

Abstract
Human L1 elements are non-LTR retrotransposons that comprise ∼17% of the human genome. Their 5′-untranslated region (5′-UTR) serves as a promoter for L1 transcription. Now we find that transcription initiation sites are not restricted to nucleotide +1 but vary considerably in both downstream and upstream directions. Transcription initiating upstream explains additional nucleotides often seen between the 5′-target site duplication and the L1 start site. A higher frequency of G nucleotides observed upstream from the L1 can be explained by reverse transcription of the L1 RNA 5′-CAP, which is further supported by extra Gs seen for full-length HERV-W pseudogenes. We assayed 5′-UTR promoter activities for several full-length human L1 elements, and found that upstream flanking cellular sequences strongly influence the L1 5′-UTR promoter. These sequences either repress or enhance the L1 promoter activity. Therefore, the evolutionary success of a human L1 in producing progeny depends not only on the L1 itself, but also on its genomic integration site. The promoter mechanism of L1 is reminiscent of initiator (Inr) elements that are TATA-less promoters expressing several cellular genes. We suggest that the L1 5′-UTR is able to form an Inr element that reaches into upstream flanking sequence.
The human genome harbors ∼17% of so-called long interspersed elements (LINEs), or L1, the vast majority of which are 5′-truncated. Full-length 6-kb elements consist of an untranslated 5′-UTR that harbors a promoter, followed by two nonoverlapping open reading frames, ORF1 and ORF2, that encode an RNA-binding protein and a protein with reverse transcriptase and endonuclease activity. The L1 element is terminated by a 3′-UTR region that contains a poly(A) signal (Ostertag and Kazazian Jr. 2001; Kazazian Jr. 2004). Novel L1 copies can be generated by retrotransposition that involves target-primed reverse transcription; L1 RNA is reverse transcribed into DNA starting from a free 3′-hydroxyl group in the DNA strand produced by L1 endonuclease cleavage. L1 endonuclease cuts one DNA strand at the genomic target site at a 5′-TT/AAAA-3′ consensus sequence, more generally, 5′-(Y)n/(R)n-3′. The second cut in the opposite DNA strand occurs 7 to 20 bp downstream from the first cleavage, but does not display similar sequence preferences (Jurka 1997; Cost et al. 2002). The length and sequence of the target site duplication (TSD) created during L1 retrotransposition is determined by the distance between the first and the second cut by L1 endonuclease. It has been estimated that 80–100 L1 elements in the average human genome are capable of retrotransposition (Brouha et al. 2003). Besides retrotransposing its own RNA, the L1 retrotransposition machinery rarely displays trans-activity that mobilizes Alu and SVA elements, and that forms processed pseudogenes (Boeke 1997; Ostertag and Kazazian Jr. 2001; Wei et al. 2001; Dewannieux et al. 2003).
Transcription of L1 elements is obligatory for L1 retrotransposition, and tightly regulated L1 RNA has been detected in a small number of cell lines, such as NTera2D1, HeLa, HL60, and 293 (Skowronski and Singer 1985; Leibold et al. 1990). Methylation of CpG sites in the L1 5′-UTR is believed to down-regulate L1 transcription (Thayer et al. 1993; Yu et al. 2001).
For human L1 elements, the first 670 nt of the 5′-UTR, more precisely, the first 100 bp, display promoter activity (Swergold 1990). However, no TATA-box is present in this region. L1 transcription was reported to initiate predominantly at, or near, nucleotide +1 of the L1 element (Swergold 1990; Minakami et al. 1992). A binding site for the transcription factor YY1 has been mapped from nucleotide +13 until +21 of the L1 element (Minakami et al. 1992; Becker et al. 1993). Because YY1 is ubiquitously expressed, it cannot be solely responsible for the observed cell-type specificity of L1 transcription, though. Transcription factors belonging to the SRY family bind to two central regions within the L1 5′-UTR (nucleotides 472–477 and 572–577), and further modulate L1 transcription (Tchenio et al. 2000). More recently, RUNX3 transcription factor was shown to bind to nucleotides 83–101. Exogenous expression of RUNX3 up-regulated L1 transcription (Yang et al. 2003). Complexes of at least two hitherto unidentified proteins, potentially regulating L1 transcription, were mapped to the extreme L1 5′-end (Mathias and Scott 1993). Interestingly, sequence regions upstream from the L1 element were also protected in DNAse footprint experiments (Mathias and Scott 1993).
An evolutionarily successful strategy of L1 to persist in the genome must ensure that L1 source elements produce significant numbers of functional progeny. To persist in the genome, a master L1 element must be able to produce other full-length elements that are themselves able to produce other full-length elements in case the first element is rendered defective by mutations. Clearly, a mechanism to produce full-length L1 RNA that subsequently can be entirely retrotransposed is essential to maintain functional L1 elements in the genome.
Various events in the L1 retrotransposition process following transcription have been clarified by recent work (Ostertag and Kazazian Jr. 2001; Deininger et al. 2003). However, the important initial event, the machinery transcribing L1 elements, still remains poorly understood. In the present study, we reveal significant variability in L1 transcription initiation sites that is reminiscent of previous findings for TATA-less cellular promoters, so-called Initiators, and provide strong supporting data for recent speculations regarding reverse transcription of capped L1 transcripts (Boeke 2003). We furthermore report an important role of upstream flanking cellular sequence for the L1 5′-UTR promoter strength that has important evolutionary implications for L1 elements to serve as master elements. These observations further our understanding of the maintenance of full-length L1 elements, and they explain sequence characteristics frequently seen in human L1s.
Results
Variable transcriptional start sites in human L1 elements
Human L1 elements frequently harbor additional nucleotides between the 5′-target site duplication (TSD) and the actual start site. For instance, previously reported full-length L1s, for which TSDs were determined (Goodier et al. 2000; Pickeral et al. 2000), show such additional nucleotides in several full-length L1 elements (Fig. 1). The longest distance between the TSD 3′-end and the 5′-start of the L1 element was 13 nt for an L1 element reported in GenBank (accession no. AC005885). In this context, we define the L1 start as 5′-GGAGGAGCC...-3′, as only those nucleotides appear conserved among human L1 elements. We examined occurrence of nucleotides in a larger number of recently published full-length L1 elements (Brouha et al. 2003). The nucleotide -1 yielded frequencies of G: 60%; A: 28%; T: 7.5%; C: 3.7%. The nucleotide frequency in position nucleotide -2 was G: 63.5%; A: 26%; T: 4%; C: 6.7%. These nucleotide positions appear much less conserved, and are therefore not included in the human L1 consensus sequence. The higher occurrence of G nucleotides is addressed below.
Figure 1.

5′-Target site duplications (TSD) for various human L1 elements as previously analyzed and annotated by Goodier et al. (2000) and Pickeral et al. (2000). Accession numbers of the GenBank entries harboring the L1 element are given on the left. Only the TSDs upstream of the L1 elements are shown. The annotated TSD 3′-ends are further separated by a /. Note that a more stringent majority-rule consensus sequence defines the L1 start sequence as 5′-GGAGGA...-3′ (see text).
Additional nucleotides between the 5′-TSD and the L1 element start could be explained by transcription initiation upstream of the L1 element. When the resulting L1 transcript was then retrotransposed in its entirety, one would observe additional upstream nucleotides in the new L1 insertion. Obviously, that hypothesis requires that L1 transcriptional start sites vary. We therefore investigated in more detail transcriptional start sites in human L1 elements by 5′-RACE. We transfected different L1 element luciferase reporter constructs (L1.3, L1C, L1D and L1M; see Table 1) into Tera-1 cells, and performed L1 reporter construct-specific 5′-RACE on cellular total RNA isolated from transfected cells. cDNA synthesis was primed using a primer located in the reporter constructs' luciferase gene. An oligo(dT) forward primer and a reverse primer located at the 3′-end of the L1 5′-UTR were used for subsequent PCRs. We found for all investigated L1 constructs that transcription initiation was not restricted to the L1 nucleotide +1. Rather, only a minority of transcripts initiated at nucleotide +1. Transcription more often initiated a few nucleotides downstream in the L1 element, as well as a few nucleotides upstream in flanking sequence. The upstreammost transcription initiation identified in our study occurred at nucleotide -9 for construct L1D. We also tested transcription initiation sites in L1 constructs that contained different upstream flanking sequences (see below). There were no obvious changes in initiation sites observable when compared with the original L1 reporter constructs (Fig. 2).
Table 1.
L1 elements used in this study
| Name | Accession no. | Length 5′-flanking sequence | Nucleotide difference to L1.3 5′-UTR | Retroposition activity (%) |
|---|---|---|---|---|
| L1.3 | N.A. | 0 | N.A. | N.A. |
| L1A | AL137191 | 295 | 5 | 0 |
| L1C | AC002980 | 387 | 2 | 132 (h) |
| L1D | AC004200 | 290 | 4 | 80 (h) |
| L1E | AC004704 | 245 | 18 | 0.1 (w) |
| L1F | AC005885 | 282 | 4 | 2.3 (w) |
| L1G | AC005908 | 57 | 8 | 0.1 (w) |
| L1H | AC006027 | 107 | 10 | 3 (w) |
| L1I | AC026113 | 472 | 16 | 0 |
| L1J | AC013759 | 38 | 12 | 0 |
| L1K | AL021069 | 403 | 11 | 0.3 (w) |
| L1L | AL022399 | 420 | 9 | 0 |
| L1M | AL031586 | 74 | 12 | 0.1 (w) |
The first column incidates the L1 element designations used in this study. The second column gives the GenBank accession numbers harboring the respective L1 elements. The length of cellular sequence flanking for a given L1 element in the upstream direction is given, as well as the nucleotide difference of each 5′-UTR sequence compared with the 5′-UTR sequence in L1.3. Retrotransposition activities (high and weak) of L1 elements, compared with the activity of L1.3, are given according to Brouha et al. (2003).
Figure 2.

Variable transcription initiation sites for human L1 elements, as determined by 5′-RACE. Different original L1 constructs (upper part) used in this study were assayed in Tera-1 cells. The determined 5′-ends of transcripts are indicated by arrows. Multiple arrows indicate multiple transcription initiation events at the respective position. No significant differences were observed in mutant constructs (lower part). The various L1 element 5′-ends and upstream flanking sequences are indicated.
Hence, the varying transcription initiation sites seen for human L1 elements are consistent with upstream nucleotides frequently found between the 5′-TSD and the L1 start. Transcription initiation in upstream flanking sequence and subsequent retrotransposition of the full-length L1 transcript can explain occurrence of extra nucleotides between the 5′-TSD and the start of an L1 element.
Reverse transcription of the L1 cap structure
Frequently seen upstream nucleotides—between the 5′-TSD and the L1 start—are less or only moderately conserved (Fig. 1). However, there is an increase of upstream G nucleotides that is statistically significant for positions from nucleotide -3 to nucleotide -1 (Fig. 3A).
Figure 3.

Results of χ2 tests for statistical significance of higher G nucleotide frequencies immediately upstream of (A) the human L1 element start. (B) A significant higher number of G nucleotides in position nucleotide -1 is shown for HERV-W pseudogenes. (C) Human Alu elements do not display such an increase. Thresholds of significance are indicated by horizontal lines.
We suggest that the upstream G nucleotides can be explained by reverse transcription of capped L1 RNA. Basically, the cap is a modified GTP. It is known that retroviral reverse transcriptases can reverse-transcribe the cap structure (Hirzmann et al. 1993; Volloch et al. 1995). As L1 RNA is most likely transcribed by RNA Polymerase II (Ostertag and Kazazian Jr. 2001), a cap structure is very likely added to the L1 RNA 5′-end. If the L1 RNA is retrotransposed in its entirety, the cap could be reverse-transcribed, resulting in an additional G upstream from the L1 element. Such a mechanism could explain frequent occurrence of additional Gs.
We found strong evidence for reverse transcription of cap structures in the course of L1-mediated retrotransposition. The human endogenous retrovirus family HERV-W displays several sequence representatives in the human genome that are not due to provirus formations but are due to L1-mediated retrotransposition. These proviruses represent (processed) pseudogenes rather than proviruses. They display hallmarks of L1-mediated retrotransposition; they lack complete 5′- and 3′-LTRs, they end in a poly(A) tail, and they display TSDs with sequence characteristics also seen in L1 elements. Although several such HERV-W loci are 5′-truncated, some loci appear as a HERV-W RNA transcript that was retrotransposed in full length (Costas 2002; Pavlicek et al. 2002a,b). As HERV-W is transcribed by RNA Pol II, its RNA could acquire a cap, which might be reverse-transcribed into an additional G. We examined reported full-length HERV-W pseudogenes regarding frequency of G nucleotides upstream of the start of the HERV-W proviral RNA. We identified additional Gs in position nucleotide -1 that cannot be attributed to the TSD generated during retrotransposition (Fig. 4). The higher frequency of a G nucleotide in position -1 was statistically significant (Fig. 3B). We did not identify HERV-W LTR variants in the human genome displaying a G in that position of the LTR sequence. Thus, there is no evidence that the additional G nucleotides were derived from the HERV-W LTR, or sequence variants for that LTR. The additional G was most likely added posttranscriptionally, strongly suggesting that L1-mediated full-length retrotransposition of HERV-W Pol II transcripts includes reverse transcription of the cap, thereby generating an additional G nucleotide upstream of the HERV-W element.
Figure 4.

Frequent occurrence of a G nucleotide upstream of L1-mediated HERV-W pseudogenes. The underlined sequence at the top is the previously reported HERV-W LTR consensus sequence, also termed LTR17. Previously reported HERV-W full-length pseudogenes (Costas 2002; Pavlicek et al. 2002a) are exemplified. GenBank accession nos. harboring the respective pseudogenes are listed on the right. Target site duplications (TSD) were determined visually and are indicated on the left. Note that an additional G nucleotide is present in position nucleotide -1 that is not present in the LTR consensus sequence, and that is not found in LTR17 variants in the human genome, supporting reverse transcription of a CAP structure by the L1 retrotransposition machinery.
We analyzed in a similar fashion human Alu elements that are likewise retrotransposed by human L1. However, Alu elements are transcribed by RNA polymerase III, and no cap structure is added to Pol III transcripts. Hence, no extra G could be generated. In full support of our hypothesis, Alu elements do not display a higher number of G nucleotides immediately upstream of the Alu (Fig. 3C).
Extra upstream Gs seen for L1 elements may also be explained by untemplated addition of nucleotides by the L1 RT. If so, 5′-truncated L1 elements, resulting from incomplete reverse transcription, are expected to display such extra nucleotides. We examined 34 randomly selected human L1 elements displaying 5′-truncations for the presence of G nucleotides immediately upstream of the L1 sequence. We found the following nucleotide frequencies: C: 4; T: 9; A: 12; G: 9. These numbers provide no evidence for a higher frequency of G nucleotides immediately upstream from the L1 sequence. Additional upstream G nucleotides are therefore unlikely to be due to untemplated addition of nucleotides by the L1 RT.
Taken together, our results provide very strong evidence for reverse transcription of the cap structure in the course of L1 retrotransposition. Extra nucleotides between the 5′-TSD and the L1 start can be explained by both upstream transcription initiation and reverse transcription of the cap.
Transcriptional activity of L1 5′-UTRs
In the course of the study of the human L1 5′-UTR, we investigated transcriptional activities of several 5′-UTRs from evolutionarily young full-length human L1 elements. Those L1 elements were recently cloned from the human genome using long PCR with primers located in upstream and downstream flanking sequence (Table 1; Brouha et al. 2003). To assay promoter activity of the L1 elements' 5′-UTRs, we subcloned respective L1 5′-UTRs, including upstream flanking sequence, into a luciferase reporter vector. We assayed transcriptional activity in the Tera-1 and T47D cell lines by transient transfection and subsequent luciferase assays for 13 different L1 constructs. All transfections were normalized using values obtained for a cotransfected pCMVβ vector.
The various 5′-UTR constructs displayed significantly different promoter strengths in both Tera-1 and T47D cells. The overall promoter activities differed by a 12- to 24-fold range in Tera-1 and T47D cells, respectively. Notably, even when the promoter strength was ∼10-fold higher in Tera-1 than in T47D, the relative transcription efficiencies were very similar in the two cell types (Supplemental Fig. A).
The various L1 element 5′-UTRs display variable numbers of sequence differences (Table 1). We examined the various 5′-UTR sequences to determine whether differences in transcriptional activities could be attributed to specific nucleotide alterations. Potential binding of transcription factors (TFs) to the L1 5′-UTR was predicted using the Transcription Element Search System (TESS; http://www.cbil.upenn.edu/tess). When comparing potential TF binding sites with variant nucleotide positions, we could not attribute different transcriptional activities to specific nucleotide variations. We therefore also considered other factors influencing the 5′-UTR promoter activity. The various L1 elements derived from diverse locations in the human genome (Brouha et al. 2003). Although the 5′-UTRs are very similar in sequence, and the few sequence differences do not explain different promoter activities, the upstream flanking sequences are obviously unrelated and therefore dissimilar in sequence. We therefore investigated the impact of the dissimilar upstream flanking sequences on the L1 5′-UTR promoter activity.
Impact of genomic 5′-flanking sequences on L1 transcription
To test the influence of the genomic flanking sequences on L1 transcription, we first examined the impact of the 387-bp, 290-bp, 282-bp, 107-bp, and 74-bp upstream flanking cellular sequences present in constructs L1C, L1D, L1F, L1H, and L1M, respectively, on the promoter activity of the L1.3 5′-UTR. We cloned the various upstream flanking sequences in their entirety immediately upstream of the L1.3 5′-UTR. In Tera-1 and T47D cells, the flanking sequences derived from L1C, L1D, L1F, and L1H significantly decreased the L1.3 5′-UTR promoter activity, with L1H flanking sequence reducing promoter activity to ∼20% of the original L1.3 construct. In contrast, the flanking sequence from L1M significantly increased the L1.3 5′-UTR activity (Fig. 5).
Figure 5.

Influence of upstream flanking cellular sequence on the L1 5′-UTR promoter activity. Luciferase reporter assays were performed in both Tera-1 (upper panel) and T47D cells (lower panel). In both panels, promoter activities of the L1.3 5′-UTR in combination with upstream flanking sequences from various other L1 elements are shown in the left part. Upstream flanking sequences from L1C, L1D, L1F, and L1H significantly reduced the L1.3 5′-UTR promoter activity. The upstream flanking sequence from L1M significantly increased the L1.3 5′-UTR promoter activity. The right part depicts promoter activities of various L1 5′-UTRs when flanking cellular sequences are included or excluded from reporter constructs. The tested L1 constructs are about a tenth as active in T47D cells as in Tera-1 cells. Note that the 5′-UTRs display some differences in transcriptional activities when assayed without flanking sequences. However, flanking sequences modulate the 5′-UTR activities considerably. Standard deviations are indicated.
Because the effect of the various upstream flanking sequences on the L1.3 5′-UTR promoter activity appeared intrinsic to each flanking sequence, we examined the influence of each flanking sequence on the promoter activity of the respective downstream 5′-UTRs. To do so, we deleted upstream flanking sequence regions from constructs L1C, L1D, L1F, L1H, and L1M, and assayed promoter activities of the respective sole 5′-UTRs in Tera-1 and T47D cells. Deletion of upstream flanking sequences in constructs L1C, L1D, L1F, and L1H considerably increased the 5′-UTR promoter activity. For example, the deletion variant for L1C was ∼60% more active than the original construct. In contrast, the deletion variant for L1M was ∼55% less active than the original construct. Similar differences were observed in both the Tera-1 and the T47D cell line (Fig. 5). The p-values for all observed differences were in the range of 0.03 to 0.0001 (mean 0.0057, standard deviation 0.0093). These results further demonstrate an important role of 5′-flanking sequence on the L1 5′-UTR promoter by acting as a repressor or enhancer.
We next asked whether sequence alterations in nucleotides immediately flanking the L1 5′-UTR influenced promoter activity. We generated three reporter constructs, derived from L1.3, for which the first 10 bp immediately upstream of the L1 start were replaced by the corresponding sequences present in L1C, L1D, and L1F. We furthermore generated four reporter constructs for which the first 5 bp immediately upstream of the L1.3 start were altered to G5, A5, T5, or C5. The different modifications did not significantly affect promoter activities when assayed in Tera-1 and T47D cells (Fig. 6). Therefore, the nucleotide composition immediately upstream from the 5′-UTR does not appear to influence L1 5′-UTR promoter activity.
Figure 6.

No influence of 10-bp and 5-bp sequence alterations immediately upstream of the L1.3 5′-UTR on the promoter activity. For instance, flC10-L1.3 is an L1.3 derivative for which the first 10 bp immediately upstream of the 5′-UTR were replaced by the corresponding sequence region in L1C. (G)5 indicates that the first 5 bp immediately upstream of the L1.3 5′-UTR have been altered to G nucleotides.
Discussion
Although transcription of L1 sequences is a crucial step in the course of L1 retrotransposition, the machinery regulating and initiating transcription of L1 is poorly understood. Our study provides important insight into the initiation of human L1 transcription. We show here that transcription initiation sites of human L1 elements are much more variable than previously thought (Swergold 1990; Minakami et al. 1992). Human L1 elements frequently display extra nucleotides between the 5′-TSD and the actual L1 start. These extra nucleotides evidently do not belong to the TSD. We hypothesized that such extra nucleotides indicated transcription initiation upstream from the L1 nucleotide +1, and subsequent retrotransposition of that longer RNA. Indeed, 5′-RACE revealed that transcription initiation occurred at nucleotide +1 of the L1 element in only a minority of cases. The majority of initiation sites were located in upstream flanking sequence and downstream L1 5′-UTR sequence, with initiation sites ranging from nucleotide -9 to nucleotide +4. Furthermore, our results can explain 5′-truncated L1 elements, lacking only a few nucleotides at the 5′-end, such as the L1 element in GenBank accession no. HS798A17 that is lacking the first 8 nt (see Fig. 1). Either, the precursor L1 RNA was not entirely reverse transcribed by the L1 RT, or, fully supported by our findings, the precursor L1 RNA only had this length because transcription had initiated at that position. Two 5′-transductions of 145 bp and 215 bp were recently identified. In these instances, transcription in the precursor element would have initiated in a more distant upstream position (International Human Genome Sequencing Consortium 2001; Symer et al. 2002). However, a cellular promoter other than the L1 5′-UTR could have produced the respective RNAs.
Furthermore, our study can explain higher frequency of extra G nucleotides between the 5′-TSD and the L1 start. In principle, extra G nucleotides could stem from reverse transcription of transcripts that were initiated in GC-rich regions upstream of the actual L1 start site. Based on our observations, we favor another explanation. We suggest that the L1 RT is able to reversetranscribe the RNA 5′-cap structure that is added upon RNA Pol II-mediated transcription. L1 RNA being transcribed by RNA Pol II is strongly indicated (see Ostertag and Kazazian Jr. 2001 for a more detailed discussion). A reverse-transcribed cap would result in an additional G for a fully retrotransposed L1 RNA. Retroviral RT has previously been reported to reverse-transcribe the cap (Hirzmann et al. 1993; Volloch et al. 1995). We also present evidence for reverse transcription of a cap in L1-mediated retrotransposition, of processed pseudogenes from the HERV-W family of human endogenous retroviruses (Costas 2002; Pavlicek et al. 2002a). Pol II-transcribed HERV-W RNAs that were retrotransposed in full-length display a significantly higher number of Gs immediately flanking the pseudogene 5′-end. In contrast, Alu elements do not display an additional G, as they are transcribed by RNA Pol III, and no cap is added. Notably, the L1 element in GenBank accession no. HS798A17 (lacking the first 8 nt) displays an extra G that is not part of the TSD or the L1 sequence (Fig. 1). The same is true for an L1 element in GenBank accession no. AL158193 (Brouha et al. 2003) that lacks the first 28 nt of the L1 sequence and displays an extra G between the 5′-TSD and the L1 start (data not shown). For both L1 elements, transcription very likely initiated at nucleotide 8 or nucleotide 28, respectively, of the precursor L1, and the cap was reverse-transcribed during retrotransposition into an extra G. In contrast, human L1 elements with severe 5′-truncations that are due to premature termination of reverse transcription do not display a higher number of Gs immediately upstream of the L1 5′-end, excluding untemplated addition of nucleotides, preferentially G. Taken together, our data strongly suggest that the L1 machinery is able to reversetranscribe cap structures, producing additional upstream G nucleotides.
Some L1 elements included in our study display several upstream G nucleotides between the 5′-TSD and the L1 start site. This could be explained by L1 insertion into a G-rich genomic region (e.g., AC002385, AC005939, and AC004554 in Fig. 1). However, the G-rich region then would be part of the TSD, which is not the case. It was recently shown that full-length L1 inserts retain the capacity for retrotransposition (Kimberland et al. 1999). In light of the postulated reverse transcription of cap structures, we suggest that stretches of G result from repeated L1 retrotransposition cycles. Here, an L1 element was precursor to a new L1 insert. The latter then included an additional G from reverse transcription of the cap. The new element then itself produced another L1 insert. When transcription initiated at the “new” G, another G was added, and so on. Boeke (2003) has also suggested a higher frequency of G nucleotides due to repeated rounds of cap reverse transcription. Our study presents further strong support for this “exotic idea” (Boeke 2003). We further conclude that slightly truncated L1 elements (e.g., the L1 in AL158193) could acquire only one extra G because they very likely are rendered transcriptionally inactive by the lack of a YY1 site that is important for promoter activity (Minakami et al. 1992; Becker et al. 1993).
To date, the L1 5′-UTR has been thought to regulate L1 transcription. Our study strongly suggests that not only L1 elements but also their 5′-flanking sequences should be regarded when studying the transcriptional regulation of L1. The various L1 5′-UTRs displayed different transcriptional activities when tested as sole 5′-UTRs. Thus, single or a combination of nucleotide differences within the 5′-UTRs influence the promoter activity. However, for each 5′-UTR, the cellular sequence, originally flanking the L1 element, modulated the 5′-UTR activity significantly. Upstream flanking cellular sequences present in L1 elements L1C, L1D, L1D, and L1H significantly reduced the L1.3 5′-UTR promoter activity, whereas the flanking sequence from L1M increased the L1.3 5′-UTR promoter activity. Repressing or enhancing effects of upstream flanking sequences were further corroborated by reporter constructs that lacked the flanking sequences.
It is currently unclear which factors, such as common motifs, influence the 5′-UTR promoter activities. At this point, we exclude the possibility that nucleotides just upstream of the L1 5′-UTR affect promoter activity, as sequence alterations of 10 bp or 5 bp immediately upstream of the L1.3 5′-UTR did not significantly alter the L1.3 5′-UTR promoter activity. Potentially, transcription factors (TFs) binding in flanking sequence could interact with the L1 5′-UTR. However, because standard predictions for potential TF binding (MatInspector) yield numerous matches—55, 39, and 69 matches for L1C, L1F, and L1D, respectively—it is currently difficult to delineate particular TFs that play a role. Also, if TFs in upstream flanking sequence are involved in the regulation, each flanking sequence could, in principle, affect the L1 5′-UTR promoter by different TF sets. As for the flanking sequence of L1M, we did not find 5′-RACE products within the flanking sequence that would indicate that its enhancing effect is caused by an internal promoter in that flanking sequence. Indeed, more detailed subsequent studies will be required to reveal factors in upstream flanking cellular sequences that interact with the L1 5′-UTR and that modulate its activity. Swergold (1990) previously investigated regulation of an L1 promoter. Deletion of ∼360 bp of plasmid backbone sequence immediately upstream of the L1 5′-UTR reduced the L1 promoter activity by 80%. In the light of our more extensive analysis, the nonspecific context effect suggested then (Swergold 1990) is rather another example of the important influence of different upstream sequences on the L1 promoter.
Nevertheless, the significant effect of upstream flanking sequences on the L1 promoter activity has important evolutionary consequences for human L1 elements. If an L1 element retrotransposes in full length into a flanking sequence context that significantly down-regulates the 5′-UTR, the L1 element will not produce L1 progeny because it is transcribed not at all or at very low levels. It could not serve as master element even though other L1 element portions, such as ORF1 and ORF2, encode active proteins. On the other hand, L1 elements landing in a beneficial flanking sequence context could produce many more offspring, and therefore become master elements. Similar suggestions were recently made for mammalian and plant short interspersed elements (SINEs). SINE loci that escape strong negative transcriptional regulation are usually associated with external enhancers. It has been proposed that the combination of internal signals in SINEs and external signals in flanking sequences can result in efficient transcription of a limited number of SINE loci, defining those loci as master SINEs (Chesnokov and Schmid 1996; Deininger et al. 1996; Arnaud et al. 2001).
Transcription of human L1 elements is mediated by RNA Pol II, yet is independent of a TATA-box. L1 elements cannot rely on a TATA-box to drive their transcription. It is very unlikely that a new L1 element will insert into a genomic location just downstream from a functional TATA-box. Several cellular genes are transcribed from TATA-less promoters, so-called initiator (Inr) elements. It is known that transcription initiates at variable positions within the Inr (Weis and Reinberg 1992). Our finding of transcription initiating at different positions of the L1 5′-end suggests that the L1 5′-UTR forms an Inr as well. Transcription frequently initiating in L1 upstream flanking sequence furthermore suggests that the Inr formed by the L1 5′-UTR reaches into upstream flanking sequence. We note that Mathias and Scott (1993) reported that not only the L1 5′-UTR 5′-end but also upstream flanking sequence, with nucleotides up to nucleotide -9, were protected in DNase footprint experiments. It was furthermore concluded that the protein complex consists of probably more than two proteins (Mathias and Scott 1993). Those findings support an Inr that is formed by the L1 5′-end and that reaches into upstream flanking sequence. However, the exact mechanism of the L1 5′-UTR promoter remains to be elucidated in more detailed analysis. Although YY1 has been described to interact with Inr elements, and also binds to the L1 5′-UTR 5′-end (Minakami et al. 1992; Becker et al. 1993), it remains to be seen whether one or several of a number of transcription factors interacting with Inr elements (Javahery et al. 1994; Manzano-Winkler et al. 1996) also bind to the L1 5′-UTR 5′-end. In addition, transcription factors binding (or not binding) in more upstream flanking sequences may further modulate that process.
One may furthermore speculate that non-LTR retrotransposons in other species as well apply an Inr-mediated mechanism to drive their transcription. Considering a promoter mechanism that makes sense in terms of evolution, an Inr could be an appropriate survival strategy for many non-LTR retrotransposons. The promoter activity of non-human elements may be similarly influenced by flanking sequences, or the elements may have evolved more autonomous mechanisms to escape that influence to become more independent from the host genome. Future work should also focus on cellular factors regulating the human L1 promoter activity; regulators within the L1 element, regulators in flanking cellular sequence, and factors involved in the suggested Inr mechanism.
Methods
5′-RACE
We used the 5′/3′-RACE Kit, 2nd generation (Roche) following the manufacturer's protocol. L1 reporter constructs (see below) were transfected into Tera-1 cells. Total RNA was isolated 24–48 h after transfection using TRIzol (Invitrogen). RNA was subsequently treated with DNase I for 15 min at 37°C. cDNA was synthesized from 1.5 μg of RNA using a primer located in the luciferase gene 5′-end (5′-TATCTCTTCATAGCCTTATGCA-3′). PCR was performed on cDNA following the conditions recommended by the manufacturer. An annealing temperature of 60°C was used for the oligo(dT) primer, provided by the manufacturer, and the primer P2 (5′-CGGGATCCCTTTGTGGTTTTATCTA CTTTT-3′).
Statistical analysis
We performed χ2 analyses for statistical evaluation of higher frequencies of individual nucleotides as described (Jurka 1997). Briefly, 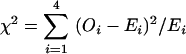 , where Oi is the individual base occurrence and Ei is the total number of bases at a given position. We considered significance levels of P < 0.01 for 3 degrees of freedom as statistically significant.
, where Oi is the individual base occurrence and Ei is the total number of bases at a given position. We considered significance levels of P < 0.01 for 3 degrees of freedom as statistically significant.
Construction of L1 reporter constructs
Full-length L1 elements, including upstream flanking cellular sequence, were recently cloned from the human genome into the pCEP4 vector multiple cloning site (Brouha et al. 2003), the latter harboring a KpnI site upstream of the cloned L1 fragment (see below).
Luciferase reporter constructs containing the L1 5′-UTR and upstream flanking cellular sequence, named, for instance, pL1.3, were generated as follows. We amplified by PCR the entire upstream flanking sequence and the L1 5′-UTR from a total of 13 different L1 constructs. PCR primers were located downstream from the pCEP4 CMV promoter (P1: 5′-CGGGATCCTCAGAT CTCTAGAAGCTGGGTAC-3′), and located at the 3′-end of the L1 5′-UTR sequence (P2: 5′-CGGGATCCCTTTGTGGTTTTATCTAC TTTT-3′). PCR products were amplified using standard PCR conditions. Both PCR primers included recognition sites for BamHI at their ends that were instrumental for cloning PCR products into the luciferase reporter vector pLuc (kindly provided by Friedrich Graesser, University of Saarland, Homburg/Saar, Germany), which had been linearized with BamHI and treated with alkaline phosphatase. Note that the L1.3 construct does not harbor upstream flanking cellular sequence because of the previously used cloning strategy for that L1 element (Sassaman et al. 1997).
To generate reporter plasmids pL1.3-fl..., we cloned upstream flanking cellular sequence from the luciferase reporter constructs L1C, L1D, L1F, L1H, and L1M immediately upstream of the L1.3 5′-UTR as follows. We amplified by PCR a sequence portion using primer P1, and a primer located within the L1 5′-UTR 5′ portion (nucleotides 87–108; P3: 5′-GATGAACC CGGTACCTCAGATG-3′). The amplified short L1 5′-UTR portion is identical in sequence for all elements, and furthermore includes a recognition site for KpnI at the 3′-end. A KpnI site, which stems from the pCEP4 multiple cloning site, is furthermore present at the PCR product 5′-end. PCR products derived from L1C–L1M were digested with KpnI and were cloned into the L1.3 luciferase reporter construct, that had been digested with KpnI and dephosphorylated.
Reporter plasmids, such as pL1C-Δfl, lack upstream flanking cellular sequence. We deleted upstream flanking sequences from the original reporter constructs L1C, L1D, L1F, L1H, L1M by replacing the upstream flanking cellular sequence with a sequence derived from the L1.3 construct. We amplified a PCR product from the L1.3 original luciferase reporter construct using primers P1 and P2. The PCR product was digested with KpnI and the short restriction fragment harboring the L1 5′-UTR 5′-end and upstream sequence was cloned into the L1D–L1M plasmids. The latter had been digested with KpnI and dephosphorylated to release the L1 5′-UTR 5′-end and upstream flanking cellular sequence. As a reminder, the L1 5′-UTR 5′-region included in the ligated fragment is identical in sequence for all L1 elements used.
In luciferase reporter constructs, such as pL1.3-flC10, we replaced the first 10 bp immediately upstream of the L1 5′-UTR by respective 10 bp present in clones L1C, L1D, and L1F. In luciferase reporter constructs, such as pL1.3-G5, -A5, and so on, the first 5 bp immediately upstream of the L1.3 5′-UTR were altered to 5 × G, 5 × A, and so on. To do so, we first digested the L1.3 luciferase reporter construct with KpnI, and cloned the resulting restriction fragment (see above) into a KpnI-digested pGEM-T vector. We altered respective sequence regions using a PCR-mediated approach. PCR primers immediately neighbored each other and pointed outward, with one primer introducing respective mutations. Primer sequences are available from the authors. PCR products were amplified with Pfu polymerase, and were subsequently treated with T4 polynucleotide kinase and religated to create functional plasmids. Clones harboring sequence alterations were digested with KpnI and the restriction fragment was cloned back into the L1.3 luciferase reporter construct linearized with KpnI.
Sequences of all constructed reporter plasmids were verified by sequencing using the SequiTherm Excel II DNA Sequencing Kit-LC (Biozym) and an automated DNA sequencer (Licor 4000-L, MWG). Raw sequence data were analyzed using the Sequencher software (Gene Codes). The various cloning strategies are summarized in Supplemental Figure B.
Transfections and luciferase assays
Tera-1 and T47D cells were cultured in proper cell culture media in a humidified incubator at 37°C and 5% CO2. We transiently transfected cells that had been grown to 60%–80% confluency in 12 well plates using Fugene6 (Roche) and following the manufacturer's instructions. Each well received 500 ng of L1 reporter contruct and 200 ng of pCMVβ. The latter is a β-galactosidase-expressing vector driven by the CMV early promoter. Cells were lysed 24 to 48 h after transfection, and luciferase and β-galactosidase activities were determined using the Luciferase Assay System and the β-galactosidase Assay System (Promega), respectively, following the manufacturer's instructions. Luciferase activity was measured with a Lumat LB 9507 luminometer (Berthold Technologies). β-Galactosidase activities were used to normalize luciferase activities. The latter are given in relation to a CMV promoter-driven luciferase expressing control vector that was included in each transfection experiment. Transcriptional activities of the various L1 constructs were determined at least in triplicate each.
Acknowledgments
This work was supported by grants from the Deutsche Forschungsgemeinschaft to J.M. (Ma2298/2-1) and E.M. (Me917/16-1). We thank Jean-Marc Deragon, Haig H. Kazazian, John L. Goodier, and Eric M. Ostertag for many helpful comments on the manuscript.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.2745804.
Footnotes
[Supplemental material is available online at www.genome.org. The following individuals kindly provided reagents, samples, or unpublished information as indicated in the paper: F. Graesser.]
References
- Arnaud, P., Yukawa, Y., Lavie, L., Pelissier, T., Sugiura, M., and Deragon, J.M. 2001. Analysis of the SINE S1 Pol III promoter from Brassica; impact of methylation and influence of external sequences. Plant J. 26: 295-305. [DOI] [PubMed] [Google Scholar]
- Becker, K.G., Swergold, G.D., Ozato, K., and Thayer, R.E. 1993. Binding of the ubiquitous nuclear transcription factor YY1 to a cis regulatory sequence in the human LINE-1 transposable element. Hum. Mol. Genet. 2: 1697-1702. [DOI] [PubMed] [Google Scholar]
- Boeke, J.D. 1997. LINEs and Alus—The polyA connection. Nat. Genet. 16: 6-7. [DOI] [PubMed] [Google Scholar]
- ———. 2003. The unusual phylogenetic distribution of retrotransposons: A hypothesis. Genome Res. 13: 1975-1983. [DOI] [PubMed] [Google Scholar]
- Brouha, B., Schustak, J., Badge, R.M., Lutz-Prigge, S., Farley, A.H., Moran, J.V., and Kazazian Jr., H.H. 2003. Hot L1s account for the bulk of retrotransposition in the human population. Proc. Natl. Acad. Sci. 100: 5280-5285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesnokov, I. and Schmid, C.W. 1996. Flanking sequences of an Alu source stimulate transcription in vitro by interacting with sequence-specific transcription factors. J. Mol. Evol. 42: 30-36. [DOI] [PubMed] [Google Scholar]
- Cost, G.J., Feng, Q., Jacquier, A., and Boeke, J.D. 2002. Human L1 element target-primed reverse transcription in vitro. EMBO J. 21: 5899-5910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costas, J. 2002. Characterization of the intragenomic spread of the human endogenous retrovirus family HERV-W. Mol. Biol. Evol. 19: 526-533. [DOI] [PubMed] [Google Scholar]
- Deininger, P.L., Tiedge, H., Kim, J., and Brosius, J. 1996. Evolution, expression, and possible function of a master gene for amplification of an interspersed repeated DNA family in rodents. Prog. Nucleic Acids Res. Mol. Biol. 52: 67-88. [DOI] [PubMed] [Google Scholar]
- Deininger, P.L., Moran, J.V., Batzer, M.A., and Kazazian Jr., H.H. 2003. Mobile elements and mammalian genome evolution. Curr. Opin. Genet. Dev. 13: 651-658. [DOI] [PubMed] [Google Scholar]
- Dewannieux, M., Esnault, C., and Heidmann, T. 2003. LINE-mediated retrotransposition of marked Alu sequences. Nat. Genet. 35: 41-48. [DOI] [PubMed] [Google Scholar]
- Goodier, J.L., Ostertag, E.M., and Kazazian Jr., H.H. 2000. Transduction of 3′-flanking sequences is common in L1 retrotransposition. Hum. Mol. Genet. 9: 653-657. [DOI] [PubMed] [Google Scholar]
- Hirzmann, J., Luo, D., Hahnen, J., and Hobom, G. 1993. Determination of messenger RNA 5′-ends by reverse transcription of the cap structure. Nucleic Acids Res. 21: 3597-3598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium. 2001. Initial sequencing and analysis of the human genome. Nature 409: 860-921. [DOI] [PubMed] [Google Scholar]
- Javahery, R., Khachi, A., Lo, K., Zenzie-Gregory, B., and Smale, S.T. 1994. DNA sequence requirements for transcriptional initiator activity in mammalian cells. Mol. Cell. Biol. 14: 116-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurka, J. 1997. Sequence patterns indicate an enzymatic involvement in integration of mammalian retroposons. Proc. Natl. Acad. Sci. 94: 1872-1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazazian Jr., H.H. 2004. Mobile elements: Drivers of genome evolution. Science 303: 1626-1632. [DOI] [PubMed] [Google Scholar]
- Kimberland, M.L., Divoky, V., Prchal, J., Schwahn, U., Berger, W., and Kazazian Jr., H.H. 1999. Full-length human L1 insertions retain the capacity for high frequency retrotransposition in cultured cells. Hum. Mol. Genet. 8: 1557-1560. [DOI] [PubMed] [Google Scholar]
- Leibold, D.M., Swergold, G.D., Singer, M.F., Thayer, R.E., Dombroski, B.A., and Fanning, T.G. 1990. Translation of LINE-1 DNA elements in vitro and in human cells. Proc. Natl. Acad. Sci. 87: 6990-6994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzano-Winkler, B., Novina, C.D., and Roy, A.L. 1996. TFII is required for transcription of the naturally TATA-less but initiator-containing Vβ promoter. J. Biol. Chem. 271: 12076-12081. [DOI] [PubMed] [Google Scholar]
- Mathias, S.L. and Scott, A.F. 1993. Promoter binding proteins of an active human L1 retrotransposon. Biochem. Biophys. Res. Commun. 191: 625-632. [DOI] [PubMed] [Google Scholar]
- Minakami, R., Kurose, K., Etoh, K., Furuhata, Y., Hattori, M., and Sakaki, Y. 1992. Identification of an internal cis-element essential for the human L1 transcription and a nuclear factor(s) binding to the element. Nucleic Acids Res. 20: 3139-3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostertag, E.M. and Kazazian Jr., H.H. 2001. Biology of mammalian L1 retrotransposons. Annu. Rev. Genet. 35: 501-538. [DOI] [PubMed] [Google Scholar]
- Pavlicek, A., Paces, J., Elleder, D., and Hejnar, J. 2002a. Processed pseudogenes of human endogenous retroviruses generated by LINEs: Their integration, stability, and distribution. Genome Res. 12: 391-399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlicek, A., Paces, J., Zika, R., and Hejnar, J. 2002b. Length distribution of long interspersed nucleotide elements (LINEs) and processed pseudogenes of human endogenous retroviruses: Implications for retrotransposition and pseudogene detection. Gene 300: 189-194. [DOI] [PubMed] [Google Scholar]
- Pickeral, O.K., Makalowski, W., Boguski, M.S., and Boeke, J.D. 2000. Frequent human genomic DNA transduction driven by LINE-1 retrotransposition. Genome Res. 10: 411-415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassaman, D.M., Dombroski, B.A., Moran, J.V., Kimberland, M.L., Naas, T.P., DeBerardinis, R.J., Gabriel, A., Swergold, G.D., and Kazazian Jr., H.H. 1997. Many human L1 elements are capable of retrotransposition. Nat. Genet. 16: 37-43. [DOI] [PubMed] [Google Scholar]
- Skowronski, J. and Singer, M.F. 1985. Expression of a cytoplasmic LINE-1 transcript is regulated in a human teratocarcinoma cell line. Proc. Natl. Acad. Sci. 82: 6050-6054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swergold, G.D. 1990. Identification, characterization, and cell specificity of a human LINE-1 promoter. Mol. Cell. Biol. 10: 6718-6729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symer, D.E., Connelly, C., Szak, S.T., Caputo, E.M., Cost, G.J., Parmigiani, G., and Boeke, J.D. 2002. Human L1 retrotransposition is associated with genetic instability in vivo. Cell 110: 327-338. [DOI] [PubMed] [Google Scholar]
- Tchenio, T., Casella, J.F., and Heidmann, T. 2000. Members of the SRY family regulate the human LINE retrotransposons. Nucleic Acids Res. 28: 411-415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thayer, R.E., Singer, M.F., and Fanning, T.G. 1993. Undermethylation of specific LINE-1 sequences in human cells producing a LINE-1-encoded protein. Gene 133: 273-277. [DOI] [PubMed] [Google Scholar]
- Volloch, V.Z., Schweitzer, B., and Rits, S. 1995. Transcription of the 5′-terminal cap nucleotide by RNA-dependent DNA polymerase: Possible involvement in retroviral reverse transcription. DNA Cell Biol. 14: 991-996. [DOI] [PubMed] [Google Scholar]
- Wei, W., Gilbert, N., Ooi, S.L., Lawler, J.F., Ostertag, E.M., Kazazian Jr., H.H., Boeke, J.D., and Moran, J.V. 2001. Human L1 retrotransposition: Cis preference versus trans complementation. Mol. Cell. Biol. 21: 1429-1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weis, L. and Reinberg, D. 1992. Transcription by RNA polymerase II: Initiator-directed formation of transcription-competent complexes. FASEB J. 6: 3300-3309. [DOI] [PubMed] [Google Scholar]
- Yang, N., Zhang, L., Zhang, Y., and Kazazian Jr., H.H. 2003. An important role for RUNX3 in human L1 transcription and retrotransposition. Nucleic Acids Res. 31: 4929-4940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, F., Zingler, N., Schumann, G., and Stratling, W.H. 2001. Methyl-CpG-binding protein 2 represses LINE-1 expression and retrotransposition but not Alu transcription. Nucleic Acids Res. 29: 4493-4501. [DOI] [PMC free article] [PubMed] [Google Scholar]