

Abstract
Disease-mediated therapeutic protein–drug interactions have recently gained attention from regulatory agencies and pharmaceutical industries in the development of new biological products. In this study, we developed a physiologically based pharmacokinetic (PBPK) model using SimCYP to predict the impact of elevated interleukin-6 (IL-6) levels on cytochrome P450 (CYP) enzymes and the treatment effect of an anti-IL-6 monoclonal antibody, sirukumab, in patients with rheumatoid arthritis (RA). A virtual RA patient population was first constructed by incorporating the impact of systemic IL-6 level on hepatic and intestinal expression of multiple CYP enzymes with information from in vitro studies. Then, a PBPK model for CYP enzyme substrates was developed for healthy adult subjects. After incorporating the virtual RA patient population, the PBPK model was applied to quantitatively predict pharmacokinetics of multiple CYP substrates in RA patients before and after sirukumab treatment from a clinical cocktail drug interaction study. The results suggested that, compared with observed clinical data, changes in systemic exposure to multiple CYP substrates by anti-IL-6 treatment in virtual RA patients have been reasonably captured by the PBPK model, as manifested by modulations in area under plasma concentration versus time curves for midazolam, omeprazole, S-warfarin, and caffeine. This PBPK model reasonably captured the modulation effect of IL-6 and sirukumab on activity of CYP3A, CYP2C9, CYP2C19, and CYP1A2 and holds the potential to be utilized to assess the modulation effect of sirukumab on the metabolism and pharmacokinetics of concomitant small-molecule drugs in RA patients.
KEY WORDS: cytochrome P450, interleukin-6, monoclonal antibody, sirukumab, therapeutic protein–drug interaction
INTRODUCTION
Some key drug-metabolizing enzymes, such as cytochrome P450s (CYPs), are known to be modulated by systemic proinflammatory cytokines released during infection or inflammation, resulting in alteration in biotransformation and elimination of small-molecule substrates of the affected CYPs (1). Systemic levels of interleukin-6 (IL-6), a potent proinflammatory cytokine, have been found elevated in patients with various systemic inflammatory diseases including psoriasis and rheumatoid arthritis (RA) (2,3) and patients with certain types of cancer (4). Several in vitro studies have demonstrated that higher (>100 pg/mL) concentrations of IL-6 suppressed the expression and activity of several CYP enzymes, such as CYP3As (including CYP3A4 and CYP3A5), CYP2C19, CYP2C9, and CYP1A2 (5–7). An in vitro study also reported that such suppressive effects could be attenuated by co-incubation with an anti-IL-6 antibody (5). Consistently, a recent in vivo drug–drug interaction (DDI) study conducted in RA patients showed that the administration of an anti-IL-6 receptor monoclonal antibody (mAb) tocilizumab reversed IL-6-induced suppression of CYP3A4 and CYP2C19 activity, as reflected by a significant decrease in systemic exposure to simvastatin (a CYP3A4 substrate) and omeprazole (a CYP2C19 substrate) following tocilizumab treatment (8,9). Another cocktail clinical DDI study conducted in RA patients also reported that treatment with an anti-IL-6 antibody sirukumab led to decreases in systemic exposure to CYP3A4 substrate midazolam, CYP2C19 substrate omeprazole, and CYP2C9 substrate S-warfarin but increases in systemic exposure to CYP1A2 substrate caffeine (10), further confirming the possibility of therapeutic protein–drug interactions (TP-DI) between anti-IL-6 therapeutic proteins (TPs) and small-molecule drugs in RA patients via modulation of CYP enzymes.
In recent years, a physiologically based pharmacokinetic (PBPK) modeling strategy has increasingly been employed during drug development and regulatory review (11,12). Physiologically based pharmacokinetic models comprise compartments based on the anatomy and physiology of the biological system. The mechanistic nature allows PBPK models to distinguish and characterize the interplay between drug-specific and biological system-specific parameters. Due to the comprehensive array of drug-independent system features, PBPK modeling may offer researchers an a priori approach to predict a compound’s pharmacokinetic (PK) behavior under a variety of clinical circumstances, such as variability in age, disease, and genetics, with prior knowledge of the biological system and the drug substance’s physicochemical characteristics (11,12). A recently published PBPK model successfully demonstrated the impact of IL-6 and the therapeutic effect of tocilizumab on CYP3A4 activity in RA patients by characterizing the changes of PK of CYP3A4 substrate simvastatin in RA virtual patients before and after treatment (13), suggesting the potential of PBPK modeling in describing the impact of RA disease and anti-IL-6 therapy on CYP enzyme activity as well as the associated changes in drug exposure.
This study’s objective was to develop a PBPK model with in vitro–in vivo extrapolation strategy that simultaneously characterized the impact of excessive exposure to IL-6 on multiple CYP enzymes and the treatment effect of 300 mg anti-IL-6 mAb sirukumab on the PK of CYP enzyme substrates midazolam (CYP3A), omeprazole (CYP2C19), S-warfarin (CYP2C9), and caffeine (CYP1A2) in patients with active RA.
MATERIALS AND METHODS
Pharmacokinetic Data
Pharmacokinetic data were obtained from the sirukumab clinical cocktail TP-DI study (10), which was an open-label, phase 1 study in men and women aged 18–65 years, inclusive, who had a diagnosis of RA and screening C-reactive protein ≥8.0 mg/L. Twelve patients, genotyped to exclude poor metabolizers of CYP2C9 and CYP2C19, were enrolled. In this study, patients received an oral cocktail of CYP probe substrates consisting of 0.03 mg/kg midazolam, 10 mg warfarin (plus 10 mg vitamin K), 20 mg omeprazole, and 100 mg caffeine at 1 week prior to (day 1) and 3 weeks after (day 29) a single subcutaneous dose of 300 mg sirukumab. Series of plasma samples were collected and analyzed for probe substrate concentrations of midazolam, S-warfarin, omeprazole, and caffeine using validated liquid chromatography coupled to mass spectrometry/mass spectrometry methods at Frontage Laboratories, Inc. (Exton, PA, USA). Pharmacokinetic parameters were described as arithmetic mean and standard deviation (SD). The geometric mean of post-/pre-sirukumab treatment ratios (day 29/day 1) for area under plasma concentration versus time curves (AUC) and peak concentration (C max) were calculated. The 90% confidence interval (CI) of the geometric mean ratio for each individual CYP probe substrate was also established.
Pooled Analysis of Systemic IL-6 Concentrations in RA Patients and Healthy Subjects
Literature data regarding systemic levels of IL-6 were pooled together to determine the population mean with SD for systemic levels of IL-6 in RA patients (8,10,14–20) and in healthy subjects (16,17,20–26). The mean or individual IL-6 plasma or serum concentration values from different studies were combined, and associated variability was calculated with R (http://www.r-project.org) and Excel (Microsoft, Redmond, WA). The impact of sample size and variability of each individual study were also included in the current analysis.
PBPK Models
The PBPK models for each individual CYP enzyme substrate (midazolam, omeprazole, S-warfarin, and caffeine) in the virtual RA patient population were developed and qualified in an absorption, distribution, metabolism, and excretion (ADME) simulator (SimCYP V13.1; SimCYP Limited, Sheffield, UK). The virtual RA patient population was characterized by incorporating the impact of systemic IL-6 level on hepatic and intestinal expression of multiple CYP enzymes of the healthy Caucasian population. Details of the general aspects of the PBPK model characteristics, enzyme dynamics, and the kinetics of victim drugs within the ADME simulator have been previously described (27,28). The simulator built-in library models of midazolam, omeprazole, S-warfarin, and caffeine were used in the current PBPK model to characterize plasma concentrations of these CYP enzyme substrates with modification through model optimization.
Modeling of IL-6 Profiles
The systemic IL-6 concentration used in the current PBPK model was simulated with the following model inputs, which are adopted from a recently published IL-6 PBPK model with modification (13): molecule weight = 21,000 g/mol systemic clearance (CLi.v.) = 1.0 L/h and volume of distribution at steady state (Vss) = 0.43 L/kg. IL-6 was introduced into the system via intravenous infusion (0.005–0.1 μg/h) for the duration of the simulation (40 days), and the resultant steady-state systemic concentrations ranged from 5 to 100 pg/mL. Simulated steady-state IL-6 concentrations were then linked to effects on multiple hepatic CYP enzyme levels, and new steady-state (SS) levels of hepatic CYP enzymes were achieved over the simulation period (depending on the setting of turnover rate of each individual CYP enzyme within Simcyp, 90% of SS was reached between 7 and 19 days after continuous exposure to IL-6). The suppressive effect of IL-6 on intestinal CYP enzymes was assumed to be the same as that on hepatic CYPs, and the intestinal CYP enzyme levels were manually modified in the virtual RA patient population (13,29,30). For each individual intestinal CYP enzyme, the remaining enzyme activity was calculated with the log (inhibitor/agonist) versus response model (details presented in “Modeling of Enzyme Dynamics” section) with the enzyme inhibition/induction information obtained from in vitro enzyme regulation studies (Table I).
Table I.
CYP Enzyme Regulation Parameters by IL-6 for PBPK Model Input
| Parameters (unit) | Values |
|---|---|
| E max_CYP1A2 a | 1.34 |
| EC50_CYP1A2 a (pg/mL) | 8.0 |
| E min_CYP2C9 b | 0.053 |
| EC50_CYP2C9 b (pg/mL) | 121.0 |
| E min_CYP2C19 b | 0.214 |
| EC50_CYP2C19 b (pg/mL) | 71.3 |
| E min_CYP3A4 c | 0.25 |
| EC50_CYP3A4 c (pg/mL) | 75.2 |
| E min_CYP3A5 b | 0.034 |
| EC50_CYP3A5 b (pg/mL) | 51.0 |
CYP cytochrome P450, PBPK physiologically based pharmacokinetic model
aValues were obtained by simultaneous fitting of effect of IL-6 on CYP1A2 activity data from literature (5) with the log (agonist) versus response model. The E max_CYP1A2 value used as PBPK model input was derived by normalizing model fitted value of E max to that of baseline activity value (E min), which was 107.5 and 80.33%, respectively
bValues were obtained by directly applying effect of interleukin-6 (IL-6) on CYP enzyme messenger RNA expression values reported in literature (5)
cValues were obtained by simultaneous fitting of effect of IL-6 on CYP3A4 activity data from literature (5) with the log (inhibitor) versus response model
Modeling of Enzyme Dynamics
Within the ADME simulator, the modulation effects of IL-6 on CYP enzymes were modeled as suppression on CYP2C9, CYP2C19, CYP3A4, and CYP3A5 and induction on CYP1A2 in the liver with the following equation modified from literature, based on the assumption that time-dependent concentration of IL-6 (IL-6t) affects the rate of hepatic enzyme production directly, and levels of IL-6 in circulation are similar to those in the liver and intestine (13):
where Enzact,H–i(t) represents the hepatic level of an active CYP isozyme at any given time and Enz0,H–i represents the basal hepatic level of the CYP isozyme (Enzact,H–i(0) = Enz0,H–i). E min/max is the minimum/maximal CYP enzyme activity (i.e., maximum suppression/induction) expressed as a fraction of vehicle control. EC50 is the IL-6 concentration that causes 50% of enzyme suppression/induction effect (E min/max); [IL-6]t represents the concentration of perpetrator (IL-6) at time t. Mean degradation rate constant (k deg,H–i) values of each specific hepatic CYP enzyme used in the simulations were the default values provided by the ADME simulator (27,28). The values of E min and EC50 for CYP2C9, CYP2C19, and CYP3A5 were taken directly from a recent in vitro study reported by Dickmann et al. (5) (Table I). The E min value for CYP3A4, the E max value for CYP1A2, and EC50 values for these two enzymes were obtained by re-analyzing the reported in vitro data from the same study (5) (Table I). The dose–response curves from different individual healthy donors (5) were first digitized using GetData software (version 2.24, http://getdata-graph-digitizer.com). EC50 and E max or E min values were derived with GraphPad Prism 6 (GraphPad Software, Inc., San Diego, CA) using the log concentration of perpetrator (IL-6) and remaining enzyme activity information with the log (inhibitor) versus response model (Y = E min + (E max − Emin)/(1 + 10[(X − logEC50) × Hill slope]), the log (agonist) versus response model (Y = E min + (E max − E min)/(1 + 10[(logEC50 − X) × Hill slope])), or the bell-shaped dose response model [Y = E max_plateau + [(E max_plateau − E max)/(1 + 10[(logEC50,1 − X) × Hill slope 1])] + [(E min − Emax_plateau)/(1 + 10[(X − logEC50,2) × Hill slope 2])]] (5).
Development and Validation of PBPK Model to Simulate IL-6–CYP Substrates Interaction in RA Patients Before and After Sirukumab Treatment
The PBPK model was developed with a stepwise strategy. First, plasma concentration profiles of individual CYP substrates in RA patients after sirukumab treatment from the sirukumab TP-DI study were simulated with the healthy European Caucasian virtual population provided by the ADME simulator. A visual prediction check was applied to evaluate the predictive accuracy of PBPK model prediction versus observed concentration-time profiles and appropriateness of curve shapes. Then, several necessary drug-specific parameters of the CYP substrates, such as fraction of absorption, absorption rate constant, single-adjusting compartment, distribution clearance, and intrinsic hepatic clearance, for all or some of the CYP substrates were optimized using the parameter estimation function and automated sensitivity analysis functions of the ADME simulator (Table II). Subsequently, plasma concentration profiles of the CYP substrates in RA patients before sirukumab treatment from the sirukumab clinical cocktail TP-DI study (10) were simulated with the developed virtual RA patient population and the optimized CYP substrate profiles. Model validation process was performed by comparing PBPK model prediction values to observed values from clinical TP-DI studies between tocilizumab and CYP3A4 substrate simvastatin and CYP2C19 substrate omeprazole, respectively, in RA patients after optimization of simvastatin drug-specific parameters (8,31).
Table II.
Modifications in Drug Specific Parameters of Each Individual CYP Enzyme Substrates by Model Optimization
| Parameters (unit) | Original values provided by SimCYP | Values after model optimization |
|---|---|---|
| faMidazolam | 1 | 0.9 |
| KaMidazolam (1/h) | 3 | 15 |
| VsacMidazolam (L/kg) | N/A | 0.3 |
| Q Midazolam (L/h) | N/A | 30 |
| Clint-CYP2C19-Omeprazole(μL/min/pmol of isoform) | 264 | 66 |
| Clint-CYP3A4-Omeprazole(μL/min/pmol of isoform) | 39.5 | 9.9 |
| FaS-Warfarin | 1 | 0.8 |
| KaS-Warfarin (1/h) | 1.85 | 15 |
| VsacS-Warfarin (L/kg) | N/A | 0.03 |
| Q S-Warfarin (L/h) | N/A | 0.5 |
| faCaffeine | 1 | 0.7 |
| KaCaffeine (1/h) | 2.18 | 10 |
| Clint-CYP3A4-Simvastatin(μL/min/pmol of isoform) | 2597 | 1800 |
Cl int intrinsic hepatic clearance, CYP cytochrome P450, fa fraction of absorption, ka absorption rate constant, Q distribution clearance, Vsac volume of single-adjusting compartment
All PK simulations were conducted using 10 trials containing 10 subjects each with CYP substrates orally administered on day 31 or day 39 of simulation to ensure the regulatory effect of IL-6 on the expression levels of all hepatic CYP enzymes reached SS at time of dosing. Mean and distribution of demographic covariates (e.g., age, sex, body weight, and genotypes) of the virtual subjects were generated via a Monte Carlo method within the ADME simulator. Interindividual variability of model parameters was incorporated within the PBPK model using the values predefined within the ADME simulator. AUC0–240 h or AUC0-24 h and C max of simulated plasma concentration profiles and the 90% CIs of simulated values were determined with the method provided by the ADME simulator. Since the model simulation suggested that at 240 h following administration, almost no dectable CYP substrates was found in the plasma, AUC0–240 h values were used to represent AUC0–infinity in the current analysis.
RESULTS
Re-analysis of In Vitro CYP3A4 and CYP1A2 Modulation Profiles from Literature (5)
The dose–response curves of 6β-hydroxytestosterone formation (CYP3A4 activity) under different IL-6 concentrations from 5 individual donors (Hu8110, Hu1242, Hu4151, Hu1001, and Hu8064) with complete dose–response information (1–10000 pg/mL) were fitted simultaneously with the log (inhibitor) versus response model (Fig. 1a). The derived E min and EC50 values of IL-6 on CYP3A4 activity were 25% and 75.2 pg/mL, respectively.
Fig. 1.
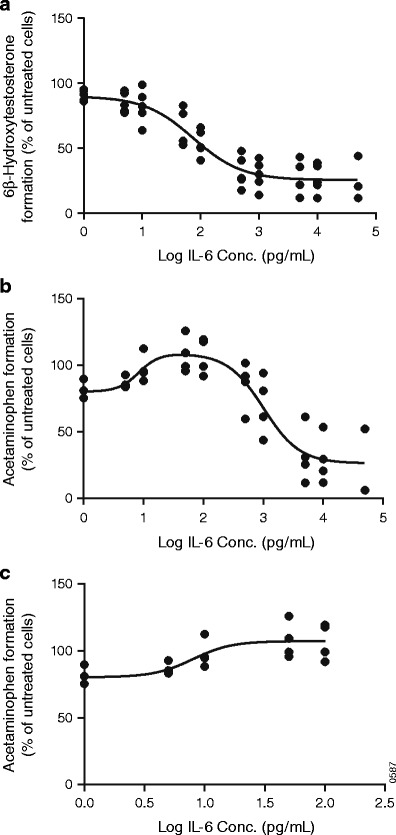
Re-analysis of the effects of interleukin (IL)-6 on modulation of CYP1A2 and CYP3A4. The inhibitory effect of IL-6 on CYP3A4 activity was presented with 6β-hydroxytestosterone formation from testosterone (a). The bell-shaped effect of IL-6 on CYP1A2 activity over the wide concentration range (1–50,000 pg/mL) (b) and the inductive effect of IL-6 on CYP1A2 activity at the lower concentration range (1–100 pg/mL) (c) were presented with acetaminophen formation from phenacetin. Symbols represent individual observed data digitized from literature (5). Data were fit to a variable slope dose–response model, and lines represent model estimation results
The dose–response curves of acetaminophen formation (CYP1A2 activity) under different IL-6 concentrations from 8 different individual healthy donors were first individually fitted with the bell-shaped dose response model. The 4 individuals’ dose response curves (Hu8110, Hu1242, Hu4151, and Hu8064) that had complete dose–response information (1–10000 pg/mL) were then fitted with the bell-shaped response model (Fig. 1b). Eventually, the lower IL-6 concentration (1–100 pg/mL) of these curves was re-analyzed simultaneously with the log (agonist) versus response model (Fig. 1c). The derived E max and EC50 values of IL-6 on CYP1A2 activity were 134% and 8.0 pg/mL, respectively.
Pooled Analyses of Systemic IL-6 in RA Patients and in Healthy Subjects
The reported baseline systemic IL-6 concentrations were highly varied, ranging from 1.24–11 pg/mL in healthy subjects (16,17,20–26) and from 3.51–119 pg/mL in RA patients (8,10,14–20). Based on the pooled analysis using data available in the literature, the estimated average systemic IL-6 concentrations in healthy subjects was 3.27 ± 2.38 pg/mL (Fig. 2a), while that in RA patients was 49.3 ± 48.5 (Fig. 2b).
Fig. 2.
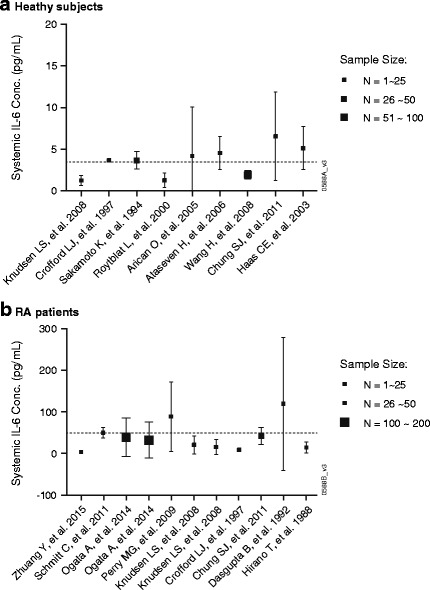
Pooled analysis of systemic (plasma or serum) interleukin (IL)-6 concentrations in healthy subjects (a) and rheumatoid arthritis (RA) patients (b) based on literature information from various sources. Symbols with standard deviation (SD) bars represented observed population mean ± SD data digitized from literature (8,10,14–26). Dashed lines represent the derived average IL-6 concentrations in healthy subjects (3.27 ± 2.38 pg/mL) and in RA patients (49.3 ± 48.5 pg/mL)
Prediction of CYP Enzyme Substrate PK in RA Patients Before and After Sirukumab Treatment
The developed PBPK model provides a consistent representation of the impact of elevated IL-6 and the treatment effect of anti-IL-6 mAb sirukumab on the activities of multiple CYP enzymes in RA patients, as manifested by the comparison of observed versus predicted PK profiles of midazolam (Fig. 3a, b), omeprazole (Fig. 3c, d), S-warfarin (Fig. 3e, f), and caffeine (Fig. 3g, h) in the absence of IL-6 (analogous to healthy subjects or RA patients treated with sirukumab) or presence of IL-6 (analogous to RA patients, where IL-6 average steady-state systemic concentration (CIL-6, SS) was assumed to be 50 pg/mL). Tables III and IV also suggest that the predicted AUC0−infinity and C max values of these CYP substrates with the presence of 50 or 0 pg/mL of IL-6, which represent pre- and post-sirukumab treatment systemic exposure, respectively, and the post-/pre-treatment AUC0−infinity and C max ratio values all reasonably captured the impact of sirukumab treatment on systemic exposure (represented by observed AUC0−infinity, C max, and the post-/pre-treatment AUC0−infinity and C max ratio values) to several CYP enzyme substrates in RA patients.
Fig. 3.
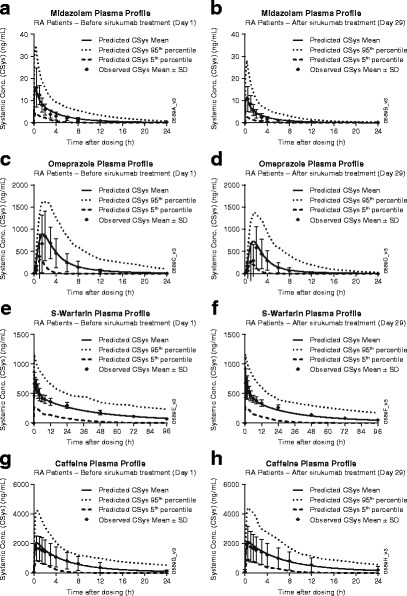
Observed versus predicted plasma concentration profiles for midazolam (a, b), omeprazole (c, d), S-warfarin (e, f), and caffeine (g, h) before and after sirukumab treatment in rheumatoid arthritis (RA) patients. Symbols represent mean ± standard deviation (SD) from CNTO136ARA1001 study (10). The solid, dashed, and dotted lines represent the predicted mean and 5 or 95% confidence interval of the current physiologically based pharmacokinetic model at respective cytochrome p450 enzyme substrates
Table III.
The AUC and AUC Ratios for Midazolam, Omeprazole, S-Warfarin, and Caffeine in RA Patients Before and After Sirukumab Treatment
| CYP substrates | Observations (10) | Predictions | ||||
|---|---|---|---|---|---|---|
| AUC_Day 1 a,c(pre-sirukumab) | AUC_Day 29 a,c(post-sirukumab) | AUC_Day29/AUC_Day1ratiob (post/pre-sirukumab) | AUC_IL-6 50 pg/mL a(pre-sirukumab) | AUC_IL-6 0 pg/mL a(post-sirukumab) | AUC_IL-6 0 pg/mL/AUC_IL-6 50 pg/mL ratiob(post/pre-sirukumab) | |
| Midazolam | 50.7 (24.3) | 32.6 (15.8) | 0.65 (0.47–0.89) | 57.5 (46.6) | 33.0 (27.4) | 0.57 (0.44–0.69) |
| Omeprazole | 3.72 (2.62) × 103 | 2.13 (1.54) × 103 | 0.59 (0.34–1.02) | 4.60 (3.91) × 103 | 3.00 (2.63) × 103 | 0.66 (0.54–0.77) |
| S-Warfarin | 2.43 (0.44) × 104 | 1.99 (0.34) × 104 | 0.82 (0.73–0.92) | 2.56 (1.81) × 104 | 1.92 (1.42) × 104 | 0.75 (0.63–0.87) |
| Caffeine | 1.40 (1.05) × 104 | 1.95 (1.58) × 104 | 1.34 (0.84–2.15) | 1.54 (1.22) × 104 | 1.98 (1.51) × 104 | 1.34 (0.99–2.08) |
AUC area under plasma concentration versus time curves, RA rheumatoid arthritis
aAUC (ng*h/mL) are presented as mean (SD) of 0 to infinity values
bAUC ratio is presented as geometric mean ratio (90% CI)
cPatients received an oral cocktail of CYP probe subs at 1 week prior to (day 1) and 3 weeks after (day 29) a single subcutaneous dose of 300 mg sirukumab
Table IV.
The C max and C max Ratios for Midazolam, Omeprazole, S-Warfarin, and Caffeine in RA Patients Before and After Sirukumab Treatment
| CYP substrates | Observations (10) | Predictions | ||||
|---|---|---|---|---|---|---|
| C max_Day 1 a,c(pre-sirukumab) | C max_Day 29 a,c(post-sirukumab) | C max_Day 29/ C max_Day 1 ratiob(post/pre-sirukumab) | C max _IL-6 50 pg/mL a(pre-sirukumab) | C max _IL-6 0 pg/mL a(post-sirukumab) | C max _IL-6 0 pg/mL/Cmax _IL-6 50 pg/mL ratiob(post/pre-irukumab) | |
| Midazolam | 17.3 (7.8) | 11.9 (5.7) | 0.69 (0.50–0.94) | 16.3 (10.4) | 11.9 (8.3) | 0.71 (0.57–0.84) |
| Omeprazole | 1069 (463.7) | 734 (466.1) | 0.62 (0.43–0.91) | 935 (432.2) | 767 (371.0) | 0.81 (0.67–0.92) |
| S-Warfarin | 780 (115.3) | 782 (147.2) | 1.00 (0.87–1.15) | 664 (285.6) | 664 (285.7) | 1.00 (0.99–1.01) |
| Caffeine | 1836 (888.0) | 2063 (1177.9) | 1.10 (0.80–1.52) | 2090 (1091.0) | 2158 (1112.1) | 1.03 (1.00–1.11) |
C max peak plasma concentration, CYP cytochrome, RA rheumatoid arthritis
a C max (ng/mL) is presented as mean (SD)
b C max ratio is presented as geometric mean ratio (90% CI)
cPatients received an oral cocktail of CYP probe subs at 1 week prior to (day 1) and 3 weeks after (day 29) a single subcutaneous dose of 300 mg sirukumab
Validation of PBPK Model Using PK Data from RA Patients Before and After Tocilizumab Treatment
In order to validate the developed PBPK model, predictions of PK profiles of CYP3A4 substrate simvastatin and CYP2C19 substrate omeprazole in RA patients before and after treatment with IL-6 receptor (IL-6R) antibody tocilizumab were conducted and compared with the observed values. The simulated simvastatin AUC0–24 h values were 95.7 ± 85.9 and 48.9 ± 42.7 ng*h/mL in RA patients with the influence of 50 pg/mL IL-6 or without any influence of IL-6, which represent pre- and post-tocilizumab treatment, respectively. The predicted mean effect ratio of tocilizumab (calculated as comparing CYP substrate systemic exposure after and before tocilizumab treatment) was 0.51 (90% CI 0.42–0.58). These values were similar to the observed AUC0–24 h and the post-/pre-treatment AUC ratio values, which were 102 ± 44 ng*h/mL, 42.3 ± 18 ng*h/mL, and 43% (90% CI 34–55%), respectively (8). The PBPK model prediction also suggested that anti-IL-6R treatment by tocilizumab may cause a 34% (90% CI 23%–46%) decrease in systemic exposure to omeprazole, which is also similar to the observed value (28%) in RA patients (31).
Sensitivity Analysis
Sensitivity analysis was conducted by simulating the treatment effect of sirukumab on the PK of midazolam, omeprazole, S-warfarin, and caffeine at different steady-state systemic IL-6 levels (5–100 pg/mL). The modulation in CYP substrates’ exposure in RA patients was expressed as the mean effect ratios of either C max or AUC0–infinity in RA patients after treatment with sirukumab—when IL-6 was neutralized—to C max or AUC0–infinity in untreated RA patients with excessive IL-6 exposure (Fig. 4). Simulations using concentration of 5 pg/mL of IL-6, which is close to the baseline levels of IL-6 reported in healthy subjects, suggested that anti-IL-6 treatment would exhibit minimal effect on CYP enzyme activity in healthy subjects. The post-/pre-treatment AUC0-infinity ratios of midazolam, omeprazole, S-warfarin, and caffeine were 0.87, 0.94, 0.97, and 1.16, respectively. Similarly, the post-/pre-treatment C max ratios of these compounds were 0.92, 0.97, 1.00, and 1.02, respectively. On the other hand, when baseline IL-6 levels elevated from 25 to 100 pg/mL, which represents the baseline levels of IL-6 reported in RA patients, our PBPK simulation results revealed that administration of sirukumab caused a remarkable drop in the post-/pre-treatment AUC ratio of midazolam (0.69–0.44), omeprazole (0.78–0.53), and S-warfarin (0.85–0.62) but an increase in that of caffeine (1.30–1.37). Similar trends were also observed in the post-/pre-treatment C max ratios of midazolam (0.80–0.62), omeprazole (0.88–0.74), and caffeine (1.03–1.04), while the post-/pre-treatment C max ratio of S-warfarin was unchanged (1.00–1.00).
Fig. 4.
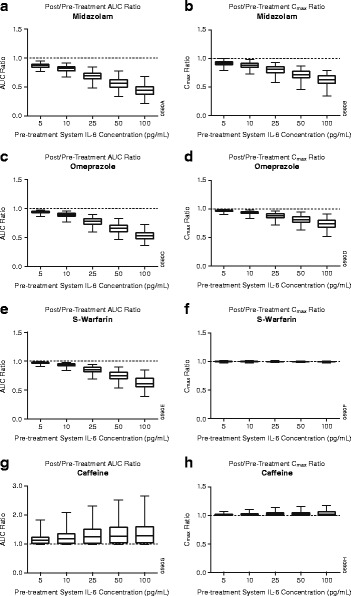
Predicted post-/pre-sirukumab treatment area under plasma concentration versus time curves (AUC0–infinity) and peak plasma concentration (C max) ratios of cytochrome p450 enzyme substrates for midazolam (a, b), omeprazole (c, d), S-warfarin (e, f), and caffeine (g, h) in patients with different pre-treatment systemic interleukin-6 (IL-6) levels. The data are represented in box and whisker plots
DISCUSSION
This PBPK model represents one of the first steps in using in vitro data to quantitatively predict the magnitude of clinical TP-DIs by simultaneously characterizing the impact of IL-6 on several important CYP enzymes (CYP3A4, CYP2C9, CYP2C19, and CYP1A2) in one integrated model using recently reported in vitro IL-6 modulation data obtained from human hepatocytes (5).
An accurate quantification of in vivo IL-6 level is critical for the development of this PBPK model, since IL-6 acts as the driving force of regulation of all CYP enzymes in vivo and, subsequently, the clinical TP-DIs between anti-IL-6 TPs and small-molecule drugs that are metabolized by these CYP enzymes. Systemic IL-6 levels are known to be highly variable in patients with RA and in healthy subjects. Our literature search revealed that the reported baseline systemic IL-6 concentrations in healthy subjects ranged from 1.24 to 6.56 pg/mL (16,17,20–26) while that in RA patients ranged from 3.51 to 119 pg/mL (8,10,14–20). Therefore, a pooled analysis was performed using the literature values, and the average IL-6 concentrations of the overall healthy subjects and RA population were obtained, which were 3.27 ± 2.38 pg/mL (Fig. 2a) in healthy subjects and 49.3 ± 48.5 pg/mL in RA patients (Fig. 2b); in the present PBPK model, the baseline IL-6 concentration was assumed to be 50 pg/mL in RA patients before sirukumab treatment. On the other hand, the IL-6 level in RA patients after sirukumab treatment was assumed to be similar as that in healthy subjects in the current PBPK model, since sirukumab binds to IL-6 with very high affinity (k d = 0.175 pM) (32), and a complete neutralization of free IL-6 in the system till day 42 after sirukumab treatment is expected. The predicted blood concentrations of sirukumab on days 15, 29, and 50 of the TP-DI study (day 7, day 21, and day 42 following a 300-mg subcutaneous sirukumab dose) are around 169, 77.6, and 27.3 nM, respectively, based on the PK information of sirukumab following a 100-mg subcutaneous dose (33), and the fact that sirukumab exhibited linear pharmacokinetics in human across a wide dosing range (0.3 to 10 mg/kg) (34). Consistently, the model prediction (Fig. 1, Tables III and IV) also reasonably characterized the PK behaviors of several CYP substrates in RA patients on days 15 and 50 of the TP-DI study (day 7 and day 42 after sirukumab treatment), which both are similar as that on day 29 (day 21 after sirukumab treatment) (10). In addition, the results from the sensitivity analysis revealed that the PBPK model prediction is sensitive to the baseline IL-6 levels (Fig. 4), which further suggested that the current choice of baseline IL-6 levels in this PBPK analysis may reasonably characterize the enzyme regulatory behavior of IL-6 in vivo.
Interleukin-6 has been generally considered as a suppressor for CYP enzymes (31). In vitro studies have shown that CYP3A4, CYP2C19, CYP2C9, and CYP1A2 expression and activities were suppressed at high IL-6 concentrations (>100 pg/mL) (5,6). At a more physiologically and pathologically relevant concentration range (1–100 pg/mL), however, it seems that IL-6 acts as a mild inducer for CYP1A2 expression and activity in some individual hepatocytes with large interindividual variability (5,7). The differentiation of IL-6’s behavior in modulation of various CYPs may be attributed to the fact that, unlike CYP3A4, CYP2C19, and CYP2C9, which are modulated by pregnane X receptor and constitutive androstane receptor (35), CYP1A2 is regulated by aryl hydrocarbon receptor (AhR) pathway (36). A study conducted in mice reported that knock-out of Ahr suppressed the induction effect of IL-6 on IL-17 (37), supporting the possibility of IL-6 in inducing CYP1A2 in human through AhR pathway. Additionally, other unknown regulation pathways may also contribute to such differentiation. In the current analysis, all individual enzyme kinetics data reported by Dickmann et al. (5) were digitized and pooled together for re-analysis. Post hoc analysis of the fitting results of the bell-shaped dose response curve suggested that the suppression effect of IL-6 on CYP1A2 did not become significant until its concentration reached 250 pg/mL. Therefore, the lower concentration part (1–100 pg/mL) of the whole dose–response curve data was used to estimate the induction effect of IL-6 on CYP1A2 in healthy subjects and in RA patients. This concentration range covered the average baseline IL-6 concentration ranges in both healthy subjects (1.24–6.56 pg/ml) (16,17,20–26) and those in RA patients (3.51–119 pg/mL) (8,10,14–20). The PBPK simulation results using the estimated E max and EC50 value of IL-6 on CYP1A2 induction reasonably captured the moderate modulation effect of IL-6 on caffeine PK in RA patients (Fig. 1, Tables III and IV). This analysis also reminds us that more attention should be drawn when integrating information and knowledge from different sources. The effect of IL-6 on CYP1A2 activity suggested that capturing the behavior of the perpetrator compounds (and/or other substances) at a physiologically and/or pathologically relevant concentration range may be critical for successful development of the PBPK model. Also, further investigation is necessary to fully delineate the impact of IL-6 on regulation of CYP1A2 expression and activity.
During the development of the PBPK model, prior knowledge of the biological system is often used to help identify the behavior of drugs in different systems, such as children and patients with decreases in renal function (12,38,39). This PBPK analysis, on the other hand, showcases the potential of using the drug information to help understand the system. In the current work, the impact of IL-6 on regulation of various CYP enzymes was identified by triggering IL-6 and downstream biological system in RA patients with the IL-6-neutralizing antibody sirukumab. As a result, the development of a PBPK model can also reasonably characterize the TP-DI in RA patients between CYP enzyme substrates and tocilizumab, an IL-6R antibody that blocks IL-6 binding to soluble and membrane-expressed IL-6R and the downstream signaling (8). It has been reported that tocilizumab treatment caused 57 and 28% decreases in system exposure to CYP3A4 substrate simvastatin and CYP2C19 substrate omeprazole, respectively (8,31). The results from our model validation process also reasonably captured such trends, which showed that blockage of IL-6 pathway caused 49% (90% CI 42–58% and 34% (90% CI 23–46%) decreases in systemic exposure to simvastatin and omeprazole, respectively. Ultimately, this PBPK model may also be applied to explore the potential impact of RA and anti-IL-6 treatment on metabolism and PK of small molecules that are metabolized by CYP enzymes.
CONCLUSION
In summary, this investigation demonstrates a successful example of practical use of PBPK modeling and simulation strategy, with a clearly defined mechanistic RA virtual population, to predict disease-mediated therapeutic protein–drug interactions. By adopting the up-to-date knowledge merging top–down (human PK data) and bottom–up (drug physicochemical properties, in vitro disposition data, and impact of disease factor) approaches, the impact of IL-6 and anti-IL-6 mAb sirukumab was well captured by the PBPK model. The model can be utilized to predict PK of small-molecule drugs in RA patients and the treatment effect of anti-IL-6. This PBPK analysis also can serve as conceptual framework and workflow process for demonstrating the applications of PBPK models as a supporting tool for development of other cytokine neutralizing antibodies. It also holds the potential to be used to explore the impact of cytokine modulation on small-molecule drug PK by bridging available in vitro and in vivo information.
ACKNOWLEDGMENTS
This study was supported by Janssen Research & Development, LLC. The authors thank Robert Achenbach of Janssen Scientific Affairs, LLC, for the manuscript preparation and submission support.
COMPLIANCE WITH ETHICAL STANDARDS
Conflict of Interest
The authors Jiang, Zhuang, Xu, Wang, and Zhou are employees of Janssen Research & Development, LLC, at the time of the study. All authors own stock in Johnson & Johnson.
REFERENCES
- 1.Morgan ET. Impact of infectious and inflammatory disease on cytochrome P450-mediated drug metabolism and pharmacokinetics. Clin Pharmacol Ther. 2009;85(4):434–438. doi: 10.1038/clpt.2008.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dowlatshahi EA, van der Voort EA, Arends LR, Nijsten T. Markers of systemic inflammation in psoriasis: a systematic review and meta-analysis. Br J Dermatol. 2013;169(2):266–282. doi: 10.1111/bjd.12355. [DOI] [PubMed] [Google Scholar]
- 3.Kishimoto T. IL-6: from its discovery to clinical applications. Int Immunol. 2010;22(5):347–352. doi: 10.1093/intimm/dxq030. [DOI] [PubMed] [Google Scholar]
- 4.Gao SP, Mark KG, Leslie K, Pao W, Motoi N, Gerald WL, et al. Mutations in the EGFR kinase domain mediate STAT3 activation via IL-6 production in human lung adenocarcinomas. J Clin Invest. 2007;117(12):3846–3856. doi: 10.1172/JCI31871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dickmann LJ, Patel SK, Rock DA, Wienkers LC, Slatter JG. Effects of interleukin-6 (IL-6) and an anti-IL-6 monoclonal antibody on drug-metabolizing enzymes in human hepatocyte culture. Drug Metab Dispos. 2011;39(8):1415–1422. doi: 10.1124/dmd.111.038679. [DOI] [PubMed] [Google Scholar]
- 6.Dallas S, Sensenhauser C, Batheja A, Singer M, Markowska M, Zakszewski C, et al. De-risking bio-therapeutics for possible drug interactions using cryopreserved human hepatocytes. Curr Drug Metab. 2012;13(7):923–929. doi: 10.2174/138920012802138589. [DOI] [PubMed] [Google Scholar]
- 7.Dickmann LJ, Patel SK, Wienkers LC, Slatter JG. Effects of interleukin 1beta (IL-1beta) and IL-1beta/interleukin 6 (IL-6) combinations on drug metabolizing enzymes in human hepatocyte culture. Curr Drug Metab. 2012;13(7):930–937. doi: 10.2174/138920012802138642. [DOI] [PubMed] [Google Scholar]
- 8.Schmitt C, Kuhn B, Zhang X, Kivitz AJ, Grange S. Disease-drug-drug interaction involving tocilizumab and simvastatin in patients with rheumatoid arthritis. Clin Pharmacol Ther. 2011;89(5):735–740. doi: 10.1038/clpt.2011.35. [DOI] [PubMed] [Google Scholar]
- 9.Actemra (package insert). South San Francisco CG, Inc; 2014.
- 10.Zhuang Y, de Vries DE, Xu Z, Marciniak SJ, Chen D, Leon F, et al. Evaluation of disease-mediated therapeutic protein-drug interactions between an anti-lnterleukin-6 monoclonal antibody (sirukumab) and cytochrome P450 activities in a phase I study in patients with rheumatoid arthritis using a cocktail approach. J Clin Pharmacol. 2015 doi: 10.1002/jcph.561. [DOI] [PubMed] [Google Scholar]
- 11.Huang SM, Rowland M. The role of physiologically based pharmacokinetic modeling in regulatory review. Clin Pharmacol Ther. 2012;91(3):542–549. doi: 10.1038/clpt.2011.320. [DOI] [PubMed] [Google Scholar]
- 12.Rowland M, Peck C, Tucker G. Physiologically-based pharmacokinetics in drug development and regulatory science. Annu Rev Pharmacol Toxicol. 2011;51:45–73. doi: 10.1146/annurev-pharmtox-010510-100540. [DOI] [PubMed] [Google Scholar]
- 13.Machavaram KK, Almond LM, Rostami-Hodjegan A, Gardner I, Jamei M, Tay S, et al. A physiologically based pharmacokinetic modeling approach to predict disease-drug interactions: suppression of CYP3A by IL-6. Clin Pharmacol Ther. 2013;94(2):260–268. doi: 10.1038/clpt.2013.79. [DOI] [PubMed] [Google Scholar]
- 14.Ogata A, Tanimura K, Sugimoto T, Inoue H, Urata Y, Matsubara T, et al. Phase III study of the efficacy and safety of subcutaneous versus intravenous tocilizumab monotherapy in patients with rheumatoid arthritis. Arthritis Care Res. 2014;66(3):344–354. doi: 10.1002/acr.22110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Perry MG, Kirwan JR, Jessop DS, Hunt LP. Overnight variations in cortisol, interleukin 6, tumour necrosis factor alpha and other cytokines in people with rheumatoid arthritis. Ann Rheum Dis. 2009;68(1):63–8. doi:10.1136/ard.2007.086561. [DOI] [PMC free article] [PubMed]
- 16.Chung SJ, Kwon YJ, Park MC, Park YB, Lee SK. The correlation between increased serum concentrations of interleukin-6 family cytokines and disease activity in rheumatoid arthritis patients. Yonsei Med J. 2011;52(1):113–120. doi: 10.3349/ymj.2011.52.1.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crofford LJ, Kalogeras KT, Mastorakos G, Magiakou MA, Wells J, Kanik KS, et al. Circadian relationships between interleukin (IL)-6 and hypothalamic-pituitary-adrenal axis hormones: failure of IL-6 to cause sustained hypercortisolism in patients with early untreated rheumatoid arthritis. J Clin Endocrinol Metab. 1997;82(4):1279–1283. doi: 10.1210/jcem.82.4.3852. [DOI] [PubMed] [Google Scholar]
- 18.Dasgupta B, Corkill M, Kirkham B, Gibson T, Panayi G. Serial estimation of interleukin 6 as a measure of systemic disease in rheumatoid arthritis. J Rheumatol. 1992;19(1):22–25. [PubMed] [Google Scholar]
- 19.Hirano T, Matsuda T, Turner M, Miyasaka N, Buchan G, Tang B, et al. Excessive production of interleukin 6/B cell stimulatory factor-2 in rheumatoid arthritis. Eur J Immunol. 1988;18(11):1797–1801. doi: 10.1002/eji.1830181122. [DOI] [PubMed] [Google Scholar]
- 20.Knudsen LS, Christensen IJ, Lottenburger T, Svendsen MN, Nielsen HJ, Nielsen L, et al. Pre-analytical and biological variability in circulating interleukin 6 in healthy subjects and patients with rheumatoid arthritis. Biomarkers. 2008;13(1):59–78. doi: 10.1080/13547500701615017. [DOI] [PubMed] [Google Scholar]
- 21.Sakamoto K, Arakawa H, Mita S, Ishiko T, Ikei S, Egami H, et al. Elevation of circulating interleukin 6 after surgery: factors influencing the serum level. Cytokine. 1994;6(2):181–186. doi: 10.1016/1043-4666(94)90040-X. [DOI] [PubMed] [Google Scholar]
- 22.Roytblat L, Rachinsky M, Fisher A, Greemberg L, Shapira Y, Douvdevani A, et al. Raised interleukin-6 levels in obese patients. Obes Res. 2000;8(9):673–675. doi: 10.1038/oby.2000.86. [DOI] [PubMed] [Google Scholar]
- 23.Arican O, Aral M, Sasmaz S, Ciragil P. Serum levels of TNF-alpha, IFN-gamma, IL-6, IL-8, IL-12, IL-17, and IL-18 in patients with active psoriasis and correlation with disease severity. Mediat Inflamm. 2005;5:273–279. doi: 10.1155/MI.2005.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ataseven H, Bahcecioglu IH, Kuzu N, Yalniz M, Celebi S, Erensoy A, et al. The levels of ghrelin, leptin, TNF-alpha, and IL-6 in liver cirrhosis and hepatocellular carcinoma due to HBV and HDV infection. Mediat Inflamm. 2006;2006(4) doi: 10.1155/MI/2006/78380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang H, Moser M, Schiltenwolf M, Buchner M. Circulating cytokine levels compared to pain in patients with fibromyalgia—a prospective longitudinal study over 6 months. J Rheumatol. 2008;35(7):1366–1370. [PubMed] [Google Scholar]
- 26.Haas CE, Kaufman DC, Jones CE, Burstein AH, Reiss W. Cytochrome P450 3A4 activity after surgical stress. Crit Care Med. 2003;31(5):1338–1346. doi: 10.1097/01.CCM.0000063040.24541.49. [DOI] [PubMed] [Google Scholar]
- 27.Jamei M, Marciniak S, Feng K, Barnett A, Tucker G, Rostami-Hodjegan A. The Simcyp population-based ADME simulator. Expert Opin Drug Metab Toxicol. 2009;5(2):211–223. doi: 10.1517/17425250802691074. [DOI] [PubMed] [Google Scholar]
- 28.Rowland Yeo K, Jamei M, Yang J, Tucker GT, Rostami-Hodjegan A. Physiologically based mechanistic modelling to predict complex drug-drug interactions involving simultaneous competitive and time-dependent enzyme inhibition by parent compound and its metabolite in both liver and gut—the effect of diltiazem on the time-course of exposure to triazolam. Eur J Pharm Sci. 2010;39(5):298–309. doi: 10.1016/j.ejps.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 29.Sanada H, Sekimoto M, Kamoshita A, Degawa M. Changes in expression of hepatic cytochrome P450 subfamily enzymes during development of adjuvant-induced arthritis in rats. J Toxicol Sci. 2011;36(2):181–190. doi: 10.2131/jts.36.181. [DOI] [PubMed] [Google Scholar]
- 30.Uno S, Kawase A, Tsuji A, Tanino T, Iwaki M. Decreased intestinal CYP3A and P-glycoprotein activities in rats with adjuvant arthritis. Drug Metab Pharmacokinet. 2007;22(4):313–321. doi: 10.2133/dmpk.22.313. [DOI] [PubMed] [Google Scholar]
- 31.Evers R, Dallas S, Dickmann LJ, Fahmi OA, Kenny JR, Kraynov E, et al. Critical review of preclinical approaches to investigate cytochrome p450-mediated therapeutic protein drug-drug interactions and recommendations for best practices: a white paper. Drug Metab Dispos. 2013;41(9):1598–1609. doi: 10.1124/dmd.113.052225. [DOI] [PubMed] [Google Scholar]
- 32.Gardner D, Lacy E, Wu S, Shealy D. Preclinical characterization of sirukumab, a human monoclonal antibody that targets human interleukin-6 signaling. Ann Rheum Dis. 2015;74:207. doi: 10.1136/annrheumdis-2015-eular.5124. [DOI] [Google Scholar]
- 33.Zhuang Y, Xu Z, de Vries DE, Wang Q, Shishido A, Comisar C, et al. Pharmacokinetics and safety of sirukumab following a single subcutaneous administration to healthy Japanese and Caucasian subjects. Int J Clin Pharmacol Ther. 2013;51(3):187–199. doi: 10.5414/CP201785. [DOI] [PubMed] [Google Scholar]
- 34.Xu Z, Bouman-Thio E, Comisar C, Frederick B, Van Hartingsveldt B, Marini JC, et al. Pharmacokinetics, pharmacodynamics and safety of a human anti-IL-6 monoclonal antibody (sirukumab) in healthy subjects in a first-in-human study. Br J Clin Pharmacol. 2011;72(2):270–281. doi: 10.1111/j.1365-2125.2011.03964.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chai X, Zeng S, Xie W. Nuclear receptors PXR and CAR: implications for drug metabolism regulation, pharmacogenomics and beyond. Expert Opin Drug Metab Toxicol. 2013;9(3):253–266. doi: 10.1517/17425255.2013.754010. [DOI] [PubMed] [Google Scholar]
- 36.Zhou SF, Wang B, Yang LP, Liu JP. Structure, function, regulation and polymorphism and the clinical significance of human cytochrome P450 1A2. Drug Metab Rev. 2010;42(2):268–354. doi: 10.3109/03602530903286476. [DOI] [PubMed] [Google Scholar]
- 37.Kimura A, Naka T, Nohara K, Fujii-Kuriyama Y, Kishimoto T. Aryl hydrocarbon receptor regulates Stat1 activation and participates in the development of Th17 cells. Proc Natl Acad Sci U S A. 2008;105(28):9721–9726. doi: 10.1073/pnas.0804231105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jiang XL, Zhao P, Barrett JS, Lesko LJ, Schmidt S. Application of physiologically based pharmacokinetic modeling to predict acetaminophen metabolism and pharmacokinetics in children. CPT Pharmacometrics Syst Pharmacol. 2013;2 doi: 10.1038/psp.2013.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hsu V, de LT Vieira M, Zhao P, Zhang L, Zheng JH, Nordmark A, et al. Towards quantitation of the effects of renal impairment and probenecid inhibition on kidney uptake and efflux transporters, using physiologically based pharmacokinetic modelling and simulations. Clin Pharmacokinet. 2014;53(3):283–293. doi: 10.1007/s40262-013-0117-y. [DOI] [PMC free article] [PubMed] [Google Scholar]