

Abstract
We have developed a new microarray technology for quantitative gene-expression profiling on the basis of randomly assembled arrays of beads. Each bead carries a gene-specific probe sequence. There are multiple copies of each sequence-specific bead in an array, which contributes to measurement precision and reliability. We optimized the system for specific and sensitive analysis of mammalian RNA, and using RNA controls of defined concentration, obtained the following estimates of system performance: specificity of 1:250,000 in mammalian poly(A+) mRNA; limit of detection 0.13 pM; dynamic range 3.2 logs; and sufficient precision to detect 1.3-fold differences with 95% confidence within the dynamic range. Measurements of expression differences between human brain and liver were validated by concordance with quantitative real-time PCR (R2 = 0.98 for log-transformed ratios, and slope of the best-fit line = 1.04, for 20 genes). Quantitative performance was further verified using a mouse B- and T-cell model system. We found published reports of B- or T-cell-specific expression for 42 of 59 genes that showed the greatest differential expression between B- and T-cells in our system. All of the literature observations were concordant with our results. Our experiments were carried out on a 96-array matrix system that requires only 100 ng of input RNA and uses standard microtiter plates to process samples in parallel. Our technology has advantages for analyzing multiple samples, is scalable to all known genes in a genome, and is flexible, allowing the use of standard or custom probes in an array.
Microarray technology has allowed the abundance of thousands of different mRNAs to be measured simultaneously and efficiently from a single biological sample (Schena et al. 1995; Lockhart et al. 1996; Lockhart and Winzeler 2000). As a result, the analysis of individual genes has given way to the analysis of large sets of genes and the discovery of patterns and relationships in their expression. This has spawned a myriad of exciting new applications that are helping to shape the emerging field of systems biology (Marton et al. 1998; Golub et al. 1999; Hughes et al. 2000; Ideker et al. 2001; van't Veer et al. 2002; Yvert et al. 2003). The microarrays that have spurred these advances can be manufactured by a variety of techniques, including spotting (Schena et al. 1995), photolithographic synthesis (Fodor et al. 1991), and inkjet synthesis (Blanchard 1998). In each case, individual probes are placed or synthesized at predefined locations on the substrate. However, conventional arrays can suffer from one or more limitations, including poor data quality, as a result of high intra- and interarray variability, often associated with spotted arrays.
We describe here a powerful and intrinsically robust alternative that substantially overcomes these limitations. Our gene-expression profiling system is based on randomly assembled arrays of beads in wells (Michael et al. 1998). Following random assembly, the location and identity of each bead, bearing an oligonucleotide probe, is determined via a sequential decoding process (Gunderson et al. 2004). An advantage of this approach is that dense packing can be achieved using simple and efficient bulk processes. Furthermore, the technology is intrinsically scalable; the arrays described in this study use beads with diameters of three microns, producing a packing density ∼400 times that of a conventional spotted microarray. Elsewhere, packing densities ∼40,000 times that of a conventional array have been achieved through the assembly of 300-nm beads (Michael et al. 1998).
The BeadArray technology has previously been shown to be a robust readout platform for single nucleotide polymorphism (SNP) genotyping, where it has demonstrated very high accuracy, call rate, and reproducibility at high multiplexing levels (Fan et al. 2003). It is being used to generate over half the genotyping data for the International HapMap Project (www.hapmap.org), which will derive a detailed map of common genetic variation across the human genome (The International HapMap Consortium 2003). In addition, the BeadArray platform has been effective for gene-expression profiling using PCR-based assays in combination with universal arrays (Yeakley et al. 2002; Fan et al. 2004). Both of these applications made use of universal arrays containing up to 1536 usable capture sequences.
Despite these successes, the use of the BeadArray technology for quantitative gene-expression profiling from complex samples by hybridization to gene-specific probes has not previously been demonstrated. Although some preliminary data suggested that the platform is capable of high sensitivity (Epstein et al. 2002), a number of significant challenges had to be overcome in order to create a robust, quantitative, high-performance system suitable for use with biological samples such as mammalian poly(A+) mRNA. We have now developed such a system. We show that it is capable of accurately and robustly reporting mRNA abundance for hundreds of genes. Our methods can be applied to many thousands of samples, a scale of experimentation that has been impractical with other technologies, and can be extended to develop arrays designed to analyze all known genes in a genome. The technology is also compatible with our SNP genotyping system, enabling genotyping and gene-expression profiling on the same platform.
Results
Design of a gene-expression probe array based on random assembly of beads in wells
The arrays used in the experiments reported here are described in Figure 1. Typically, each array has up to 1536 different bead types, each represented on average by ∼30 copies in any array. Each bead type has ∼700,000 copies of a particular oligonucleotide probe covalently attached to it (Fig. 1). Because the population of beads in an array is a random sampling of a starting bead pool containing 1536 bead types, the representation of the bead types in the array is effectively Poisson. That is, there is a variable number of each of the 1536 bead types both within and between arrays (Gunderson et al. 2004). Thus, two important issues must be addressed to ensure that the random arrays can be used for quantitative measurements of mRNA abundance.
Figure 1.

Design of a randomly assembled gene-specific probe array. (A) Representation of an individual bead lodged in a well. Attached to the bead by its 5′ end is a chimeric oligonucleotide ∼75 nucleotides in length, comprising an ∼25-nucleotide identifier sequence and a 50-nucleotide gene-specific probe. The bead identifier sequence is decoded using an algorithm described previously (Gunderson et al. 2004). We tested gene-specific probes of 25 and 50 bp in length and found that the 50 mers showed superior performance, consistent with prior findings (Hughes et al. 2001). The drawing is not to scale; the relative size of the oligonucleotide has been vastly exaggerated to show its features. (B) There are ∼50,000 beads in an ∼1.4-mm diameter optical fiber bundle, each bead lodged in a well at the end of an individual fiber in the bundle. (C) The bundles are arranged in a 96-array matrix matching the format of a standard microtiter plate.
Firstly, because each array is unique, how can we compare results from array to array? By virtue of the ∼30-fold oversampling (50,000 beads/1536 bead types), we can ensure that decoded arrays have greater than or equal to five beads of each type in the array, so that all sequences are represented (Gunderson et al. 2004). Furthermore, the randomness and redundancy provide us with considerable advantages; randomness minimizes the effects of spatially localized artifacts, and redundancy increases measurement precision and robustness. These factors combine to increase measurement accuracy.
Secondly, because each probe has associated with it an identifier sequence (Fig. 1), how do we ensure that this sequence doesn't interfere with the analysis of the target mRNA? This is done in two ways. The identifier sequences are computationally screened to avoid similarity to the human and mouse genomes. The probability of cross-hybridization to other genomes is also low, and for the analysis of any particular genome, it is simple to omit a small number of identifier sequences if needed. Also, the identifier sequences are only half the length of the gene-specific probes and have correspondingly lower Tm's (52.0 ± 2.3°C vs. 70.7 ± 1.7°C). By hybridizing labeled total mammalian poly(A+) mRNA samples to arrays containing the identifier sequences but lacking the gene-specific probe sequences, we estimated that the identifier sequences contribute an average of up to five counts over background, with only a few sequences giving higher signals (data not shown). This is a small amount of signal relative to the gene-specific probes and is not expected to have any significant effect on the analysis.
Array formats designed for a variety of gene-expression applications
The experiments described in this study all make use of the Sentrix array matrix format shown in Figure 1. However, the basic concept of placing beads in wells to form a randomly ordered array can be used to create a variety of array formats suitable for a range of applications. In addition to the format shown in Figure 1, which is read using a custom high-resolution reader (Barker et al. 2003), we have developed silicon substrates that have the dimensions of a 2.5 × 7.5-cm microscope slide and can be read on a 5-μm resolution Axon GenePix scanner by virtue of larger well spacing (T. Dickinson, G. Smith, H. Bennett, and R. Barrett, unpubl.). Yet other silicon substrates have been used to develop two designs of whole-genome array, with probes for ∼24,000 and ∼48,000 gene sequences (G. Wang, G. Smith, S. Barnard, and D. Che, unpubl.; further information is available on www.illumina.com). These higher density arrays can be read on a BeadArray scanner. All of these formats make use of 3-μm silica beads; the same bead pools can be loaded into the different substrates, and give similar quantitative performance. Therefore, substantially similar results to those obtained below can be obtained using a variety of bead-based array formats suitable for a range of experimental designs and detection systems.
Dose-response study using spiked mRNAs of known concentration
We designed a dose-response study to estimate the limit of detection, dynamic range, and precision of the 96-array matrix gene-specific probe system for the analysis of a mammalian mRNA sample. We prepared a series of samples that consisted of labeled human liver cell line RNA spiked with known quantities of individually labeled mRNAs synthesized in vitro. This approach has been described previously for microarray performance characterization (Lockhart et al. 1996). We used as spikes nine mRNAs, produced by in vitro transcription (IVT) of cloned bacterial and viral genes whose sequences are absent from the human genome. Twelve samples, representing 12 concentrations, were each replicated eight times to give a total of 96 samples. Each sample contained all nine spikes at a given concentration ranging from zero to 200 pM. (Fig. 2).
Figure 2.

Arrangement of spiked samples for hybridization. Each sample was produced by adding labeled spike controls to labeled complex RNA derived from human HepG2 poly(A+) RNA. The spike controls were added at the pM concentrations indicated in the figure. All nine spiked mRNAs were present at the same concentration within a given sample (e.g., 200 pM in sample a1). Samples were arranged in a staggered fashion to avoid the possibility of row/column positional bias. Hybridization was performed using 1 μg of each sample at a final concentration of 25 ng/μL.
Each sample was hybridized to eight different arrays in a 96-array matrix. This provided eight technical replicates, sufficient data to allow a statistical analysis of noise in the quantitative readout step. The dose response curves and the resolvable fold change across the tested concentration range, generated for each of the nine genes, are shown in Figure 3.
Figure 3.

Dose-response curves. Data points represent the mean of eight arrays. Signal intensities are plotted in blue vs. target concentration. Error bars represent the two-sided symmetric 90% confidence intervals for a single reading, calculated on the basis of the spread of eight separate readings. All points contain error bars, but some are too small to be resolved at the plotted scale. The resolvable fold change is plotted in red and green vs. target concentration. Each data point estimates the ability to distinguish concentration fold change for a single reading. Concentration levels are defined as resolved when estimated one-sided 95% confidence intervals do not overlap. Values below twofold are colored green, whereas those greater than twofold are colored red.
Reproducibility of quantitative measurements and dependence on sample input
The dose-response results were reproducible across different manufacturing lots of array matrices and hybridization days. We obtained similar results from 15 independent trials of the experiment, hybridized on five separate days using a total of 720 arrays manufactured on seven different dates (Fig. 4). The quantitative performance of the system based on this significant amount of replication is summarized in Table 1.
Figure 4.

Dynamic range, detectable fold change, and limit of detection for 15 array matrices. The array matrices, manufactured on five separate days, were used to perform dose-response experiments identical to that described above, except in these experiments, we used four replicates per concentration instead of eight. Dynamic range corresponds to the concentration range over which twofold concentration changes can be distinguished with 95% confidence (represented by the green portions of the lines in Fig. 3); the values plotted in the graph (blue diamonds, left axis) are determined by dividing the upper concentration limit of this range by the lower limit for the given experiment. Precision (orange squares, left axis) corresponds to the distinguishable fold change across the determined dynamic range. Limit of detection (green triangles, right axis) corresponds to 0.99 detection p-value generated using normal model of intensities of 20 negative control probes that have no corresponding target in the sample. All performance values given represent the median value for the nine spike targets used in the experiment.
Table 1.
Performance metrics
| Metrica | Value | Confidence |
|---|---|---|
| Input Requirement (Total RNA) | 100 ng | n/a |
| Limit of Detectionb | ∼0.13 pM | 99% |
| Specificityc | ∼1:250,000 | 99% |
| Precisiond | ∼1.3-fold | 95% |
| Dynamic Rangee | ∼3.2 Logs | n/a |
| Array-to-Array %CVf | <10% | n/a |
The median value of nine spike controls is given for each performance metric. All metrics listed derive from measurements made using two probe sequences per gene
Limit of detection is determined by a negative control detection model (see Methods)
Specificity is determined by dividing the number of molecules detected at the limit of detection by the number of molecules present in 1 μg of sample background. The average mammalian transcript length is estimated at 2000 nt
Precision is the smallest change in concentration that can be detected with 95% confidence. Value given is the median across the dynamic range
Dynamic range is defined by the ability to detect twofold changes in target concentration across the specified concentration range (see Methods)
Array-to-array %CV is determined using a common population of all detectable bead-types across the 96 arrays of an array matrix
In addition to the probes used to measure the dose responses, the arrays used in the experiments summarized in Figure 4 contained probes for 587 human genes. We analyzed the data generated by these experiments to assess array-to-array hybridization signal variation and how it is influenced by gene intensity. We selected the 380 genes that were reproducibly expressed at detectable levels and plotted their coefficient of variation (standard deviation divided by intensity, abbreviated as CV) as a function of hybridization signal. As shown in Figure 5, and consistent with expectations, the CV increases inversely as gene signals approach the limit of detection. The median CV for background-subtracted, un-normalized intensity across 48 arrays in a representative experiment was 6.5%.
Figure 5.

Array signal variation as a function of gene hybridization intensity. Each blue dot represents a gene and the red line represents a smoothed function for the data on the basis of a robust best-fit function for standard deviation vs. intensity. All values are based on background-subtracted raw data from 48 replicate hybridizations. The data shown are from one experiment of the 15 described in the legend to Figure 4. This experiment was chosen to represent the others on the basis of its measurement precision, which is the median of measurement precisions for all 15 experiments.
Additional performance measurements of a microarray platform include (1) reproducibility across multiple sample labeling reactions, and (2) sensitivity to sample input variation. To test these aspects of our system's reproducibility, we performed 20 sample labeling reactions, four each, using 10, 20, 50, 150, or 500 ng of total RNA derived from mouse spleen. For the 10- and 20-ng inputs, only three of the four replicates produced adequate material for array hybridization. One microgram of biotinylated cRNA from each successful reaction was hybridized to a separate array in an array matrix. Each array in the matrix contained probes to 540 mouse genes. Each cRNA sample was present at a final concentration of 25 ng/μL.
To obtain a quantitative estimate of reproducibility, linear correlations were calculated for all pairwise combinations of the replicates at each input concentration. The means and ranges of these correlations are plotted in Figure 6A. All correlations (R2) exceeded 0.99. As further evidence of robustness, the scatter plot in Figure 6B shows the correlation for signals between sample labeling replicates using 50 and 500 ng of starting material; the high correlation (R2 > 0.99) demonstrates the reproducibility of the assay even with input material concentrations differing by 10-fold.
Figure 6.

Sample labeling reproducibility. (A) Twenty sample labeling reactions were processed using our standard conditions with 10, 20, 50, 150, or 500 ng of total mouse spleen RNA as input material (four replicates each). For each input amount, correlation values (R2) of gene signals were determined for all pairwise comparisons of all successful replicates. The median R2 values, with ranges, are plotted. (B) Representative scatter plot of the intensity values for all genes measured in one of the 50-ng samples vs. those in the 500-ng sample. Whereas initial sample input varied for each labeling reaction, final array hybridization was performed using 1 μg of each labeled sample at a final concentration of 25 ng/μL.
Concordance with real-time quantitative PCR
The experiments described above measured the quantitative performance of the system and demonstrated that we could obtain quantitative data in a reproducible way. We next wanted to perform measurements on a true biological sample and to evaluate these results by comparison with a different technology. Concordance with measurements obtained using a different technology is a strong indicator that measurements are correct. Therefore, we performed an experiment that compared differential expression patterns obtained on the randomly assembled arrays with those obtained from TaqMan quantitative real-time PCR (qPCR).
The genes selected for this analysis came from a comparison of human liver with human brain. Labeled cRNA from both tissues was hybridized to separate arrays containing probes for 633 human genes. From the hybridization results, we selected a panel of 21 genes for the comparison, using the following criteria: (1) the genes showed a range of liver/brain expression ratios ranging from 0.005 to 175; and (2) every gene was expressed significantly over background, even in the tissue showing the lower amount of expression. This second criterion was necessary to avoid inaccurate expression ratios resulting from the influence of system noise.
For each of these 21 genes, we performed qPCR assays on aliquots of the same starting material. Twenty of the 21 primer pairs gave products and the log-transformed expression ratios obtained for each of the 20 genes were plotted against the corresponding values obtained on the randomly assembled arrays (Fig. 7). The measurements determined by the two systems showed good correlation (R2 = 0.98 for log-transformed ratios). Furthermore, the slope of the best-fit line was 1.04, indicating that the ratios obtained by the two methods are similar in magnitude. For highly expressed genes, the array produced somewhat compressed fold-change ratios compared with those produced by qPCR. For the five genes whose array intensities exceeded 10,000 counts in either tissue, the array-measured ratio was 0.77 ± 0.24 versus 1.04 ± 0.35 for all genes. This compression is likely due to probe saturation of highly expressed targets, a predicted feature, as the array platform has a dynamic range of ∼3 logs compared with ∼5 logs for qPCR. (Heid et al. 1996) This overall high level of concordance with qPCR validated the performance of the randomly assembled array system.
Figure 7.

Correlation of array matrix data to quantitative real-time PCR. Labeled RNA samples were made from human and brain total RNA. These were hybridized to separate array matrices containing 633 human genes. Six technical replicates were included for each sample. Twenty-one genes from this list were selected for analysis by TaqMan quantitative real-time PCR. A scatter plot of hybridization intensities of the liver and brain samples on the array matrix is shown in A. Genes selected for further analysis are shaded orange. A scatter plot of log-transformed hybridization signal ratios as determined by the two methods is shown in B.
Validation of results in a model biological system
Finally, we assessed the ability of the random arrays to generate data consistent with results previously published for a well-characterized biological system. The model system we selected was mouse B and T cells, both of which contain large numbers of cell-type specific transcripts documented in the biological literature. Our experimental design was to make a series of seven samples containing different ratios of R1.1 (T cell lymphoma) and A20 (B cell lymphoma) mRNA mixed together. This series ranged from 100% B/0% T to 0% B/100% T. Each of the seven samples was independently labeled six times, and the resulting 42 cRNA samples were hybridized to separate arrays of an array matrix, each containing probes to 540 different mouse genes. After hybridization and analysis, we identified 59 genes that were determined as detected in the 100% B cell sample, but not in the 100% T cell sample or vice versa (Fig. 8). Upon generating this list of 59 genes, we performed literature searches to establish whether there was prior evidence of T- or B-specific expression. Forty three of the 59 genes had prior literature support for their tissue specificity. We found no genes miscategorized by our array. Table 2 shows a list of all tissue-specific genes identified in our analysis.
Figure 8.

B Cell/T Cell experimental results. Seven RNA samples were prepared containing mixtures of B- and T-cell lymphoma cell-line mRNA. The samples contained 0%, 5%, 25%, 50%, 95%, and 100% B-cell RNA, with the balance in all cases being T-cell RNA. These samples were labeled by our standard protocol and hybridized to six separate arrays each. (A) The hybridization intensities of the 59 most tissue-specific genes are plotted. Each vertical stripe represents a sample and each horizontal row a gene. Boxes represent the mean hybridization intensities measured in six replicate array hybridizations. The intensity scale is shown in the legend at the bottom. Genes are labeled according to their RefSeq or UniGene ID numbers. The colors of these labels indicate prior evidence in the literature of B- or T-cell-specific expression (red refers to B-cell-specific expression; blue refers to T-cell-specific; see Table 2 for references). (B) The dose responses of two representative genes identified in this experiment are plotted. Points represent the mean intensities for each concentration. Error bars represent 90% two-sided confidence intervals calculated from six replicate hybridizations.
Table 2.
Array-based determination of tissue-specific gene expression
| Gene RefSeq or UniGene ID | HUGO Gene Symbol | Gene Name | T/B Cell Ratio by arraya | B or T-specific by literatureb | Refc |
|---|---|---|---|---|---|
| NM_010545 | Ii | Ia-associated invariant chain | 0.003 | B | 1 |
| NM_152839 | Igj | immunoglobulin joining chain | 0.004 | B | 1 |
| NM_008339 | Cd79b | CD79B antigen | 0.006 | B | 1 |
| Mm.780 | Igl-V1 | immunoglobulin λ chain, variable 1 | 0.007 | B | 1 |
| NM_010381 | H2-Ea | histocompatibility 2, class II antigen E α | 0.008 | B | 1 |
| NM_008860 | Prkcz | protein kinase C, ζ | 0.010 | — | |
| NM_010379 | H2-Ab1 | histocompatibility 2, class II antigen A, β 1 | 0.010 | B | 1 |
| NM_008528 | Blnk | B-cell linker | 0.010 | B | 1 |
| NM_025282 | Mef2c | myocyte enhancer factor 2C | 0.011 | B | 2 |
| NM_010389 | H2-Ob | histocompatibility 2, O region β locus | 0.011 | B | 1 |
| NM_009844 | Cd19 | CD19 antigen | 0.011 | B | 1 |
| NM_007643 | Cd36 | CD36 antigen | 0.015 | — | |
| NM_010387 | H2-DMb1 | histocompatibility 2, class II, locus Mb1 | 0.024 | B | 1 |
| NM_010388 | H2-DMb2 | histocompatibility 2, class II, locus Mb2 | 0.037 | B | 1 |
| NM_007549 | Blk | B lymphoid kinase | 0.038 | B | 1 |
| NM_029494 | Rsb30 | RAB30, member RAS oncogene family | 0.041 | — | |
| NM_009845 | Cd22 | CD22 antigen | 0.051 | B | 1 |
| NM_011611 | Tnfrsf5 | tumor necrosis factor receptor superfamily, member 5 | 0.062 | B | 1 |
| NM_010747 | Lyn | Yamaguchi sarcoma viral (v-yes-1) oncogene homolog | 0.065 | B | 3 |
| NM_007575 | C2ta | class II transactivator | 0.088 | B | 1 |
| NM_008533 | Ly78 | lymphocyte antigen 78 | 0.088 | B | 1 |
| NM_009744 | Bcl6 | B-cell leukemia/lymphoma 6 | 0.106 | — | |
| NM_007758 | Cr2 | complement receptor 2 | 0.107 | B | 4 |
| XM_354702 | none | similar to immunoglobulin ε | 0.110 | B | 1 |
| NM_021893 | Pdcd1lg1 | programmed cell death 1 ligand 1 | 0.126 | — | |
| NM_010557 | Il4ra | interleukin 4 receptor, α | 0.142 | — | |
| NM_007778 | Csf1 | colony stimulating factor 1 (macrophage) | 0.226 | — | |
| NM_009852 | Cd6 | CD6 antigen | 4.70 | — | |
| NM_013653 | Ccl5 | chemokine (C-C motif) ligand 5 | 7.72 | T | 5 |
| XM_284241 | Tnfrsf7 | tumor necrosis factor receptor superfamily, member 7 | 10.35 | T | 1 |
| NM_008453 | Klf3 | Kruppel-like factor 3 (basic) | 10.70 | — | |
| NM_009400 | Tnfrsf18 | tumor necrosis factor receptor superfamily, member 18 | 14.82 | T | 6 |
| NM_009192 | Sla | src-like adaptor | 16.72 | — | |
| NM_008353 | Il12rb1 | interleukin 12 receptor, β 1 | 20.91 | T | 1 |
| NM_019436 | Sit | SHP2 interacting transmembrane adaptor | 26.66 | T | 1 |
| NM_008859 | Prkcq | protein kinase C, θ | 27.05 | T | 7 |
| NM_010742 | Ly6d | lymphocyte antigen 6 complex, locus D | 31.17 | — | |
| Mm.123831 | Tcrb-V8.2 | T-cell receptor β, variable 8.2 | 35.75 | T | 1 |
| NM_008091 | Gata3 | GATA binding protein 3 | 40.45 | T | 1 |
| NM_010693 | Lck | lymphocyte protein tyrosine kinase | 40.51 | T | 1 |
| NM_010278 | Gfi1 | growth factor independent 1 | 42.67 | T | 8 |
| NM_011487 | Stat4 | signal transducer and activator of transcription 4 | 49.24 | T | 1 |
| NM_011701 | Vim | Vimentin | 52.19 | — | |
| NM_009122 | Satb1 | special AT-rich sequence binding protein 1 | 56.90 | T | 9 |
| NM_009850 | Cd3g | CD3 antigen, γ polypeptide | 59.09 | T | 1 |
| NM_009344 | Phlda1 | pleckstrin homology-like domain, family A, member 1 | 63.57 | T | 10 |
| NM_007642 | Cd28 | CD28 antigen | 67.64 | T | 1 |
| NM_011346 | Sell | selectin, lymphocyte | 72.79 | — | |
| NM_007648 | Cd3e | CD3 antigen, ε polypeptide | 93.03 | T | 1 |
| NM_008979 | Ptpn8 | protein tyrosine phosphatase, non-receptor type 8 | 113.63 | — | |
| NM_009345 | Dntt | deoxynucleotidyltransferase, terminal | 118.86 | — | |
| NM_010583 | Itk | IL2-inducible T-cell kinase | 120.75 | T | 11 |
| NM_011609 | Tnfrsf1a | tumor necrosis factor receptor superfamily, member 1a | 132.72 | — | |
| Mm.123831 | Tcrb-V13 | T-cell receptor β, variable | 167.67 | T | 1 |
| NM_010689 | Lat | linker for activation of T cells | 168.24 | T | 12 |
| NM_009382 | Thy1 | thymus cell antigen 1, θ | 171.82 | T | 1 |
| NM_009829 | Ccnd2 | cyclin D2 | 242.81 | — | |
| Mm.210785 | T cell receptor γ chain | 424.47 | T | 1 | |
| NM_013487 | Cd3d | CD3 antigen, δ polypeptide | 1532.15 | T | 1 |
T/B Ratio is the array hybridization signal measured in the 100% R1.1 sample divided by that of the 100% A20 sample. Each measurement is the mean of four separate arrays
According to literature. Blanks indicate that we could not find literature-based evidence for T- or B-cell-specific expression. For 14 of the 17 genes for which there was no literature support for the expression pattern, the same samples have been examined by a reverse transcription-based RNA quantitation assay (Fan et al. 2004); in all cases examined, the results from this orthogonal assay agreed with the results determined in this array experiment (J. Yeakley, J.-B. Fan, and E. Chudin, unpubl.)
Discussion
We developed a powerful and robust new microarray technology for gene-expression profiling on the basis of randomly assembled arrays of beads in wells. The high information density of these arrays (∼50,000 beads/∼1.4-mm diameter array) reduces sample consumption and makes them well suited for integration into sophisticated systems such as the array matrix device described herein. Each probe is replicated a minimum of five times and on average ∼30 times on every array. This built-in redundancy increases measurement precision and makes for an intrinsically robust measurement platform. We optimized the system for hybridization specificity and sensitivity, integrated the various components into a scalable system for gene-expression quantitation, and showed that accurate and reproducible data are generated from complex biological samples.
The 96-array matrix format and associated protocols make it straightforward to analyze many samples with relatively little labor and high reproducibility. We consider this a significant advance because sources of noise and error, such as intra- and interarray variability, process variability, and biological sample variability, can confound microarray experiments (Brody et al. 2002). An effective way of identifying, characterizing, and minimizing variation is to apply well-known statistical tools. Unfortunately, the ease-of-handling, and in many cases, the reproducibility of current microarray technologies makes it difficult to replicate experiments adequately. This has severely limited the ability to generate and analyze large data sets. As a consequence, the use of microarrays in applications requiring the analysis of large numbers of samples, such as epidemiological, toxicological, and pharmacological screening, has been limited mostly to proof-of-concept studies. Meaningful application of high-throughput microarray technology to large sample sets is now more practical as a result of the system described here.
Samples can be processed in standard micro-plate formats, either manually or robotically. The entire system is designed for compatibility with automation and LIMS tracking, and hence, is suitable for use in applications that require a highly reproducible process with accurate sample tracking throughout. The technology is flexible. It can be used to analyze the expression of hundreds of genes, as described in this study, as well as whole-genome sets of many thousands of genes, which will be described elsewhere. The ability to assemble large numbers of arrays from a single bead pool on the basis of a common chemistry helps to minimize interarray variability. Flexibility in array design is provided by the ability to supplement standard bead pools with sequences of the user's choosing or to make custom bead pools.3
We also developed software for array imaging and gene-expression data analysis (E. Chudin and I. Mikouliteh, unpubl.). Because of the robustness of the system, the user has to pay less attention to the data extraction process than typical with spotted arrays, and can instead focus on analysis of results. AnEx, a gene-expression data analysis program that organizes sample data and incorporates statistical and visualization tools, is commercially available as part of the gene-expression analysis system. AnEx is MIAME-compliant (www.mged.org) and generates a flat-file format that is accepted by many third-party analysis software applications.
Finally, an advantage of the system we have developed is that it uses the same technology platform as our SNP genotyping system (Fan et al. 2003) and our PCR-based gene-expression assay system (Fan et al. 2004). As a result, SNP genotyping and gene-expression profiling can now be carried out on a single microarray platform, scalable from the analysis of hundreds of genes to all known genes in a genome.
Methods
Samples
Human brain and liver total RNA were purchased from Ambion (Cat. #7962, Brain; 7960, Liver). Human HepG2 Poly(A+) mRNA was purchased from Ambion (Cat. #7849). Mouse spleen total RNA was purchased from Ambion (Cat. #7920). A20 and R1.1 cell lines were purchased from the American Type Culture Collection (ATCC; A20, Cat. #TIB-208, R1.1, Cat. #TIB-42, R1.1) and were grown according to supplier's recommendations. A20 cells were grown in RPMI 1640 medium with 2 mM L-glutamine, and supplemented with 1.5 g/L NaHCO3, 1.0 mM Na pyruvate, 10 mM HEPES, and 10% fetal bovine serum (Hyclone). R1.1 cells were grown in DMEM high-glucose medium with glutamate supplemented with 1.5 g/L NaHCO3 and 10% horse serum. Total RNA was harvested from ∼108 cells using the RNeasy Midi kit (QIAGEN) according to the manufacturer's instructions.
Labeling
Although our platform is amenable to a number of standard sample labeling techniques, our preferred approach is based on the modified Eberwine protocol (Eberwine et al. 1992), by which messenger RNA is converted to cDNA, followed by an amplification/labeling step mediated by T7 DNA polymerase. The linear amplification step reduces the amount of starting material needed. We adapted the protocol to a microtiter plate format in order to match the array matrix format, which permits 96 array hybridizations to be performed in parallel. Labeling and amplification of the total RNA samples were performed according to the MessageAmp aRNA kit (Ambion Cat. #1750) with the following modifications. Because the hybridization requirements are so modest (1 μg labeled cRNA), the standard reaction was cut down to 1/4 size and total RNA inputs were generally limited to 100 ng. The use of smaller reactions allowed us to perform 80 reactions per kit as opposed to the standard 20 reactions. This necessitated the use of additional cleanup columns for both the RT and IVT steps. QIAquick PCR Purification and RNeasy 96 well kits (QIAGEN) were used according to the manufacturer's instructions for RT and IVT cleanup, respectively. Additionally, all components of the first-strand cDNA synthesis were combined in a single step, because a separate annealing of the T7 oligo(dT) primer was found to be unnecessary (data not shown). During the IVT reaction, a 1:1 ratio of labeled bio-16-UTP (Roche Cat. #1388908) to unlabeled UTP was used with a final combined concentration of 7.5 mM.
Preparation of labeled spikes
Nine bacterial and viral genes were used to prepare RNA controls as follows: bla (pBluescriptSK+; Stratagene) cat (pCAT3-control; Promega), cre (Escherichia coli DH10B-Zip; Life Technologies), e1a (Homo sapiens HEK-293; ATCC), gfp (pEGFP; Clontech), gst (pGEX-5x-3; Amersham-Pharmacia), gus (E. coli GM48), lux (E. coli GM48), and neo (pGT-N28; New England Biolabs). The genes were cloned into the PCRII cloning vector using the TA Cloning kit (Invitrogen, Cat. #K205001-TA). Full-length sense transcripts were generated using the MEGA-script T3 kit from Ambion (Cat. #1338). Labeled antisense targets were then generated using the MessageAmp aRNA kit and were spiked into labeled Human HepG2 cRNA at the 12 concentrations shown in Figure 2.
Hybridization/washing/signal detection
All steps of hybridization, washing, blocking, and signal generation were performed by sequential transfer of a Sentrix array matrix from one 384-well plate (ThermoLab Systems; Cat. #95040000) to the next with the wells of each step containing 40 μL of the appropriate solution. All incubations were carried out without agitation and, with the exception of the hybridization, at room temperature. Amplified, biotin-labeled human or mouse RNA samples were prepared in a solution of Hyb E1 buffer (Illumina, Part #11166381) and 25% (v/v) formamide at a final concentration of 25 ng/μL. An array matrix was then mated to the hybridization plate using a sealed alignment fixture. Hybridization proceeded at 55°C, for 16 to 20 h. After hybridization, the array matrix was washed by a 5-min incubation in Illumina Wash E1 buffer, followed by a 10-min wash in fresh Wash E1 buffer (Illumina, Part #11165898). Arrays were then blocked for 5 min in 1% (w/v) casein-PBS, Hammerstein grade (Pierce, Cat. #37528). Array signal was developed by a 10-min incubation in a 1-μg/mL solution of Streptavidin-Cy3 (Amersham; Cat. #PA43001) in 1% casein-PBS blocking solution. The array matrix was washed a final time for 5 min in Wash E1 buffer. Each array was then dried with an air gun.
Imaging and signal extraction
Arrays were scanned on the BeadArray Reader, a confocal-type imaging system with ∼0.8 μm resolution and 532 and 635 nm laser illumination (Barker et al. 2003). Scans were performed in the 532-nm channel. The total scan time per array matrix (i.e., 96 arrays) was 1.5 h, roughly 1 min per array. Image analysis and data extraction software were as described previously (Fan et al. 2003). Briefly, each sequence type is represented by an average of 30 beads on the array. Bead signals were computed with weighted averages of pixel intensities, and local background was subtracted. Array images are registered by a previously described algorithm (Galinsky 2003). This algorithm supplies the position of a bead center that serves as a center for a virtual pixel. To compute bead signal, we use four real pixels covering the virtual one and combine their signals in the following way: S = A1S1+A2S2+A3S3+A4S4, where S is bead signal, Ai is area of overlap between ith pixel and the virtual pixel, and Si is 3 × 3 average taken around ith pixel after sharpening with following Laplacian:
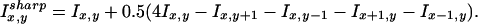 |
Here x,y are pixel coordinates and Ix,y are pixel intensities. The choice of coefficient in front of Laplacian was made after optimization of data obtained with calibrated set of Spherotech 3-micron rainbow beads (Cat. #RCP-30-5, Spherotech, Inc.). Finally, we subtract local background as average of five dimmest pixels in the 17 × 17 box centered in the pixel having maximum overlap with the virtual pixel. Sequence-type signal was calculated by averaging corresponding bead signals with outliers removed (using median absolute deviation).
Data analysis
We developed a suite of algorithms for analysis of gene expression data from microarrays (E. Chudin and I. Mikoulitch, pers. comm.). These have been incorporated into AnEx, a commercial software package for gene-expression data analysis. Array data were normalized using quantiles to fit a cubic spline. The approach is similar to a previously reported method (Workman et al. 2002). Alternatively, a robust least-squares fit (iteratively re-weighted least squares using Tukey's biweight functions) of intensities of a rank invariant set of probes (relative rank change of <0.05) was used. Detection p-values were computed using a dynamically constructed normal model based on intensities of 20 negative controls. To determine minimal resolvable fold change, we used piecewise linear approximation of intensity versus concentration. Concentration levels were considered resolvable if corresponding one-sided 95th percent confidence intervals, as computed from t-distribution did not overlap. Piecewise linear interpolation was used for both intensities and standard deviations.
Array design
Probes were designed by a custom-built pipeline that will be described in detail elsewhere (P. Rigault, in prep.). Each gene sequence for which probes were to be synthesized was subjected to a filtering process that masked regions unsuitable for probe design, based on complexity and cross-homology thresholds, as determined by DUST (D. Lipman, National Center for Biotechnology Information, pers. comm.) and BLAST (Altschul et al. 1990) algorithms, respectively. All possible 50-mer probes were identified within unmasked regions, and these were ranked by a formula that takes into account distance from the 3′ end of the transcript, melting temperature, and self-complementarity. The two highest scoring probes were then linked to 23-nt identifier sequences by use of a sequence-matching program that minimizes the probability of interactions between the probe and identifier sequence and prevents the creation of junction sequences with cross-homology to the genome in question.
Our use of two probes per gene was based on the results of pilot experiments, in which five informatically chosen probes were synthesized for each of 10 in vitro-synthesized genes. Dose response was determined for each synthetic gene using all five probes or four, three, two, or one arbitrarily selected probes. We found that we could reach our targeted performance metrics (Table 1) with two or more probes per gene, but not one (data not shown). The results of recent functional screening suggests that one probe per gene is sufficient if the probes are selected with a functional screen (T. McDaniel, B. Kermani, S. Baker, S. Oeser, and S. Kruglyak, unpubl.).
Quantitative PCR
Assays-on-Demand quantitative gene expression primers and TaqMan universal PCR master mix (Cat. #4304437) were purchased from Applied Biosystems. All PCR reactions were performed following the manufacturer's instructions.
Acknowledgments
We thank Steven Barnard, Chanfeng Zhao, Paul Kitabjian, Michael Graige, and Semyon Kruglyak for devising methods to prepare beads with gene-specific probes and for providing bead pools used in these experiments. We also thank Chan Tsan for technical assistance, Lixin Zhou for help with analysis of the B and T cell experiments, and the array manufacturing group at Illumina for providing gene-specific probe arrays.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.2739104.
Footnotes
Standard, semicustom and fully custom bead sets are provided commercially by Illumina. Semicustom bead sets are made by supplementing standard sets with sequences of the customer's choosing. Fully custom bead sets can be made from any desired set of sequences.
References
- Abbas, A.K., Lichtman, A.H., and Pober, J.S. 2003. Cellular and molecular immunology. W.B. Saunders, Philadelphia, PA.
- Altschul, S.F., Gish, W., Miller, W., Myers, E.W., and Lipman, D.J. 1990. Basic local alignment search tool. J. Mol. Biol. 215: 403-410. [DOI] [PubMed] [Google Scholar]
- Barker, D.L., Therault, G., Che, D., Dickinson, T., Shen, R., and Kain, R. 2003. Self-assembled random arrays: High-performance imaging and genomics applications on a high-density microarray platform. Proc. SPIE 4966: 1-11. [Google Scholar]
- Blanchard, A. 1998. Synthetic DNA arrays. Plenum Press, New York.
- Brody, J.P., Williams, B.A., Wold, B.J., and Quake, S.R. 2002. Significance and statistical errors in the analysis of DNA microarray data. Proc. Natl. Acad. Sci. 99: 12975-12978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson, L.A., Joh, T., Kohwi, Y., and Kohwi-Shigematsu, T. 1992. A tissue-specific MAR/SAR DNA-binding protein with unusual binding site recognition. Cell 70: 631-645. [DOI] [PubMed] [Google Scholar]
- Eberwine, J., Yeh, H., Miyashiro, K., Cao, Y., Nair, S., Finnell, R., Zettel, M., and Coleman, P. 1992. Analysis of gene expression in single live neurons. Proc. Natl. Acad. Sci. 89: 3010-3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein, J.R., Lee, M., and Walt, D.R. 2002. High-density fiber-optic genosensor microsphere array capable of zeptomole detection limits. Anal. Chem. 74: 1836-1840. [DOI] [PubMed] [Google Scholar]
- Fan, J.-B., Oliphant, A., Shen, R., Kermani, B.G., Garcia, F., Gunderson, K.L., Hansen, M., Steemers, F., Butler, S.L., Deloukas, P., et al. 2003. Highly parallel SNP genotyping. Cold Spring Harbor Symp. Biol. 68: 69-78. [DOI] [PubMed] [Google Scholar]
- Fan, J.B., Yeakley, J.M., Bibikova, M., Chudin, E., Wickham, E., Chen, J., Doucet, D., Rigault, P., Zhang, B., Shen, R., et al. 2004. A versatile assay for high-throughput gene expression profiling on universal array matrices. Genome Res. 14: 878-885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fingeroth, J.D. 1990. Comparative structure and evolution of murine CR2. The homolog of the human C3d/EBV receptor (CD21). J. Immunol. 144: 3458-3467. [PubMed] [Google Scholar]
- Fodor, S.P.A., Read, J.L., Pirrung, M.C., Stryer, L., Lu, A.T., and Solas, D. 1991. Light-directed, spatially addressable parallel chemical synthesis. Science 251: 767-773. [DOI] [PubMed] [Google Scholar]
- Galinsky, V.L. 2003. Automatic registration of microarray images. II. Hexagonal grid. Bioinformatics 19: 1832-1836. [DOI] [PubMed] [Google Scholar]
- Golub, T.R., Slonim, D.K., Tamayo, P., Huard, C., Gaasenbeek, M., Mesirov, J.P., Coller, H., Loh, M.L., Downing, J.R., Caligiuri, M.A., et al. 1999. Molecular classification of cancer: Class discovery and class prediction by gene expression monitoring. Science 286: 531-537. [DOI] [PubMed] [Google Scholar]
- Gunderson, K., Kruglyak, S., Graige, M.S., Garcia, F., Kermani, B.G., Zhao, C., Che, D., Milewski, M., Yang, R., Siegmund, C., et al. 2004. Decoding randomly ordered arrays. Genome Res. 14: 870-877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heid, C.A., Stevens, J., Livak, K.J., and Williams, P.M. 1996. Real time quantitative PCR. Genome Res. 6: 986-994. [DOI] [PubMed] [Google Scholar]
- Hermanson, G.G., Eisenberg, D., Kincade, P.W., and Wall, R. 1988. B29: A member of the immunoglobulin gene superfamily exclusively expressed on β-lineage cells. Proc. Natl. Acad. Sci. 85: 6890-6894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes, T.R., Marton, M.J., Jones, A.R., Roberts, C.J., Stoughton, R., Armour, C.D., Bennett, H.A., Coffey, E., Dai, H., He, Y.D., et al. 2000. Functional discovery via a compendium of expression profiles. Cell 102: 109-126. [DOI] [PubMed] [Google Scholar]
- Hughes, T.R., Mao, M., Jones, A.R., Burchard, J., Marton, M.J., Shannon, K.W., Lefkowitz, S.M., Ziman, M., Schelter, J.M., Meyer, M.R., et al. 2001. Expression profiling using microarrays fabricated by an ink-jet oligonucleotide synthesizer. Nat. Biotechnol. 19: 342-347. [DOI] [PubMed] [Google Scholar]
- Ideker, T., Thorsson, V., Ranish, J.A., Christmas, R., Buhler, J., Eng, J.K., Bumgarner, R., Goodlett, D.R., Aebersold, R., and Hood, L. 2001. Integrated genomic and proteomic analyses of a systematically perturbed metabolic network. Science 292: 929-934. [DOI] [PubMed] [Google Scholar]
- The International HapMap Consortium. 2003. The International HapMap Project. Nature 426: 789-796. [DOI] [PubMed] [Google Scholar]
- Isakov, N. and Altman, A. 2002. Protein kinase C(theta) in T cell activation. Annu. Rev. Immunol. 20: 761-794. [DOI] [PubMed] [Google Scholar]
- Lockhart, D.J. and Winzeler, E.A. 2000. Genomics, gene expression and DNA arrays. Nature 405: 827-836. [DOI] [PubMed] [Google Scholar]
- Lockhart, D.J., Dong, H., Byrne, M.C., Follettie, M.T., Gallo, M.V., Chee, M.S., Mittmann, M., Wang, C., Kobayashi, M., Horton, H., et al. 1996. Expression monitoring by hybridization to high-density oligonucleotide arrays. Nat. Biotechnol. 14: 1675-1680. [DOI] [PubMed] [Google Scholar]
- Marton, M.J., DeRisi, J.L., Bennett, H.A., Iyer, V.R., Meyer, M.R., Roberts, C.J., Stoughton, R., Burchard, J., Slade, D., Dai, H., et al. 1998. Drug target validation and identification of secondary drug target effects using DNA microarrays. Nat. Med. 4: 1293-1301. [DOI] [PubMed] [Google Scholar]
- Michael, K.L., Taylor, L.C., Schultz, S.L., and Walt, D.R. 1998. Randomly ordered addressable high-density optical sensor arrays. Anal. Chem. 70: 1242-1248. [DOI] [PubMed] [Google Scholar]
- Nocentini, G., Giunchi, L., Ronchetti, S., Krausz, L.T., Bartoli, A., Moraca, R., Migliorati, G., and Riccardi, C. 1997. A new member of the tumor necrosis factor/nerve growth factor receptor family inhibits T cell receptor-induced apoptosis. Proc. Natl. Acad. Sci. 94: 6216-6221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, C.G., Lee, S.Y., Kandala, G., and Choi, Y. 1996. A novel gene product that couples TCR signaling to Fas(CD95) expression in activation-induced cell death. Immunity 4: 583-591. [DOI] [PubMed] [Google Scholar]
- Schall, T.J., Jongstra, J., Dyer, B.J., Jorgensen, J., Clayberger, C., Davis, M.M., and Krensky, A.M. 1988. A human T cell-specific molecule is a member of a new gene family. J. Immunol. 141: 1018-1025. [PubMed] [Google Scholar]
- Scheijen, B., Jonkers, J., Acton, D., and Berns, A. 1997. Characterization of pal-1, a common proviral insertion site in murine leukemia virus-induced lymphomas of c-myc and Pim-1 transgenic mice. J. Virol. 71: 9-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schena, M., Shalon, D., Davis, R.W., and Brown, P.O. 1995. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 270: 467-470. [DOI] [PubMed] [Google Scholar]
- Siliciano, J.D., Morrow, T.A., and Desiderio, S.V. 1992. itk, a T-cell-specific tyrosine kinase gene inducible by interleukin 2. Proc. Natl. Acad. Sci. 89: 11194-11198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van't Veer, L.J., Dai, H., van de Vijver, M.J., He, Y.D., Hart, A.A., Mao, M., Peterse, H.L., van der Kooy, K., Marton, M.J., Witteveen, A.T., et al. 2002. Gene expression profiling predicts clinical outcome of breast cancer. Nature 415: 530-536. [DOI] [PubMed] [Google Scholar]
- Workman, C., Jensen, L.J., Jarmer, H., Berka, R., Gautier, L., Nielser, H.B., Saxild, H.H., Nielsen, C., Brunak, S., and Knudsen, S. 2002. A new non-linear normalization method for reducing variability in DNA microarray experiments. Genome Biol. 3: research0048. [DOI] [PMC free article] [PubMed]
- Yamanashi, Y., Kakiuchi, T., Mizuguchi, J., Yamamoto, T., and Toyoshima, K. 1991. Association of B cell antigen receptor with protein tyrosine kinase Lyn. Science 251: 192-194. [DOI] [PubMed] [Google Scholar]
- Yeakley, J.M., Fan, J.B., Doucet, D., Luo, L., Wickham, E., Ye, Z., Chee, M.S., and Fu, X.D. 2002. Profiling alternative splicing on fiber-optic arrays. Nat. Biotechnol. 20: 353-358. [DOI] [PubMed] [Google Scholar]
- Yvert, G., Brem, R.B., Whittle, J., Akey, J.M., Foss, E., Smith, E.N., Mackelprang, R., and Kruglyak, L. 2003. Trans-acting regulatory variation in Saccharomyces cerevisiae and the role of transcription factors. Nat. Genet. 35: 57-64. [DOI] [PubMed] [Google Scholar]
- Zhang, W., Sloan-Lancaster, J., Kitchen, J., Trible, R.P., and Samelson, L.E. 1998. LAT: The ZAP-70 tyrosine kinase substrate that links T cell receptor to cellular activation. Cell 92: 83-92. [DOI] [PubMed] [Google Scholar]
WEB SITE REFERENCES
- www.hapmap.org; International HapMap Project.
- www.illumina.com; Illumina, Inc.
- www.mged.org; Microarray Gene Expression Data Society.