples taken at 4 h (range 2–12 h) and 7 days (range 6–8 days) after drug administration, or two blood samples taken at 3 days (range 2–4 days) and 14 days (range 12–16 days) after drug administration (half of the patients were randomly allocated to the first sampling schedule, and the other half to the second sampling schedule); (iii) at week 4 prior to drug administration; (iv) between the sixth and seventh administrations (weeks 20–24), using the same sampling schedule as that established between the first and second administrations; and (v) at all treatment visits prior to study drug administration including at completion or withdrawal.
Levels were quantified using a radioimmunoassay (SGS Cephac, Saint Benoit, France), with a lower limit of quantification of 0.078 ng/mL, an intra‐assay precision of 2.7–5.8%, and an inter‐assay precision of 3.5–6.5%.
Tumor progression and CgA levels were assessed every 12 weeks during year 1 and every 24 weeks during year 2. Tumor progression was assessed using RECIST v1.0 (20) (preferably by CT, alternatively by MRI). An increase (>20%) of the sum of the longest tumor diameters or the appearance of a new lesion was deemed disease progression. In patients with elevated CgA levels at week 48, serum CgA levels were assessed every 12 weeks during year 2 using a solid‐phase two‐site immunoradiometric assay (Cisbio Bioassays, Codolet, France) with a lower limit of quantitation assessed by internal validation of 10 ng/mL, an intra‐assay precision of 4.2%, and an inter‐assay precision of 6.8–8.3%.
Data Analyses
A nonlinear mixed effect modeling (NLME), also known as the population approach (21), was used to analyze LAN, CgA, and PFS data. An NLME model consists of a structural model, a random effects model, and a covariate model. Interpatient variability was assumed to follow a log-normal distribution. The SAEM algorithm, implemented in NONMEM v7.2 (22), was used to estimate model parameters.
Analyses were performed sequentially: the PK model was selected and then the corresponding empirical Bayes parameter estimates were used to describe the time course of CgA and PFS.
Lanreotide Pharmacokinetics
The LAN pharmacokinetic properties were characterized as part of a pooled analysis of four clinical trial including patients with functioning and nonfunctioning GEP-NETs (23). The popPK model selected consisted on a one-compartment disposition model with an absorption process characterized by two parallel absorption pathways, following first- and zero-order kinetics, and was used to predict the corresponding serum profiles in LAN to develop the models for CgA dynamics and PFS using the model parameters represented in Supplementary Table S1.
Disease Progression Model—CgA Dynamics
A total of 1298 CgA measurements (placebo, n = 632; LAN, n = 666) were included in the analysis. Each patient contributed a median of seven samples (range 1–11).
CgA measurements followed a heavily right skewed distribution and were Box-Cox transformed (24) for the analysis (leading to a more normal distribution-like) according to Eq. 1:
| 1 |
where CgAi′ are the individual Box-Cox-transformed CgA measurements and λ is the power transform parameter, which was estimated to be −0.215 using the ‘powertransform’ function of the car package in R (25).
The first step in the model building process was to describe a disease progression model using only CgA levels from patients in the placebo arm. Disease progression models explored included the following: (i) empirical models where CgA levels increase either linearly, exponentially with time, or following a Gompertz equation; and (ii) semimechanistic models in which indirect response (26) or an unobserved tumor mass drives CgA synthesis (27).
After establishing the disease progression model, the effect of LAN on CgA levels was assessed. Several models (linear, E MAX, or sigmoidal E MAX) were tested to describe the relationship between individual predicted LAN levels and CgA dynamics. In addition, models including an effect compartment approach to account for any delays between LAN concentrations and reduction in CgA levels (28) or considering the development of resistance mechanisms were also explored.
The base population model, which better described CgA dynamics, was characterized by a linear (Box-Cox scale) increase of CgA levels over time (representing disease progression) and an inhibitory E MAX model (accounting for LAN effects as represented by Eq. 2):
| 2 |
where CgA0 is the CgA plasma concentration at the time of the start of the clinical trial, λ is the rate constant describing the linear increase of CgA levels, E MAX is the maximum inhibitory drug effects (i.e., maximum decrease in CgA levels due to LAN), C LA is the predicted individual LAN concentrations in serum, and C 50 is the LAN concentration required to obtain half of maximum CgA inhibition.
Due to the study characteristics (first CgA measurement was obtained after 12 weeks of the start of the study and continuous treatment with LAN along the study without off-drug periods), selection of more mechanistic models (considering synthesis and degradation rates of CgA (29)) was not feasible.
PFS—Informative Dropout
PFS was defined as time to disease progression or death within 96 weeks after the first study treatment. Study withdrawals due to disease progression were considered informative dropouts. Study withdrawals due to other reasons (e.g., protocol violation, consent withdrawal) were analyzed as censored information. We modeled informative dropouts simultaneously with CgA to describe the link between CgA dynamics and probability of having disease progression (30).
A parametric time-to-event model was used to describe PFS, allowing identification of the underlying hazard function [h(t), i.e., instantaneous rate of event], from which the survivor function (i.e., probability of remaining in the study) can be easily obtained by integrating the hazard with respect to time (31). Different distributions (exponential, Weibull, log-logistic, and Gompertz) were explored to describe the baseline hazard rate of progression. Parametric time-to-event models allow predictors to be included directly in the hazard function (both categorical/continuous and time-constant or variable variables). Different expressions of CgA were explored to relate the base hazard rate and CgA levels, which included the following: (i) full time course of CgA (CgAt), (ii) CgA levels at baseline (CgA0), and (iii and iv) the difference and ratio between CgA levels at each time and CgA levels at baseline (CgAt − CgA0 and CgAt/CgA0, respectively).
A Weibull distribution better described the underlying hazard. The Weibull model includes a scale parameter (β) and a shape parameter (γ). If γ = 1, then the hazard is constant over time, whereas values different than 1 allow the hazard to change over time. The inclusion of the ratio between CgA levels and CgA0 in the hazard function provided a better prediction of PFS. Therefore, the base model for PFS followed Eq. 3:
| 3 |
where β and γ are the base and shape parameters of a Weibull model, and the parameter α modulates the CgA effects.
Model Selection Criteria
Selection among models was based on the following: (i) the minimum value of the objective function provided by NONMEM, equal to −2 × Log(likelihood) (denoted –2LL); −2LL differences of 3.84 and 6.67 are considered significant at the 5% and 1% levels, respectively, for nested models differing in one parameter; (ii) precision of parameter estimates; and (iii) results from model performance judged by visual exploration of goodness of fit plots and predictive checks.
Covariate Selection
Once the base population models for CgA and progression-free survival were developed, a covariate analysis was performed. The following patient characteristics were considered for inclusion as covariates in the models: age; sex; weight; total number of target lesions at baseline; primary tumor location (categorized as pancreas vs. other locations, due to predominant pancreas location in both groups: 41.6% and 47.6% of the patients in the LAN and placebo groups, respectively); baseline hepatic tumor load, categorized either as mean <25% or ≥25% or by using five different categories: (i) 0%, (ii) 0 to ≤10%, (iii) 10 to ≤25%, (iv) 25 to ≤50%, and (v) >50%); and progressive status at baseline, defined according to RECIST using a screening CT scan obtained within a maximum of 14 weeks of baseline (Table I).
Covariate selection was performed using the stepwise covariate modeling implemented in the PsN software (32) by means of the −2LL ratio test with significance levels of 0.05 and 0.001 for the forward inclusion and for backward deletion approaches, respectively. For the case of continuous covariates, linear and nonlinear relationships between model parameters and covariates were evaluated as part of the stepwise selection procedure.
Model Evaluation
Evaluation of the final model was based mainly on simulation-based diagnostics. A total of 500 datasets with the same study design characteristics as the original were simulated based on the simultaneous biomarker-dropout model. For the evaluation of the CgA model, the 5th, 50th, and 95th percentiles of the simulated observations in each dataset were computed for the different time intervals. Informative dropout was included in model simulations. The 90% prediction interval of each calculated percentile was obtained and plotted against the 5th, 50th, and 95th percentiles of raw CgA levels. For the PFS model, simulated event (i.e., disease progression) times were obtained following the MTIME method (33) to create Kaplan-Meier visual predictive checks (VPC). Precision of parameter estimates was obtained from the analysis of 200 bootstrap datasets.
RESULTS
Figure 1a shows the individual profiles of LAN, CgA, and the empirical Kaplan-Meier curves describing PFS for the two treatment arms. The model schematically represented in Fig. 1b successfully described LAN time profiles, CgA dynamics, and PFS.
Fig. 1.
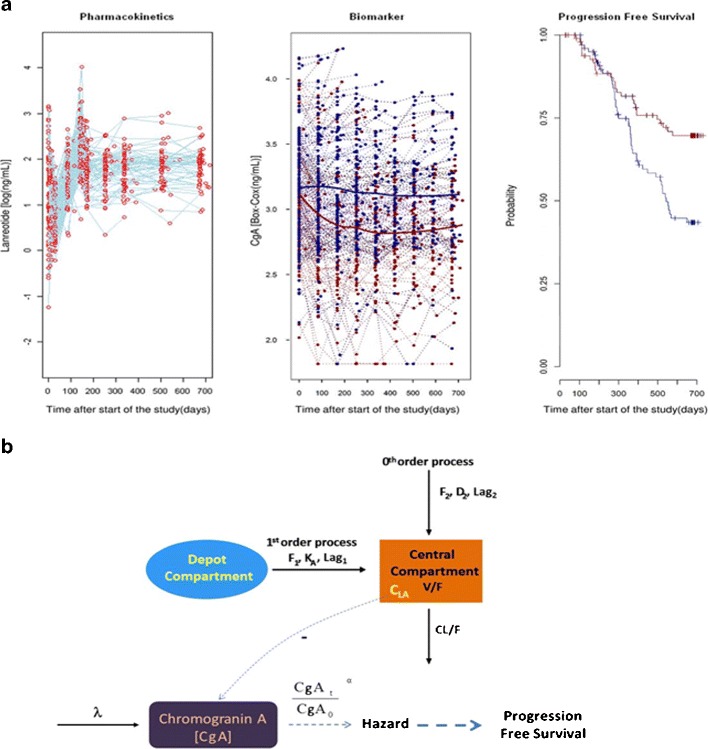
a Representation of available data included in the analysis: time profile of LAN concentrations (left), serum CgA biomarker profiles (center), and Kaplan-Meier of PFS (right). Dots in the left and center panels correspond to individual observations. Blue and red lines in the center and right panels depict the median time profiles for placebo- and LAN-treated patients, respectively. b Schematic representation of the PK/PD model for LAN, CgA, and PFS
LAN concentrations were adequately described by the population PK model (23). The estimated half-life of LAN was 0.59 h, and the model predicted trough (predose) value (2.5th–97.5th prediction intervals corresponding to the 120-mg dose administered subcutaneously every 4 weeks) was 6 (3–11) ng/mL. As shown in ref. (23), the selected population pharmacokinetic model provided a good descript of the concentrations vs. time data (non- and steady state).
CgA dynamics were characterized by a linear disease progression (Box-Cox scale) and an inhibitory E MAX model induced by LAN concentrations. The disease progression model for CgA dynamics during the study period (2 years) was characterized by a linear increase of CgA (Box-Cox transformed) over time; however, caution is recommended at the time of extrapolation beyond that period. Supplementary Fig. S1 shows the distributions of CgA levels under natural, logarithmic, and Box-Cox transformation. Data supported the estimation of interpatient variability in CgA0, disease progression rate (λ), and E MAX. As E MAX and C 50 parameters were not estimated precisely, reparameterization after defining C 50 as the ratio between E MAX and a slope parameter was used (34). Values of η-shrinkage were found to be 14.7%, 25.6%, and 42.7% for CgA0, λ, and slope, respectively. The remaining parameters were constrained to have a small degree of interpatient variability to facilitate estimation via the SAEM algorithm. In addition, interpatient variability was also included in the residual error variability, which resulted to be additive on the Box-Cox domain.
Parameters were estimated with adequate precision (Table II); in no case did the 95% confidence intervals (obtained by bootstrapping) include zero. Interpatient variability for rate of disease progression rate (λ) and the slope were high (around 150%) despite precision of those parameter estimates being adequate.
Table II.
Population PD Parameter Estimates of CgA and PFS Models
| Parameter/covariate model | Estimates | 5th–95th percentilea |
|---|---|---|
| θ CgA0 = 3.13 | 3.08–3.18 | |
| θ NLES = 2.4 × 10−2 | (1.8–3.2) × 10−2 | |
| θ AGE = 4.9 × 10−3 | (3.5–6.1) × 10−3 | |
| λ (ng/mL/day)b | 1.7 × 10−4 | (1.2–2.1) × 10−4 |
| E MAX (ng/mL)b | 5.3 × 10−1 | (4.6–6.7) × 10−1 |
| Slope | 9.5 × 10−2 | 6.6 × 10−2–1.5 × 10−1 |
| C 50 (ng/mL)c | 5.53 | 4.39–7.00 |
| Residual error (ng/mL)b | 6.5 × 10−2 | (6.0–7.1) × 10−2 |
| IIV_CgA0 [CV(%)]b | 11.6 | 10.4–12.6 |
| IIV_λ [CV(%)]b | 127 | 106–151 |
| IIV_Slope [CV(%)] | 153 | 116–217 |
| IIV_Res. error [CV(%)] | 57.6 | 50.4–65.0 |
| β | 7.6 × 10−4 | (6.7–8.9) × 10−4 |
| γ | 1.72 | 1.52–1.88 |
| α | 16.0 | 12.4–18.1 |
| θ HLOAD > 25% | 1.84 | 0.74–3.44 |
| θ TLOC(PANCREAS) | 1.21 | 0.34–2.36 |
CgA 0 CgA levels at baseline, λ disease progression rate, E MAX maximum effect on CgA decrease induced by LAN concentrations, Slope parameter used to estimate C 50 as the ratio between E MAX and Slope C 50, LAN concentration required to exhibit half of maximum inhibitory effect, IIV interpatient variability, β base parameter in Weibull model, γ shape parameter in Weibull model, α parameter governing the link between CgA and PFS
a90% confidence intervals calculated from 200 bootstrap datasets
bParameters in the Box-Cox domain
cSecondary parameters (i.e., derived from C 50 = E MAX / θ C50)
Covariate analysis identified the number of lesions at baseline (NLES) to be the most significant covariate in CgA0 (32-point reduction in −2LL; i.e., P < 0.001) among all covariates tested in the population CgA model. In addition to the number of lesions affecting CgA0, patients’ age was also identified as significant (30-point reduction in −LL; i.e., P < 0.001). Although primary tumor location and age were found to have a significant effect on the rate of disease progression (λ) initially (P < 0.05), they were removed during the backward deletion step (P < 0.001) and therefore were not kept in the final model. Supplementary Fig. S2 shows the relationship between baseline CgA levels (on the Box-Cox scale) and the aforementioned covariates found to be statistically significant. The selected covariates were included in the model through linear functions as shown in Eq. 4:
| 4 |
where θ CgA0 is the typical population estimate for CgA0, 4 is the median number of lesions at baseline, and 63 is the median age in the population studied.
Figure 2a shows the individual predictions of CgA dynamics in eight randomly selected patients receiving either placebo or LAN. Data for CgA were analyzed simultaneously with the informative dropouts and consequently were included in the construction of the VPCs. As shown in Fig. 2b, the model performs adequately in capturing the central trend and the spread of the data.
Fig. 2.
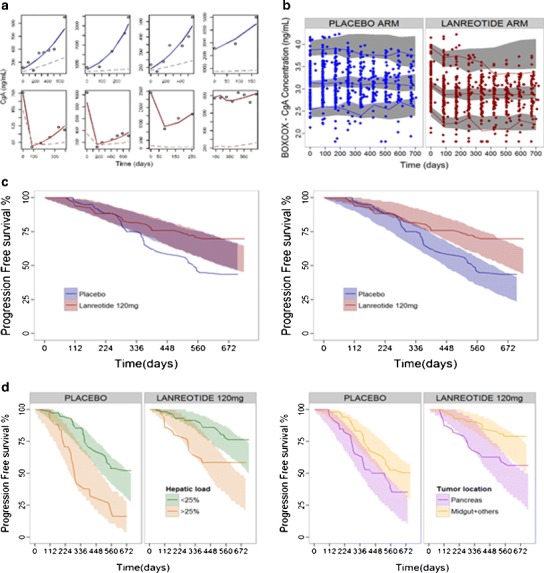
a Individual CgA observations (points) and CgA model predictions (light blue lines) vs. time from patients receiving placebo (top panel) or LAN (bottom panel). Dashed lines represent typical model predictions. b VPC corresponding to the selected final population PD model for CgA effects (including the model for dropout). Dots depict observations; lines correspond to 2.5th, 50th, and 97.5th percentiles of the observations; and gray shaded areas represent the 95% prediction intervals of the 2.5th–50th–97.5th percentiles of 500 simulated datasets. c Kaplan-Meier plot of observed progression-free survival in placebo (blue) and LAN arms (red) and 95% prediction intervals (shaded areas) based on 500 simulations for base hazard following a Weibull distribution (left panel) and hazard influenced by the ratio of CgA levels from baseline (right panel). d Kaplan-Meier plot of the final population model for PFS, stratified by the two main prognostic factors found in the model: hepatic tumor load (left panel) and primary tumor location (right panel). Lines depict observed PFS and shaded areas represent 95% prediction intervals based on 500 simulations
Inclusion of the ratio CgA/CgA0 on the hazard (Eq. 3) decreased the −2LL by 38 points (P < 0.001) and was found to be a better predictor than the other expressions tested relating CgA and PFS. In addition, the inclusion of the CgAt/CgA0 ratio on the hazard significantly improved model diagnostics of both CgA (not shown) and PFS (Fig. 2c).
Among the covariates tested as potential predictors of the PFS, hepatic tumor load and primary tumor location were found to be significant (P < 0.001). The inclusion of five different hepatic tumor load categories (i.e., four parameters) provided no significant improvement over using a single parameter for the two categories tested (<25% or ≥25%). Both covariates, hepatic tumor load (>25%) and pancreatic tumor, were associated with a higher hazard rate and were included in the hazard model according to Eq. 5:
| 5 |
where are explained in Eq. 3, and θ HLOAD and θ PTLOC represent the parameters accounting for the h(t) increase in patients with hepatic tumor load >25% and patients with pancreatic tumors, respectively (both parameters were set to 0 in case of baseline hepatic tumor load lower than 25% and primary tumor location outside the pancreas).
Table II also lists the parameters associated to the PFS models, which as in case of the model for CgA dynamics, were estimated with high precision. The observed Kaplan-Meier curve (stratified by significant covariates) was comprised within the 95% confidence intervals of the model-based simulated median Kaplan-Meier, suggesting that the hazard model described in Eq. 5 successfully described the probabilities of disease progression observed in the studied population (Fig. 2d).
DISCUSSION
We have developed a population model to describe the PK/PD of LAN administered by deep subcutaneous injection to patients with nonfunctioning GEP-NETS. The PD model included the description of CgA profiles and the clinical endpoint PFS. Figure 3 explores the link between LAN concentrations (simulated C trough concentrations representing typical and 5th–95th percentile profiles given interpatient variability in the pharmacokinetic model), CgA levels, and PFS. Of note, different LAN concentrations (Fig. 3a) lead to notable differences in the CgA time profiles (Fig. 3b) and, consequently, a drastic change in PFS (Fig. 3c).
Fig. 3.
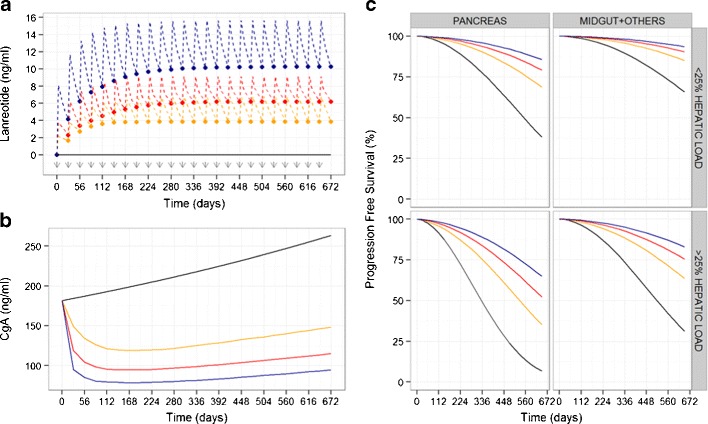
Simulated profiles to explain the link between lanreotide concentrations, CgA levels, and PFS. a Simulated lanreotide C trough concentrations after 120 mg subcutaneous injections every 28 days (time of administrations represented by gray arrows). Black line depicts LAN concentrations in patients receiving placebo; red line represents typical LAN profile in the population studied; blue and yellow lines depict 95th and 5th percentiles of possible C trough LAN concentrations given interpatient variability in the PK model. b Simulated CgA time course levels corresponding to LAN concentrations shown in a. c Simulated PFS curves according to the predose CgA levels shown in b for the main prognostic factors included in the final model (pancreatic tumors and hepatic tumor load >25%)
According to parameter estimates (Table II), typical CgA0 corresponds to 3.13 ng/mL on the Box-Cox scale, which translates to 181.5 ng/mL on the linear scale. The covariate effect results in a predicted CgA0 of 96.4 or 382.9 ng/mL, corresponding to a 63-year-old patient with one or seven target lesions at baseline (5th and 95th percentile of number of lesions in the studied population), respectively. A 1-year change from the median population age (63 years) correlates with a 5% change in CgA0.
The typical LAN concentration required to produce one half of the maximum effect was 5.53 ng/mL, corresponding approximately to the typical predose steady-state LAN concentration at steady-state in GEP-NET patients receiving 120 mg subcutaneous LAN every 4 weeks (Fig. 3a, red dashed line). The profiles shown in Fig. 3b indicate that LAN slows disease progression over the time period studied. On the contrary, CgA levels in an untreated individual would be increased by 20% after 1 year.
During the development of the model, it was confirmed that inclusion of informative dropouts in the biomarker analysis improved model diagnostics significantly. Note that in Fig. 1a, the central tendency of CgA levels in patients in the placebo group appears to be constant over time—giving the illusion of lack of disease progression. However, this can be explained by informative dropout: patients with CgA levels higher than baseline are more likely to drop out of the study; therefore, those patients remaining in the study at later time points will be those with smaller increases in CgA. In addition, it has been shown that ignoring informative dropout can potentially bias biomarker parameters (35).
The probability of disease progression in GEP-NETs was successfully described by an underlying Weibull model modified by three predictors. The ratio between predicted CgA levels and individual CgA at baseline (CgA0) was found to be the most significant predictor for PFS and accounted for the difference in PFS curves observed between the treatment and placebo arm (Fig. 2c). Interestingly, the treatment arm was not included as a covariate on the hazard since that information was implicitly included in the link between the CgA ratio and the PFS: CgA levels were typically reduced with respect to baseline in patients receiving LAN, whereas the main tendency in placebo patients was an increase of CgA levels from baseline. We found that PFS was significantly longer for patients receiving LAN than those patients receiving placebo, thus corroborating previous findings (6). The other two predictors of PFS were hepatic tumor load >25% at baseline and primary tumor located in the pancreas. These results are consistent with previous knowledge, which correlate hepatic tumor load and pancreatic tumors with worse prognosis in GEP-NETs (3,9,11,16,18,36).
To visualize the effect of CgA ratio on PFS, we performed simulations of median expected time to event (MTTE) given the observed range of CgA ratios at steady state (Fig. 4) in the different subpopulations (hepatic tumor load and pancreatic tumors). Assuming stable biomarker levels (i.e., CgAt/CgA0 = 1), hepatic tumor load >25% is predicted to be associated with 44% lower MTTE relative to hepatic tumor load <25%. Similarly, pancreatic tumor MTTE is 38% relative to primary tumors in other locations without treatment or disease progression.
Fig. 4.
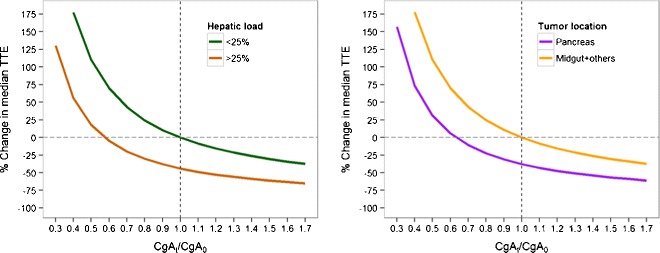
Relationship between CgAt/CgA0 ratio and median time to event (MTTE, i.e., time to disease progression) for different hepatic tumor loads (left panel) and tumor locations (right panel), assuming constant CgA levels at steady state
The general trend in all populations is that increasing levels of biomarkers leads to a reduction in PFS. The required level of biomarker inhibition to achieve a specified increase in MTTE is also dependent on the subpopulation. However, when considering substantial increases of MTTE (e.g., of more than 100%), the required inhibition is similar between populations. For example, to increase median time to event by 100, 48% and 65% inhibition of CgA levels is required for patients with hepatic tumor load <25% and hepatic tumor load >25%, respectively. Similarly, 48% and 61% inhibition of CgA levels is required for increasing median time to event by 100% for patients with pancreatic tumors and nonpancreatic tumors, respectively. This suggests that although hepatic tumor load and tumor location significantly affect PFS, LAN may be suitable for a broad population of patients if substantial biomarker inhibition can be achieved.
Currently, CgA is the most commonly accepted biomarker for monitoring patients with GEP-NET. Although CgA has been evaluated as surrogate marker of response (previous studies found that an early decrease of CgA levels is linked with favorable outcomes (37) and elevated CgA levels with poor overall prognosis (3,11,38)), it is deemed category 3 (i.e., “based upon any level of evidence, there is major disagreement”) by the National Comprehensive Cancer Network (NCCN) (39). None of these studies included either longitudinal analysis of CgA levels or a quantitative relationship integrating CgA time profiles with clinical outcome. In the present work, we used NLME modeling to assess the putative use of CgA as a marker for patient follow-up. The use of NLME models allows the integration of different sources of knowledge to describe the underlying time course of the disease. Indeed, the use of mathematical models to assess the predictive performance of circulating biomarkers has been highlighted previously (40–42). Certainly, there are several recent examples where mathematical models have been used to describe the time course of tumor markers and their link with clinical outcomes in different cancer indications. Some include human chorionic gonadotropin as an early predictor of methotrexate resistance in low-risk gestational trophoblastic neoplasia patients (43), mathematical models to personalize vaccination regimens to stabilize prostate-specific antigen (PSA) levels (42,44), soluble VEGF receptor 3 to monitor adverse events and clinical response in patients with imatinib-resistant gastrointestinal stromal tumors (45,46), a semimechanistic model involving lactate dehydrogenase (LDH) and neuron-specific enolase (NSE) dynamics to individualize disease monitoring in small cell lung cancer patients (27,47), and CA-125 as an early predictive biomarker of recurrent ovarian cancer (48).
Circulating tumor markers such as CgA are easily measured in peripheral blood and do not present the same limitations of imaging procedures regarding the frequency of measurement and, therefore, in conjunction with imaging techniques (i.e., CT scans), provide a powerful strategy to monitor disease. Indeed, the search for emerging tumor markers that can be used as prognostic and predictive factors of clinical outcome has increased substantially in the last decades. This urge has been driven by the ultimate objective to attain personalized medicine. In order to achieve this personalized approach to cancer management, the identification of significant prognostic and predictive factors that allow us to reliably separate, for example, those patients with more aggressive diseases or more likely to respond to certain treatments, is strictly required.
A strength of this investigation is the availability of biomarker and clinical outcome data from untreated patients in the CLARINET study (this is not frequent in the oncology field). Data on placebo patients allowed us to estimate the λ parameter which corresponds to the natural increase of CgA over time in the absence of treatment. Although published works in which NLME models have been applied to data from randomized placebo-controlled clinical trials in oncology are scarce, a recent example that includes data from placebo patients is the mathematical model of tumor growth kinetics in renal cell carcinoma patients after treatment either with placebo or pazopanib (49). Modeling tumor growth or biomarker dynamics data from untreated patients provide additional knowledge of the underlying disease proliferation and therefore enable a more realistic description of the behavior of the disease.
The results of the current investigation suggest that the change in CgA over time is a relevant covariate/predictor of PFS in GEP-NETs at the population level, in both untreated and treatment-naive patients. In addition, we found that patients with a primary tumor in the pancreas and patients with a baseline hepatic tumor load >25% are likely to have a worse prognosis.
The relationship established in this work between the biomarker CgA and PFS is limited by its restriction to treatment-naive patients. Further studies to identify how CgA levels affect clinical outcomes at the individual level are needed. In addition to the likely contribution of CgA to PFS, factors such as time elapsed from diagnosis, previous treatment with LAN or another somatostatin analog, and duration of treatment should be expected to show predictive effects.
CONCLUSIONS
Our results provide confirmatory evidence of the efficacy of LAN in GEP-NETs. To the best of our knowledge, this is the first analysis which develops a framework linking PK of LAN to biomarker dynamics and uses the latter to describe PFS. This framework offers a better understanding of the effect of treatment on a surrogate endpoint of PFS (CgA) and ultimately the clinical endpoint (PFS). One of the main advantages of this type of model-based framework combining LAN, CgA, and PFS is that models can be used to conduct simulations to predict PFS in new settings, predict long-term clinical outcome in phase III trials (50), or explore different dosing schedules.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
(DOCX 16.7 kb)
{kind=link}
Observed CgA levels distributions represented by histograms (upper panels) and Q-Q plots (lower panels) for nontransformed (natural scale), log-transformed and Box-Cox transformed CgA levels. (GIF 196 kb)
{kind=link}
Relationship between observed baseline CgA levels and main covariates that showed statistical significance in the CgA model, including patient age, primary tumor location and number of lesions. Gray line shows the smooth curve fitted by the loess function. (GIF 266 kb)
Acknowledgments
The authors would like to thank Nicholas Brown, Senior Publications Officer, Ipsen Biopharm Ltd.
Compliance with Ethical Standards
Conflict of Interest
This work was funded by Ipsen Pharma. Núria Buil-Bruna was supported by a predoctoral fellowship from Asociación de Amigos de la Universidad de Navarra. Marion Dehez, Amandine Manon, and Quyen Nguyen are employees of Ipsen which is the marketing authorization holder of Somatuline®, and Iñaki F. Trocóniz received research funding from Ipsen.
References
- 1.Yao JC, Hassan M, Phan A, Dagohoy C, Leary C, Mares JE, et al. One hundred years after “carcinoid”: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. 2008;26:3063–72. doi: 10.1200/JCO.2007.15.4377. [DOI] [PubMed] [Google Scholar]
- 2.Ramage JK, Davies AH, Ardill J, Bax N, Caplin M, Grossman A, et al. Guidelines for the management of gastroenteropancreatic neuroendocrine (including carcinoid) tumours. Gut. 2005;54(Suppl 4):iv1–16. doi: 10.1136/gut.2004.053314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arnold R, Wilke A, Rinke A, Mayer C, Kann PH, Klose K, et al. Plasma chromogranin A as marker for survival in patients with metastatic endocrine gastroenteropancreatic tumors. Clin Gastroenterol Hepatol. 2008;6:820–7. doi: 10.1016/j.cgh.2008.02.052. [DOI] [PubMed] [Google Scholar]
- 4.Kaltsas GA, Besser GM, Grossman AB. The diagnosis and medical management of advanced neuroendocrine tumors. Endocr Rev. 2004;25:458–511. doi: 10.1210/er.2003-0014. [DOI] [PubMed] [Google Scholar]
- 5.Modlin IM, Lye KD, Kidd M. A 5‐decade analysis of 13,715 carcinoid tumors. Cancer. 2003;97:934–59. doi: 10.1002/cncr.11105. [DOI] [PubMed] [Google Scholar]
- 6.Caplin ME, Pavel M, Ćwikła JB, Phan AT, Raderer M, Sedláčková E, et al. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N Engl J Med. 2014;371:224–33. doi: 10.1056/NEJMoa1316158. [DOI] [PubMed] [Google Scholar]
- 7.SOMATULINE DEPOT labeling revision 12/22/2014 reference ID: 3677425. http://www.accessdata.fda.gov/drugsatfda_docs/label/2014/022074s010lbl.pdf.
- 8.Somatuline® autogel®. Summary of product characteristics (SmPC). Available from: https://www.medicines.org.uk/emc/medicine/25104.
- 9.Oberg K, Akerstrom G, Rindi G, Jelic S, ESMO Guidelines Working Group Neuroendocrine gastroenteropancreatic tumours: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2010;21(Suppl 5):v223–7. doi: 10.1093/annonc/mdq192. [DOI] [PubMed] [Google Scholar]
- 10.Eriksson B, Oberg K, Stridsberg M. Tumor markers in neuroendocrine tumors. Digestion. 2000;62(Suppl 1):33–8. doi: 10.1159/000051853. [DOI] [PubMed] [Google Scholar]
- 11.Janson ET, Holmberg L, Stridsberg M, Eriksson B, Theodorsson E, Wilander E, et al. Carcinoid tumors: analysis of prognostic factors and survival in 301 patients from a referral center. Ann Oncol. 1997;8:685–90. doi: 10.1023/A:1008215730767. [DOI] [PubMed] [Google Scholar]
- 12.Granberg D, Wilander E, Stridsberg M, Granerus G, Skogseid B, Oberg K. Clinical symptoms, hormone profiles, treatment, and prognosis in patients with gastric carcinoids. Gut. 1998;43:223–8. doi: 10.1136/gut.43.2.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kulke MH, Siu LL, Tepper JE, Fisher G, Jaffe D, Haller DG, et al. Future directions in the treatment of neuroendocrine tumors: consensus report of the National Cancer Institute neuroendocrine tumor clinical trials planning meeting. J Clin Oncol. 2011;29:934–43. doi: 10.1200/JCO.2010.33.2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Italiano A. Prognostic or predictive? It’s time to get back to definitions! J Clin Oncol. 2011;29:4718. doi: 10.1200/JCO.2011.38.3729. [DOI] [PubMed] [Google Scholar]
- 15.Sargent DJ, Conley BA, Allegra C, Collette L. Clinical trial designs for predictive marker validation in cancer treatment trials. J Clin Oncol. 2005;23:2020–7. doi: 10.1200/JCO.2005.01.112. [DOI] [PubMed] [Google Scholar]
- 16.Pape UF, Berndt U, Muller-Nordhorn J, Bohmig M, Roll S, Koch M, et al. Prognostic factors of long-term outcome in gastroenteropancreatic neuroendocrine tumours. Endocr Relat Cancer. 2008;15:1083–97. doi: 10.1677/ERC-08-0017. [DOI] [PubMed] [Google Scholar]
- 17.Klimstra DS, Modlin IR, Coppola D, Lloyd RV, Suster S. The pathologic classification of neuroendocrine tumors: a review of nomenclature, grading, and staging systems. Pancreas. 2010;39:707–12. doi: 10.1097/MPA.0b013e3181ec124e. [DOI] [PubMed] [Google Scholar]
- 18.Johanson V, Tisell LE, Olbe L, Wangberg B, Nilsson O, Ahlman H. Comparison of survival between malignant neuroendocrine tumours of midgut and pancreatic origin. Br J Cancer. 1999;80:1259–61. doi: 10.1038/sj.bjc.6690494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rinke A, Muller HH, Schade-Brittinger C, Klose KJ, Barth P, Wied M, et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID study group. J Clin Oncol. 2009;27:4656–63. doi: 10.1200/JCO.2009.22.8510. [DOI] [PubMed] [Google Scholar]
- 20.Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92:205–16. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 21.Lindstrom ML, Bates DM. Nonlinear mixed effects models for repeated measures data. Biometrics. 1990;46:673–87. doi: 10.2307/2532087. [DOI] [PubMed] [Google Scholar]
- 22.Bauer R. NONMEM users guide introduction to NONMEM 7.2. 0. ICON Development Solutions Ellicott City, MD. 2011.
- 23.Buil-Bruna N, Garrido M, Dehez M, Manon A, Nguyen T, Trocóniz I. Population pharmacokinetic analysis of lanreotide depot/autogel in the treatment of neuroendocrine tumors: pooled analysis of four clinical trials. Clin Pharmacokinet. 2016. doi:10.1007/s40262-015-0329-4. [DOI] [PubMed]
- 24.Box GEP, Cox DR. An analysis of transformations. J R Stat Soc Ser B Methodol. 1964;26:211–52. [Google Scholar]
- 25.Fox J, Weisberg S. An R companion to applied regression. 2. Thousand Oaks: Sage; 2011. [Google Scholar]
- 26.Post TM, Freijer JI, DeJongh J, Danhof M. Disease system analysis: basic disease progression models in degenerative disease. Pharm Res. 2005;22:1038–49. doi: 10.1007/s11095-005-5641-5. [DOI] [PubMed] [Google Scholar]
- 27.Buil-Bruna N, López-Picazo J, Moreno-Jiménez M, Martín-Algarra S, Ribba B, Trocóniz IF. A population pharmacodynamic model for lactate dehydrogenase and neuron specific enolase to predict tumor progression in small cell lung cancer patients. AAPS J. 2014;16:609–19. doi: 10.1208/s12248-014-9600-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sheiner LB, Stanski DR, Vozeh S, Miller RD, Ham J. Simultaneous modeling of pharmacokinetics and pharmacodynamics: application to d-tubocurarine. Clin Pharmacol Ther. 1979;25:358–71. doi: 10.1002/cpt1979253358. [DOI] [PubMed] [Google Scholar]
- 29.Dayneka NL, Garg V, Jusko WJ. Comparison of four basic models of indirect pharmacodynamic responses. J Pharmacokinet Biopharm. 1993;21:457–78. doi: 10.1007/BF01061691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu C, Sale M. A joint model for nonlinear longitudinal data with informative dropout. J Pharmacokinet Pharmacodyn. 2003;30:83–103. doi: 10.1023/A:1023249510224. [DOI] [PubMed] [Google Scholar]
- 31.Collett D. Modelling survival data in medical research. Boca Raton: CRC; 2003. [Google Scholar]
- 32.Lindbom L, Pihlgren P, Jonsson N. PsN-toolkit—a collection of computer intensive statistical methods for non-linear mixed effect modeling using NONMEM. Comput Methods Prog Biomed. 2005;79:241–57. doi: 10.1016/j.cmpb.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 33.Nyberg J, Karlsson KE, Jönsson S, Simonsson USH, Karlsson MO, Hooker AC. Simulating large time-to-event trials in NONMEM. PAGE 23 (2014) Abstr 3166 [www.page-meeting.org/?abstract=3166].
- 34.Schoemaker RC, Van Gerven JM, Cohen AF. Estimating potency for the E max-model without attaining maximal effects. J Pharmacokinet Biopharm. 1998;26:581–93. doi: 10.1023/A:1023277201179. [DOI] [PubMed] [Google Scholar]
- 35.Bonate PL, Suttle B. Effect of censoring due to progressive disease on tumor size kinetic parameter estimates. AAPS J. 2013;15:832–9. doi: 10.1208/s12248-013-9487-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Panzuto F, Nasoni S, Falconi M, Corleto VD, Capurso G, Cassetta S, et al. Prognostic factors and survival in endocrine tumor patients: comparison between gastrointestinal and pancreatic localization. Endocr Relat Cancer. 2005;12:1083–92. doi: 10.1677/erc.1.01017. [DOI] [PubMed] [Google Scholar]
- 37.Kouvaraki MA, Ajani JA, Hoff P, Wolff R, Evans DB, Lozano R, et al. Fluorouracil, doxorubicin, and streptozocin in the treatment of patients with locally advanced and metastatic pancreatic endocrine carcinomas. J Clin Oncol. 2004;22:4762–71. doi: 10.1200/JCO.2004.04.024. [DOI] [PubMed] [Google Scholar]
- 38.Nikou G, Marinou K, Thomakos P, Papageorgiou D, Sanzanidis V, Nikolaou P, et al. Chromogranin a levels in diagnosis, treatment and follow-up of 42 patients with non-functioning pancreatic endocrine tumours. Pancreatology. 2008;8:510–9. doi: 10.1159/000152000. [DOI] [PubMed] [Google Scholar]
- 39.NCCN clinical practice guidelines in oncology/neuroendocrine tumors. Version 1.2015 [Internet]. [Cited accessed March 2015]. Available from: http://www.nccn.org/professionals/physician_gls/PDF/neuroendocrine.pdf.
- 40.Almufti R, Wilbaux M, Oza A, Henin E, Freyer G, Tod M, et al. A critical review of the analytical approaches for circulating tumor biomarker kinetics during treatment. Ann Oncol. 2014;25:41–56. doi: 10.1093/annonc/mdt382. [DOI] [PubMed] [Google Scholar]
- 41.Keizer RJ, Schellens JH, Beijnen JH, Huitema AD. Pharmacodynamic biomarkers in model-based drug development in oncology. Curr Clin Pharmacol. 2011;6:30–40. doi: 10.2174/157488411794941368. [DOI] [PubMed] [Google Scholar]
- 42.Kogan Y, Halevi-Tobias K, Elishmereni M, Vuk-Pavlović S, Agur Z. Reconsidering the paradigm of cancer immunotherapy by computationally aided real-time personalization. Cancer Res. 2012;72:2218–27. doi: 10.1158/0008-5472.CAN-11-4166. [DOI] [PubMed] [Google Scholar]
- 43.You B, Harvey R, Henin E, Mitchell H, Golfier F, Savage P, et al. Early prediction of treatment resistance in low-risk gestational trophoblastic neoplasia using population kinetic modelling of hCG measurements. Br J Cancer. 2013;108:1810–6. doi: 10.1038/bjc.2013.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kronik N, Kogan Y, Elishmereni M, Halevi-Tobias K, Vuk-Pavlović S, Agur Z. Predicting outcomes of prostate cancer immunotherapy by personalized mathematical models. PLoS One. 2010;5 doi: 10.1371/journal.pone.0015482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hansson E, Ma G, Amantea M, French J, Milligan P, Friberg L, et al. PKPD modeling of predictors for adverse effects and overall survival in sunitinib-treated patients with GIST. CPT Pharmacometrics Syst Pharmacol. 2013;2 doi: 10.1038/psp.2013.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hansson E, Amantea M, Westwood P, Milligan P, Houk B, French J, et al. PKPD modeling of VEGF, sVEGFR-2, sVEGFR-3, and sKIT as predictors of tumor dynamics and overall survival following sunitinib treatment in GIST. CPT Pharmacometrics Syst Pharmacol. 2013;2 doi: 10.1038/psp.2013.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Buil-Bruna N, Sahota T, Lopez-Picazo JM, Moreno-Jimenez M, Martin-Algarra S, Ribba B, et al. Early prediction of disease progression in small cell lung cancer: toward model-based personalized medicine in oncology. Cancer Res. 2015;75:2416–25. doi: 10.1158/0008-5472.CAN-14-2584. [DOI] [PubMed] [Google Scholar]
- 48.Wilbaux M, Hénin E, Oza A, Colomban O, Pujade-Lauraine E, Freyer G, et al. Dynamic modeling in ovarian cancer: an original approach linking early changes in modeled longitudinal CA-125 kinetics and survival to help decisions in early drug development. Gynecol Oncol. 2014;133:460–6. doi: 10.1016/j.ygyno.2014.04.003. [DOI] [PubMed] [Google Scholar]
- 49.Bonate PL, Suttle AB. Modeling tumor growth kinetics after treatment with pazopanib or placebo in patients with renal cell carcinoma. Cancer Chemother Pharmacol. 2013;72:231–40. doi: 10.1007/s00280-013-2191-0. [DOI] [PubMed] [Google Scholar]
- 50.Claret L, Girard P, Hoff PM, Van Cutsem E, Zuideveld KP, Jorga K, et al. Model-based prediction of phase III overall survival in colorectal cancer on the basis of phase II tumor dynamics. J Clin Oncol. 2009;27:4103–8. doi: 10.1200/JCO.2008.21.0807. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX 16.7 kb)
Observed CgA levels distributions represented by histograms (upper panels) and Q-Q plots (lower panels) for nontransformed (natural scale), log-transformed and Box-Cox transformed CgA levels. (GIF 196 kb)
Relationship between observed baseline CgA levels and main covariates that showed statistical significance in the CgA model, including patient age, primary tumor location and number of lesions. Gray line shows the smooth curve fitted by the loess function. (GIF 266 kb)