

Abstract
Increasing evidence indicates a strong link between intestinal health and bone health. For example, inflammatory bowel disease can cause systemic inflammation, weight loss, and extra-intestinal manifestations, such as decreased bone growth and density. However, the effects of moderate intestinal inflammation without weight loss on bone health have never been directly examined; yet this condition is relevant not only to IBD but to conditions of increased intestinal permeability and inflammation, as seen with ingestion of high-fat diets, intestinal dysbiosis, irritable bowel syndrome, metabolic syndrome, and food allergies. Here, we induced moderate intestinal inflammation without weight loss in young male mice by treating with a low dose of dextran sodium sulfate (1%) for 15 days. The mice displayed systemic changes marked by significant bone loss and a redistribution of fat from subcutaneous to visceral fat pad stores. Bone loss was caused by reduced osteoblast activity, characterized by decreased expression of osteoblast markers (runx2, osteocalcin), histomorphometry, and dynamic measures of bone formation. In addition, we observed a reduction in growth plate thickness and hypertrophic chondrocyte matrix components (collagen X). Correlation analyses indicate a link between gut inflammation and disease score, but more importantly, we observed that bone density measures negatively correlated with intestinal disease score, as well as colon and bone TNF-α levels. These studies demonstrate that colitis-induced bone loss is not dependent upon weight loss and support a role for inflammation in the link between gut and bone health, an important area for future therapeutic development.
Keywords: intestine; inflammation bone density; growth, osteoblast; cartilage
increasing evidence indicates a strong link between intestinal health and bone health. This is clearly evident in inflammatory bowel disease (IBD). IBD affects over 1.6 million Americans. IBD patients experience chronic intestinal inflammation marked by disease flares characterized by weight loss, diarrhea, fatigue, and abdominal pain (7, 8). Additionally, IBD patients can display systemic inflammation and extra-intestinal manifestations, such as low bone density (observed before treatment with corticosteroids) (6, 11, 15, 25, 56, 62, 72–74, 77, 78). Since the onset of disease often occurs before maximum bone growth and density are attained (between the ages of 20 and 30), both bone growth and density can decrease and lead to short stature and increased risk of osteoporosis and fractures later in life (7, 8, 29, 66, 71). The mechanisms accounting for IBD-induced bone loss are thought to relate to a variety of factors that include weight loss (20, 54, 61, 63, 67, 68, 76) and systemic inflammation (21, 51).
Similar to humans, animal models of IBD display lower bone density (26, 28), as well as decreased bone growth (2, 28, 65). While many animal models of IBD exist [spontaneous, inducible, genetic, adoptive transfer (48, 84)], chemically induced colitis is most commonly used because it allows control over disease onset, duration, and inflammation severity (49, 55). Dextran sodium sulfate (DSS)-induced colitis is a well-established and frequently used model. DSS forms complexes with medium-chain fatty acids present in the colon to activate inflammation, cause barrier integrity breakdown, and increased mucosal permeability, similar to what is observed in IBD patients (35, 41, 83). Doses of DSS between 2 and 5% are used to produce colitis (49, 55, 83), which is associated with weight loss and intestinal inflammation and permeability.
Previously, we studied the effects of 5% DSS on bone health in 5-wk-old male mice. The resulting inflammation was severe, causing significant weight loss (greater than 15% body weight), bone loss, and reduced bone growth (28). In the current study, we examined whether a lower level of intestinal inflammation, one that did not cause weight loss, could cause bone loss. The effects of moderate intestinal inflammation (without weight loss) on bone health have never been directly examined; however, the condition is relevant not only to IBD patients but also to conditions of increased intestinal permeability and inflammation seen with ingestion of a high-fat diet, high-fructose diet, obesity, intestinal dysbiosis, irritable bowel syndrome, metabolic syndrome, and food allergies (10, 23). Our findings indicate that treatment of young mice with 1% DSS causes intestinal inflammation without weight loss, although we did see redistribution of fat stores. More importantly, the DSS-treated mice lost a significant amount of bone and displayed reduced growth plate thickness, demonstrating that colitis-induced bone changes are not dependent upon weight loss.
MATERIALS AND METHODS
Mouse model.
Male 5–6-wk-old C57BL/6 mice were given 1% (wt/vol) dextran sodium sulfate salt (DSS) (36,000–50,000 MW, MP Biomedical, Santa Anna, CA) in sterile drinking water for 15 days to induce colitis. This DSS concentration was chosen on the basis of pilot dose response studies (ranging from 0.1 to 5% wt/vol DSS) that revealed weight loss when 2% DSS or more was used. Thus, we selected 1% DSS as the highest dose without weight loss. Control mice were given sterile drinking water. Both groups were housed in a 12:12-h light-dark cycle room at 23°C and were allowed to drink and eat (standard chow, Teklad 8640) ad libitum. Body weights of all mice were measured throughout the experiment. The Michigan State University Animal Use Committee approved all of the protocols.
Histological confirmation of colitis.
To confirm colitis, a scoring system was used that incorporates live mouse measures, as well as cecal histology. Disease in live mice is evaluated by fecal pellet consistency (0–10, ranging from normal to loose bloody diarrhea), appearance of rectal bleeding, and weight loss. For cecal histomorphometric evaluation, paraffin-embedded cecum sections were stained with hematoxylin and eosin and were imaged at ×4, ×10, and ×25 magnification. At high magnification (×25), three areas were imaged, coded, and analyzed by researchers blind to the condition of the mouse. Active disease was evaluated by measurements of mucosal crypt length; crypt structure distortion; quantity of lymphoid aggregates, lymphoid cells, and neutrophils; and mucosal inflammation and ulceration severity. Mucosal length was measured from the top of the epithelial cells to the top of the muscularis layer. Crypt distortion measurements were obtained at the ileocecocolic junction and were scored as 0, straight crypts; 2, crypts with slight distortion; 4, distorted crypts with mild hyperchromasia with fewer goblet cells; 6, larger areas of pronounced crypt distortion, with more pronounced hyperchromasia with very few goblet cells; or 8, crypt drop-out, marked hyperchromasia, and no goblet cells. In each section, lymphoid aggregates were counted, and the abundance of lymphoid cells was counted and scored as 0, no cells; 2, 11–30 cells; 4, 31–100; or 6, more than 100 lymphoid cells and counted within the submucosal layer of the cecum. Neutrophils were scored as 0, no neutrophils present; 5, few neutrophils; or 10, numerous neutrophils in the section. Disease severity was scored as 0, normal; 2, mild, small focal areas (1–5 crypts) of disease/inflammation limited to lamina propria; 4, moderate, multifocal with areas of disease/inflammation extended into the submucosa; 6, severe with ulcers covering large areas of mucosa less than 20 crypts wide; 8, very severe with ulcers cover large areas of mucosa more than 20 crypts; or 10, extremely severe with ulcers covering large areas of mucosa more than 20 crypts wide and signs of bacteria or fungal invasion observed. Total overall disease score was obtained by the summation of the live mouse scores with the histological scores.
Bone density and architecture measurement.
Tibias were fixed in 10% formalin for 24 h and scanned using a GE Explore Locus microcomputed tomography (μCT) system at a voxel of 20 μm obtained from 720 views. The beam angle increment was 0.5, and beam strength was set at 80 peak kV and 450 μA. Each run consisted of control and DSS-treated mouse bones and a calibration phantom to standardize grayscale values and maintain consistency. On the basis of auto-threshold and iso-surface analyses of multiple bone samples, a fixed threshold of 700 was used to separate bone from bone marrow. Bone measurements were blinded. Tibial bone analyses were performed in a region of trabecular bone defined at 1% of the total length (~0.17 mm) proximal to the growth plate extending 2 mm toward the diaphysis, excluding the outer cortical bone. Trabecular bone mineral content, bone volume fraction, thickness, spacing, and number values were computed by GE Healthcare MicroView software application for visualization and analysis of volumetric image data. Cortical measurements were performed in a 2 × 2 × 2 mm cube centered midway down the length of the bone using a threshold of 1,000 to separate bone from bone marrow.
Bone static and dynamic histomorphometry.
Tibias were fixed in 10% formalin and transferred to 70% ethanol after 24 h. Fixed samples were processed for embedding in paraffin. Paraffin blocks were sectioned at 5 μm on a rotary microtome, and slides of sections were stained for tartrate-resistant acid phosphatase (TRAP) activity and counterstained with hematoxylin, according to the manufacturer protocol (387A-1KT, Sigma, St. Louis, MO). Osteoblast and osteoclast surface area were measured and expressed as a percentage of total bone surface in the tibial trabecular region ranging from the growth plate to 2 mm distal.
For dynamic histomorphometric measures of bone formation, mice were injected intraperitoneally with 200 μl of 10 mg/ml calcein (Sigma Aldrich, St. Louis, MO) at 7 and 2 days before harvest. L3 and L4 vertebrae were fixed in formalin at time of harvest and then transferred to 70% ethanol 48 h later. Vertebrae were embedded, sectioned, and examined under a UV light. Five images were taken in the trabecular bone region, and the distance between calcein lines (bone formation rate, BFR) and their length along the bone surface was measured to calculate mineral apposition rate (MAR).
Bone and colon RNA analysis.
Immediately following euthanasia, tibias and colon were cleaned of connective tissue and luminal contents, respectively; snap frozen in liquid nitrogen; and stored at −80°C. Frozen tibias were crushed under liquid nitrogen conditions with a Bessman tissue pulverizer. RNA was isolated using TriReagent (Molecular Research Center, Cincinnati, OH). RNA integrity was assessed by formaldehyde-agarose gel electrophoresis. cDNA was synthesized by reverse transcription using Superscript II reverse transcription kit and oligo dT (12–18) primers (Life Technologies, Grand Island, NY) and was amplified by real-time PCR with iQ SYBR Green Supermix (Bio-Rad, Hercules, CA). All gene-specific primers (Integrated DNA Technologies, Coralville, IA) have been published [Runx2 (47), osteocalcin (47), IL-6 (47), IFNγ (45), TNF-α (81), and IL-22 (https://mouseprimerdepot.nci.nih.gov)]. Hypoxanthine guanine phosphoribosyltransferase (79) mRNA levels were used as an internal control since they do not fluctuate with treatment. Real-time PCR was carried out for 40 cycles using the iCycler, and data were evaluated using the iCycler software. Each cycle consisted of 95°C for 15 s, 60°C for 30 s (except for osteocalcin, which has an annealing temperature of 65°C), and 72°C for 30 s. cDNA-free samples, a negative control, did not produce amplicons. Melting curve and gel analysis (sizing, isolation, and sequencing) were used to verify single amplicon products of the appropriate base pair size.
Statistical analysis.
All measurements are presented as the means ± SE. Student’s two tail t-tests were used to determine significance. Histopathology was statistically analyzed using the Mann-Whitney U-test, where noted.
RESULTS
Intestinal inflammation was induced by providing male mice with 1% DSS in the drinking water for 15 days. Control littermate mice received sterile drinking water only. The concentration of DSS was chosen on the basis of pilot dose-response studies indicating that 1% DSS caused intestinal inflammation without weight loss, unlike higher DSS doses. Accordingly, in this study, the body weights of the 1% DSS-treated mice did not significantly differ from control mice throughout the experiment and at the time of harvest (Fig. 1A). Similarly, the increase in body weight from the start to the end of the experiment did not significantly differ between groups (data not shown). Interestingly, at harvest, it was evident that the DSS treatment caused a redistribution of fat pad weight. While the level of inguinal (subcutaneous) fat mass decreased by 30%, the retroperitoneal (visceral) fat depot increased (by 233%) (Fig. 1, B and C). Effects of the DSS on the gastrointestinal tract were assessed on the basis of live mouse observations and intestine histology (Fig. 2). An extended scoring system was used to obtain greater sensitivity/range for assessing the degree of disease severity; this resulted in scores being spread out and allowed for better assessment of correlations to bone outcome measures (described later). Mice treated with DSS displayed softer stools and in some cases diarrhea at day 7 or 8; however, after this time, diarrhea was not observed, and symptoms were mild compared with those seen in past studies using higher DSS doses (28). Histologically, as expected, mice treated with 1% DSS displayed some pathology in cecum crypt structure, number of lymphoid aggregates, and number of cells counted within the aggregates. The individual histopathology scores for the DSS-treated mice were approximately twice the levels seen in control mice. Accordingly, the total disease score, the sum of live and histological parameters, was significantly higher in DSS mice compared with controls (Fig. 2).
Fig. 1.
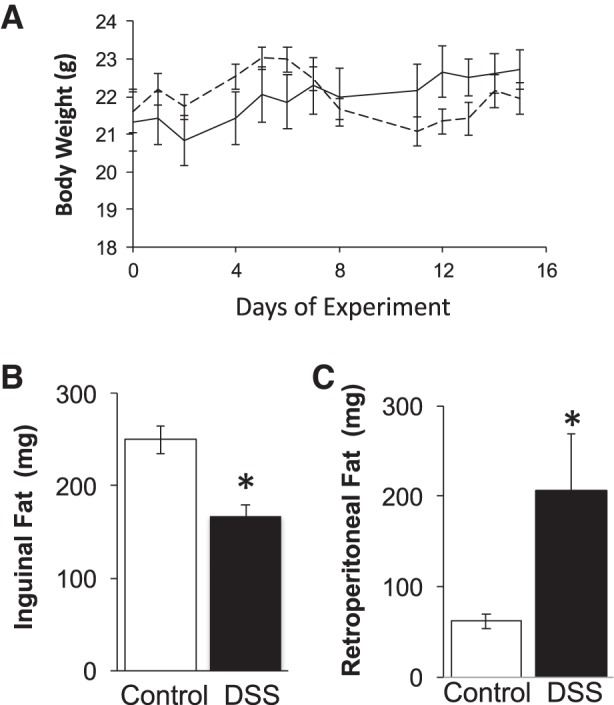
Mouse body and organ parameters. Male C57BL/6 mice were treated with 1% (wt/vol) DSS for 15 days to induce colitis. Control mice received drinking water only. Body weights were measured throughout the experiment [control, solid line; dextran sodium sulfate (DSS)-treated, dashed line]. At the time of harvest, organ weights were measured. Values represent averages ± SE *P ≤ 0.05, n ≥ 9 per group.
Fig. 2.
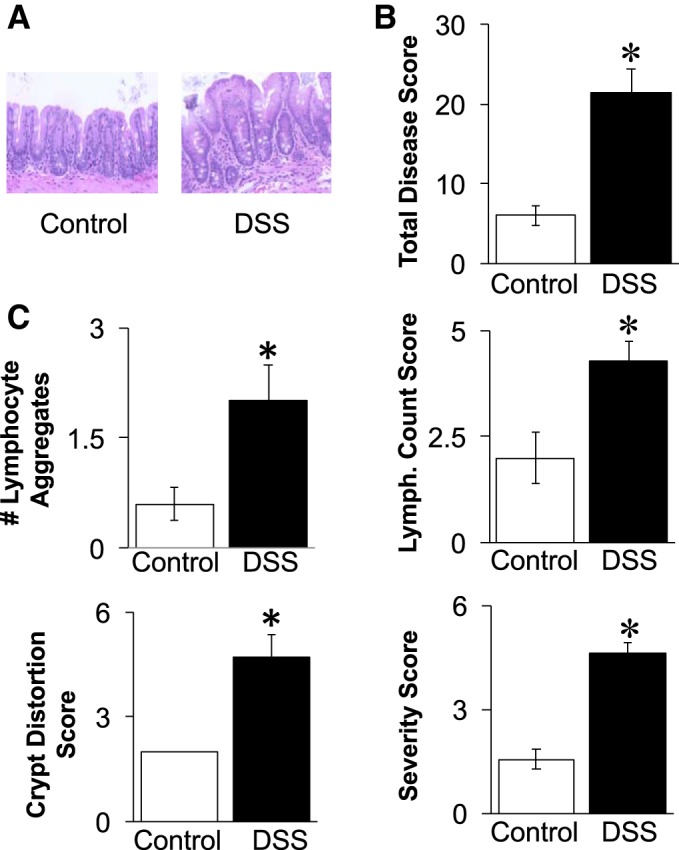
Assessment of colitis by a broad-range scoring system. A: representative cecum sections from control and DSS treated mice. B: total disease score, which is the sum of all live mouse health scores and histological scores. C: individual histological scores. Values represent averages ± SE. Statistical analyses used the Mann-Whitney U-two-tailed t-test. *P ≤ 0.01, n ≥ 9 per group.
To determine whether the previously reported bone loss associated with DSS-induced intestinal inflammation was dependent upon weight loss, we examined the long bone density and structure by microcomputed tomography. Surprisingly, mice treated with the low-dose DSS displayed a significant loss (nearly 50%) of tibial trabecular bone volume compared with controls (11.9 ± 2% vs. 22 ± 3%, respectively) (Fig. 3, Table 1) despite not losing weight. DSS-treated mice also displayed a 36% decreased bone mineral content (0.42 ± 0.03 mg compared with 0.27 ± 0.02 mg), as well as 34% decrease in bone mineral density (138 ± 11 mg/ml compared with 90 ± 8 mg/ml). Consistent with the bone volume analyses, DSS-treated mice displayed significantly increased (407%) trabecular spacing (133 ± 14 μm compared with 674 ± 19 μm) and decreased trabecular thickness (42.0 ± 2.4 μm compared with 34.3 ± 1.4 um). Cortical parameters were not affected (Table 1) nor were vertebral trabecular bone parameters (data not shown); these results are consistent with the typically longer time required to affect bone density at these sites.
Fig. 3.

Tibial bone parameters are decreased in 1% DSS-treated mice. A: representative bone isosurfaces from the proximal tibial trabecular bone region of control and DSS-treated mice. B: quantified bone volume fraction (BV/TV), bone mineral content, and bone mineral density measurements. C: trabecular bone characteristics: Tb. N., trabecular number; Tb. Sp., trabecular spacing; and Tb. Th., trabecular thickness. Values represent averages ± SE. *P ≤ 0.05, n ≥ 9 per group.
Table 1.
Cortical parameters
| Control | 1% DSS | |
|---|---|---|
| Tibia length, mm | 16.6 ± 0.2 | 16.5 ± 0.2 |
| Tt. Ar., mm2 | 1.03 ± 0.03 | 1.01 ± 0.03 |
| Ct. Ar., mm2 | 0.90 ± 0.02 | 0.86 ± 0.02 |
| Ma. Ar., mm2 | 0.13 ± 0.01 | 0.14 ± 0.01 |
| Ct. Ar./Tt. Ar., % | 87 ± 11 | 86 ± 1 |
| Ct. Th., mm | 0.37 ± 0.01 | 0.36 ± 0.01 |
| Inner Per., mm | 1.43 ± 0.06 | 1.49 ± 0.05 |
| Outer Per., mm | 4.17 ± 0.05 | 4.15 ± 0.10 |
Values are averages ± SE; n ≥ 12 per group. DSS, dextran sodium sulfate; Tt. Ar., total area; Ct. Ar., cortical area; Ma. Ar., marrow area; Ct. Ar./Tt. Ar., cortical area percent; Ct. Th., cortical thickness; Inner Per., inner perimeter; Outer Per., outer perimeter.
Given that the mice are undergoing skeletal growth during DSS treatment, we examined bone growth parameters. DSS treatment did cause a significant decrease (by 15%) in growth plate thickness (quantitated in saphenous O-stained bone sections) and in bone collagen X mRNA levels (Fig. 4). Collagen X expression is associated with chondrocyte maturation and cartilage development, and correspondingly, we observed a highly significant positive correlation between collagen X mRNA levels and growth plate thickness (Fig. 4B). Analysis of tibial bone length at this time point (Table 1) did not show a significant difference between DSS-treated and control mice (16.5 vs. 16.6 mm, respectively), which is not surprising. Given the short time frame of the study, one would only expect to see early markers of growth change that have not yet impacted full bone length.
Fig. 4.
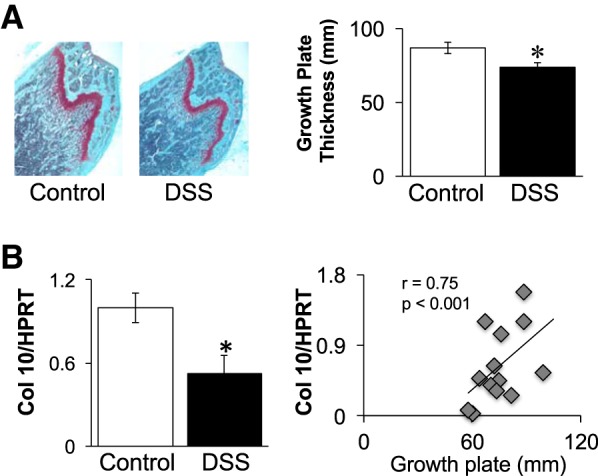
Mice treated with 1% DSS display significantly decreased growth plates width and collagen X expression. A: representative saphaneous-O-stained tibial sections and growth plate width measurements. B: collagen X (10) mRNA levels determined in whole bone RNA and expressed relative to a nonmodulated housekeeping gene, HPRT (hypoxanthine guanine phosphoribosyltransferase). Pearson’s correlation graph relates collagen X RNA levels to growth plate thickness. Values represent averages ± SE. *P ≤ 0.05, n ≥ 9 per group.
To determine whether an anabolic, bone formation defect (decreased osteoblast activity) could be contributing to the DSS-induced bone changes, we assessed bone formation by pulsing mice with calcein. The calculated bone formation rate was significantly lower (38%) in DSS-treated mice (0.44 ± 0.06 μm2·μm−3·day−1) compared with control mice (0.61 ± 0.06 μm2·μm−3·day−1) (Fig. 5A). Mineral apposition rate was also significantly decreased in DSS-treated mice compared with controls (1.37 ± 0.07 μm/day DSS, 1.83 ± 0.07 μm/day control). Consistent with the dynamic measures, static histomorphometric analyses demonstrated a decrease in the percentage of osteoblast surface/total bone surface (controls: 9.82 ± 1.22 vs. DSS: 7.11 ± 0.58; Fig. 5). Further confirmation of suppressed osteoblast activity in DSS-treated mice is demonstrated by significant decreases in Runx2 and osteocalcin mRNA levels, early- and late-stage markers of osteoblasts, respectively (Fig. 5B). We also examined expression of cytokines (including TNF, IL-1, IL-6, and IFNγ) in whole bone RNA to assess whether there were any detectable changes in the DSS-treated compared with control mice. We observed a significant increase in expression of only one proinflammatory cytokine, TNF-α, in the DSS-treated mouse bones (shown in Fig. 5B). Next, we examined whether catabolic, bone resorption parameters were elevated. In contrast to osteoblast measures, DSS treatment did not change the percentage of osteoclast surface or markers of osteoclast maturation (TRAP5 serum and RNA levels, Fig. 6). Similarly, we did not observe a change in the expression of regulators of osteoclast activation, such as RANKL (Fig. 6) and OPG (not shown). To further assess inflammation in the intestine, we examined distal colon mRNA to determine the proinflammatory cytokine expression profile in this organ. Similar to bone, we found TNF-α mRNA levels were elevated (Fig. 7A). In contrast to bone, we found elevated expression of several additional cytokines, including IL-6, IFNγ, and IL-22. The increase in colon TNF-α, IFNγ, and IL-22 mRNA levels strongly correlated with total disease score (r = 0.79, 0.65, and 0.78, respectively, P < 0.05; Fig. 7B), indicating the strong link between intestinal inflammation and cytokine expression with intestinal pathology. Given that previous studies demonstrate a link between increased intestinal TpH1 levels and bone loss, we also examined intestinal Tph1 mRNA levels in our mice. DSS-treated mice displayed lower TpH1 expression (data not shown), suggesting that increased serotonin synthesis is not related to the bone loss in this model.
Fig. 5.
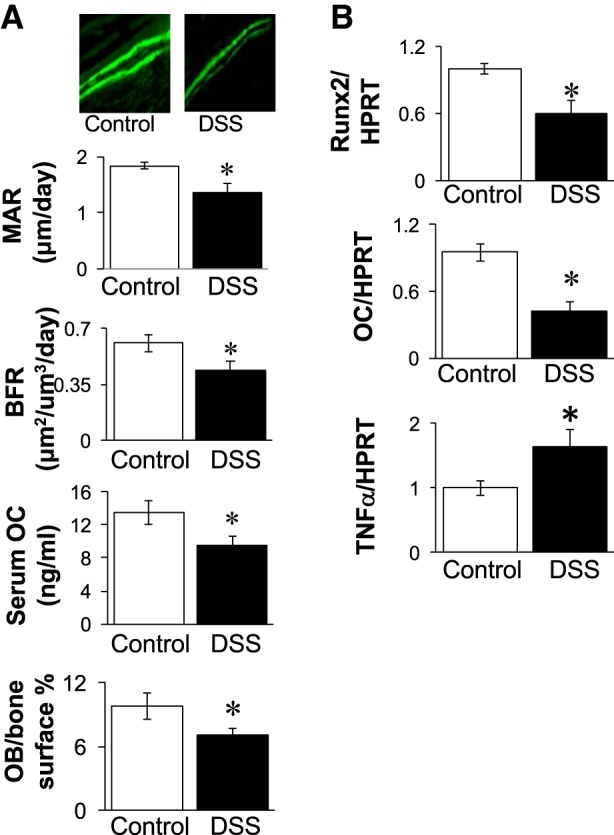
DSS treatment decreases osteoblast markers and activity and increases bone TNF expression. A: osteoblast bone formation and histomorphology analyses. Top: representative photos depicting two calcein pulses near bone surface in control and DSS-treated mice. Graphs show calculated bone formation rate (BFR) and mineral apposition rate (MAR), serum osteocalcin (OC), and osteoblast (OB) surface/bone surface. B: tibial RNA was analyzed for mRNA expression levels of osteoblast markers: Runx2 and osteocalcin (OC), as well as TNF-α mRNA levels. RNA values are expressed relative to HPRT, a nonmodulated housekeeping gene. Values represent averages ± SE. *P ≤ 0.05, n ≥ 9 per group.
Fig. 6.
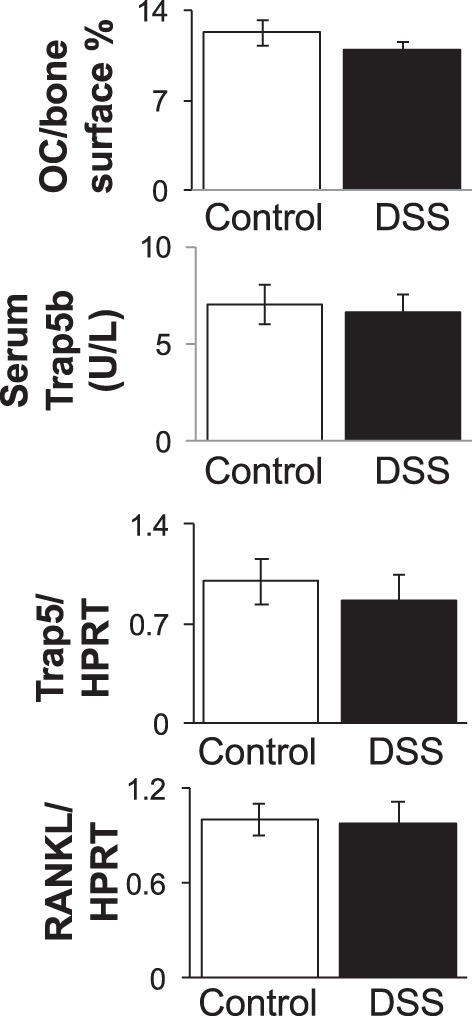
Markers of osteoclast activity are not altered by 1% DSS treatment. Osteoclast parameters were examined in control and DSS-treated male mice (15 days). Osteoclast (OC) surface/bone surface and serum-active TRAP5b levels did not differ between conditions. Tibial RNA was analyzed for mRNA expression levels of an osteoclast marker (TRAP5) and for a critical osteoclastogenic factor (RANKL). RNA values are expressed relative to HPRT, a nonmodulated housekeeping gene. Values represent averages ± SE. n ≥ 9 per group.
Fig. 7.
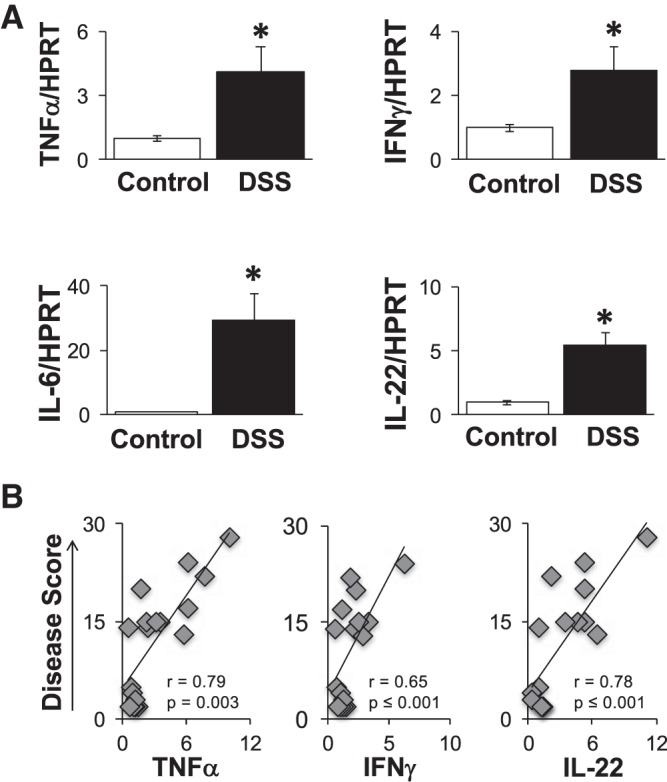
Colon cytokine gene expression is elevated in DSS-treated mice. A: cytokine mRNA levels relative to housekeeping gene expression (HPRT) in control (open bars) and DSS-treated (solid bars) colon. Values are averages ± SE. B: Pearson’s correlation graphs and analyses comparing cytokine expression levels relative to disease score. *P ≤ 0.05, n ≥ 8 per group
To assess the link between intestinal inflammation and bone pathology, we examined, by Pearson’s correlation analyses, the dependence between bone and intestinal cytokine changes. We found that colon TNF-α levels, whose levels strongly correlate with disease severity, also strongly inversely correlate with tibial bone volume (r = −0.79, P < 0.0001) and bone runx2 gene expression (r = −0.66, P ≤ 0.05) (Fig. 8). Similarly, bone volume fraction is negatively correlated with disease score (r = −0.76, P = 0.002) and bone TNF-α (r = −0.64, P < 0.0001) expression (Fig. 8). Disease score also negatively correlated with bone runx2 levels. As expected, bone TNF-α levels negatively correlate with bone osteocalcin levels (Fig. 8). However, bone osteocalcin levels did not significantly correlate with other parameters (data not shown).
Fig. 8.
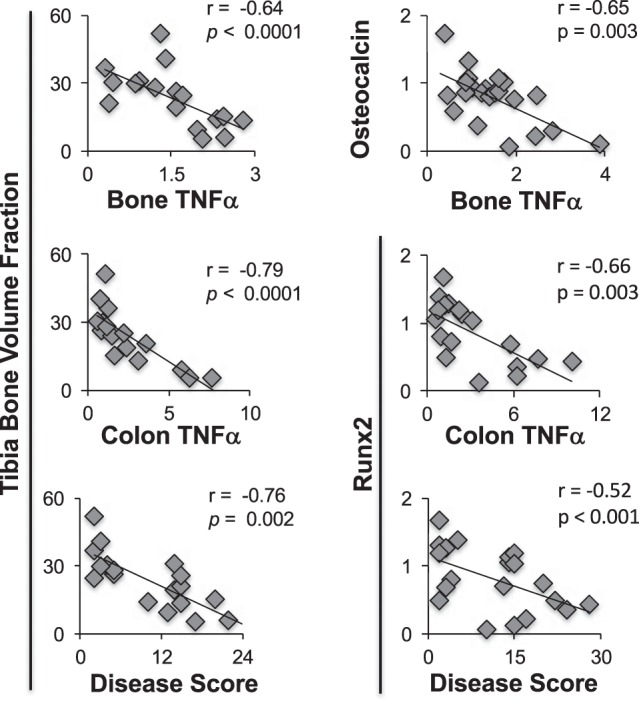
Pearson’s analyses demonstrate significant correlations between bone and colon parameters. n ≥ 8 per group.
DISCUSSION
Increasing evidence supports the importance of gut health for the maintenance of bone health. Gut-bone cross-talk is thought to involve a variety of signals, including changes in intestinal nutrient absorption, endocrine factors, and inflammation. It is known that severe intestinal inflammation, as seen in active IBD, is associated with decreased nutrient absorption and significant weight loss (3, 19, 34, 59), as well as bone loss (6, 9, 11, 15, 25, 33, 36, 53, 56, 61, 62, 72, 74, 77, 78). Similarly, animal models of IBD often display a loss of greater than 10% of body weight and significant bone loss (26, 28, 32, 40). Our laboratory previously treated 5–6-wk-old mice with 5% DSS and observed severe weight and bone loss. Once DSS treatment was ended, the mice eventually regained body weight and bone density similar to control mouse levels (28), suggesting a link between intestinal inflammation, weight loss, and osteoporosis. Interestingly, the effects of intestinal inflammation without weight loss on bone health have not been previously studied directly. Yet, it is a very relevant condition that can occur with food allergies, a high-fat diet, a high-fructose diet, obesity, and in IBD patients during remission when subclinical mucosal inflammation is present (10, 23, 46, 60). In the current study, we addressed the role of weight loss in intestinal inflammation-induced bone loss and found that intestinal inflammation effectively reduces bone density and early markers of growth regardless of weight status. We further demonstrate strong correlations between gut inflammation and bone parameters.
In the current study, although overall body weight did not differ between groups, DSS-treated mice lost a significant amount of subcutaneous fat stores, while gaining visceral fat stores. Crohn’s disease patients can experience decreased fat stores due to weight loss and increased utilization of lipids as a fuel substrate (16). Similarly, mouse models of colitis have been reported to lose subcutaneous fat (28). Even in acute colitis models, mice can display fat mass loss (39), although specific fat stores were not studied. Loss of fat mass has been shown to affect bone density (28), since adipocytes can secrete adipokines, such as leptin and adiponectin, that can increase bone density and linear growth (24). On the other hand, it is interesting that we identified an increase in visceral fat pad weight. Visceral fat is thought to be proinflammatory and associated with disease pathologies in contrast to subcutaneous fat (27). The increase in visceral fat, therefore, could contribute to bone loss through its inflammatory activity in correlation with studies, suggesting that visceral fat levels are negatively correlated with bone density (88).
As noted in our studies, only TNF-α expression was elevated in the bone of DSS-treated mice, though we only looked at one time and cannot exclude roles for other cytokines in the bone pathology. TNF-α is a key proinflammatory cytokine that is involved in bone remodeling and can stimulate osteoclastogenesis and inhibit osteoblast function (50). In our studies, we only observed a decrease in osteoblast activity. The increased expression in TNF-α can negatively regulate Runx2, as well as osteocalcin expression. We found a negative correlation between bone TNF-α expression and osteocalcin expression and bone volume. This is consistent with a role for TNF-α in decreasing osteoblast activity, as found in the decreased MAR and BFR, as determined by calcein visualization in the bone. These findings are supported by past studies demonstrating a reduction in bone formation in mouse models of IBD (26, 28). In contrast, osteoclast activity was not affected by intestinal inflammation. While inflammation and elevated TNF-α expression can stimulate osteoclast activity (4, 75), there are many reports of reduced osteoclast activity in inflammation models. Previously, using a higher dose of DSS, we reported increased inflammation and decreased osteoclast activity in 5% DSS-treated mice (28). Similarly, we and others have shown that Type 1 diabetes is associated with increased inflammation without an increase in osteoclast activity (38). In the current study, consistent with unaltered osteoclast activity, we do not see an increase in the level of a key/critical osteoclast activator, RANKL; this suggests a potential requirement for increased RANKL to promote osteoclast activation in an inflammatory environment. In addition, we used young mice that are in a stage of bone modeling, during which osteoblast activity is greater compared with osteoclast activity; this may also contribute to the inflammation affecting osteoblast activity predominantly.
DSS-induced colitis increases the levels of numerous cytokines in the colon, including those that we identified in the colon of mice in our study, ΙL-6, TNF-α, and IFNγ, which are thought to contribute to IBD pathogenesis (30). IL-6 has also been shown to be a central regulator of immune responses in different inflammatory diseases, including IBD (80), and it is upregulated in animal models of colitis (5, 85). Increased TNF-α protein and mRNA levels, as well as elevated IFNγ, were found in mucosal biopsies from patients with CD (52). IFNγ and TNF-α can induce changes in permeability (14), although there is some controversy about a specific role for IFNγ in chronic inflammatory diseases (31), since IFNγ was not produced significantly by intestinal lymphoid cells in IBD patients (13, 37, 42). Our studies found a significant correlation between colon TNF-α, as well as IFNγ levels and disease score, but only colon TNF-α levels negatively correlated with bone Runx2 expression and bone density. We did not see correlations between colon IFNγ or IL-6 levels and any bone parameters.
IL-22 expression in the colon of mice and humans is increased in inflammatory conditions and is known to contribute toward improving colitis by stimulating cell migration, epithelial restitution, and promotes intestinal barrier integrity in vitro (12, 44, 69, 86, 87). We find a correlation between cecum disease score and IL-22 in colon. This could result from the increase in proinflammatory cytokines, such as TNF-α and IFNγ, which, in turn, causes an increase in IL-22 to reduce intestinal damage.
On the basis of our results, we hypothesize a key role for TNF-α in mediating bone loss in IBD patients. The role of TNF-α in mediating bone loss is supported by several clinical studies in which IBD patients were treated with infliximab, and markers of bone remodeling were assessed. In these studies, improvements in bone metabolism were observed in the infliximab-treated patients (1, 22, 43). However, this does not prove that TNF-α is the direct cause because TNF-α has multiorgan effects. For example, inhibition of TNF-α will reduce intestinal inflammation, which will benefit bone. Further studies are needed, potentially using genetically modified mice, to address whether IBD induced increases in TNF-α directly cause bone loss.
Consistent with our findings, intestinal inflammation as seen in IBD children, can affect bone growth (7, 8, 29, 57, 64, 66). During growth, long bone is formed predominantly through endochondral ossification (EO) (18, 70), which relies on organized, sequential maturation of chondrocytes toward hypertrophy, and then replacement by trabecular bone and marrow (17, 70). As chondrocyte size increases, there is an increase in extracellular matrix production of collagen X (58, 70, 82). As EO proceeds, the cartilaginous growth plate is the site of longitudinal bone growth (70). In the current study, DSS-treated mice did not exhibit a difference in tibial bone length; however, we observed a decrease in growth plate thickness and decreased expression of collagen X. This suggests the possibility of a noticeable decrease in bone length at later time points. Chondrocytes also secrete collagen type II, IX, and XI (82); however, we did not notice a difference in the levels of the other collagens between conditions. It should be noted that Rosati et al. (58) found that collagen X-deficient mice have normal growth plate development and bone growth. Considering that our mouse model has reduced growth plate thickness and only collagen X expression, we expect that alterations in other unidentified collagens or components of the extracellular matrix may also be occurring that contribute to the growth plate defect. This is consistent with the lack of an association between suppressed collagen X and colon inflammation or disease score.
Perspectives and Significance
Taken together, our results indicate that intestinal inflammation without weight loss causes bone loss by reducing osteoblast activity, as characterized by decreased osteoblast markers (runx2, osteocalcin), histomorphometry, and dynamic measures of bone formation. In addition, we observed a reduction in growth plate thickness and a hypertrophic chondrocyte matrix component, collagen X. Correlation analyses indicate a link between gut inflammation and disease score, but more importantly, we observed negative correlations between intestinal disease score and TNF-α levels with bone density levels. Understanding the links between gut inflammation and bone loss is important for therapeutic development. A role for inhibitors of intestinal inflammation may benefit bone density under conditions of low-grade gastrointestinal pathology.
GRANTS
These studies were supported by funding from the National Institutes of Health, Grants RO1 DK-101050 and RO1 AT-007695-01.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
R.I. and L.R.M. conception and design of research; R.I. and S.R. performed experiments; R.I., S.R., N.P., and L.R.M. analyzed data; R.I., S.R., N.P., and L.R.M. interpreted results of experiments; R.I. and L.R.M. prepared figures; R.I. and L.R.M. drafted manuscript; R.I., S.R., N.P., and L.R.M. edited and revised manuscript; R.I., S.R., N.P., and L.R.M. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank the Investigative Histology Laboratory in the Department of Physiology, Division of Human Pathology and the Biomedical Imaging Center at Michigan State University for their assistance with histology and imaging, respectively. The authors also thank Fraser Collins for his critical review of the manuscript.
REFERENCES
- 1.Abreu MT, Geller JL, Vasiliauskas EA, Kam LY, Vora P, Martyak LA, Yang H, Hu B, Lin YC, Keenan G, Price J, Landers CJ, Adams JS, Targan SR. Treatment with infliximab is associated with increased markers of bone formation in patients with Crohn’s disease. J Clin Gastroenterol 40: 55–63, 2006. doi: 10.1097/01.mcg.0000190762.80615.d4. [DOI] [PubMed] [Google Scholar]
- 2.Ahmed SF, Farquharson C, McGrogan P, Russell RK. Pathophysiology and management of abnormal growth in children with chronic inflammatory bowel disease. World Rev Nutr Diet 106: 142–148, 2013. [DOI] [PubMed] [Google Scholar]
- 3.Alastair F, Emma G, Emma P. Nutrition in inflammatory bowel disease. J Parenter Enteral Nutr 35: 571–580, 2011. doi: 10.1177/0148607111413599. [DOI] [PubMed] [Google Scholar]
- 4.Alnaeeli M, Park J, Mahamed D, Penninger JM, Teng YT. Dendritic cells at the osteo-immune interface: implications for inflammation-induced bone loss. J Bone Miner Res 22: 775–780, 2007. doi: 10.1359/jbmr.070314. [DOI] [PubMed] [Google Scholar]
- 5.Atreya R, Mudter J, Finotto S, Müllberg J, Jostock T, Wirtz S, Schütz M, Bartsch B, Holtmann M, Becker C, Strand D, Czaja J, Schlaak JF, Lehr HA, Autschbach F, Schürmann G, Nishimoto N, Yoshizaki K, Ito H, Kishimoto T, Galle PR, Rose-John S, Neurath MF. Blockade of interleukin 6 trans signaling suppresses T-cell resistance against apoptosis in chronic intestinal inflammation: evidence in Crohn's disease and experimental colitis in vivo. Nat Med 6: 583–588, 2000. doi: 10.1038/75068. [DOI] [PubMed] [Google Scholar]
- 6.Bartram SA, Peaston RT, Rawlings DJ, Walshaw D, Francis RM, Thompson NP. Mutifactorial analysis of risk factors for reduced bone mineral density in patients with Crohn’s disease. World J Gastroenterol 12: 5680–5686, 2006. doi: 10.3748/wjg.v12.i35.5680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baumgart DC. The diagnosis and treatment of Crohn’s disease and ulcerative colitis. Dtsch Arztebl Int 106: 123–133, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bernstein CN, Fried M, Krabshuis JH, Cohen H, Eliakim R, Fedail S, Gearry R, Goh KL, Hamid S, Khan AG, LeMair AW, Malfertheiner P, Ouyang Q, Rey JF, Sood A, Steinwurz F, Thomsen OO, Thomson A, Watermeyer G. World Gastroenterology Organization Practice Guidelines for the diagnosis and management of IBD in 2010. Inflamm Bowel Dis 16: 112–124, 2010. doi: 10.1002/ibd.21048. [DOI] [PubMed] [Google Scholar]
- 9.Bernstein CN, Leslie WD, Leboff MS. AGA technical review on osteoporosis in gastrointestinal diseases. Gastroenterology 124: 795–841, 2003. doi: 10.1053/gast.2003.50106. [DOI] [PubMed] [Google Scholar]
- 10.Bischoff SC, Barbara G, Buurman W, Ockhuizen T, Schulzke JD, Serino M, Tilg H, Watson A, Wells JM. Intestinal permeability—a new target for disease prevention and therapy. BMC Gastroenterol 14: 189, 2014. doi: 10.1186/s12876-014-0189-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bjarnason I, Macpherson A, Mackintosh C, Buxton-Thomas M, Forgacs I, Moniz C. Reduced bone density in patients with inflammatory bowel disease. Gut 40: 228–233, 1997. doi: 10.1136/gut.40.2.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brand S, Beigel F, Olszak T, Zitzmann K, Eichhorst ST, Otte JM, Diepolder H, Marquardt A, Jagla W, Popp A, Leclair S, Herrmann K, Seiderer J, Ochsenkühn T, Göke B, Auernhammer CJ, Dambacher J. IL-22 is increased in active Crohn’s disease and promotes proinflammatory gene expression and intestinal epithelial cell migration. Am J Physiol Gastrointest Liver Physiol 290: G827–G838, 2006. doi: 10.1152/ajpgi.00513.2005. [DOI] [PubMed] [Google Scholar]
- 13.Breese E, Braegger CP, Corrigan CJ, Walker-Smith JA, MacDonald TT. Interleukin-2- and interferon-gamma-secreting T cells in normal and diseased human intestinal mucosa. Immunology 78: 127–131, 1993. [PMC free article] [PubMed] [Google Scholar]
- 14.Bruewer M, Luegering A, Kucharzik T, Parkos CA, Madara JL, Hopkins AM, Nusrat A. Proinflammatory cytokines disrupt epithelial barrier function by apoptosis-independent mechanisms. J Immunol 171: 6164–6172, 2003. doi: 10.4049/jimmunol.171.11.6164. [DOI] [PubMed] [Google Scholar]
- 15.Burnham JM, Shults J, Semeao E, Foster B, Zemel BS, Stallings VA, Leonard MB. Whole body BMC in pediatric Crohn's disease: independent effects of altered growth, maturation, and body composition. J Bone Miner Res 19: 1961–1968, 2004. doi: 10.1359/jbmr.040908. [DOI] [PubMed] [Google Scholar]
- 16.Capristo E, Mingrone G, Addolorato G, Greco AV, Gasbarrini G. Metabolic features of inflammatory bowel disease in a remission phase of the disease activity. J Intern Med 243: 339–347, 1998. doi: 10.1046/j.1365-2796.1998.00254.x. [DOI] [PubMed] [Google Scholar]
- 17.Chan D, Jacenko O. Phenotypic and biochemical consequences of collagen X mutations in mice and humans. Matrix Biol 17: 169–184, 1998. doi: 10.1016/S0945-053X(98)90056-7. [DOI] [PubMed] [Google Scholar]
- 18.Chung UI, Kawaguchi H, Takato T, Nakamura K. Distinct osteogenic mechanisms of bones of distinct origins. J Orthop Sci 9: 410–414, 2004. doi: 10.1007/s00776-004-0786-3. [DOI] [PubMed] [Google Scholar]
- 19.Day AS, Ledder O, Leach ST, Lemberg DA. Crohn’s and colitis in children and adolescents. World J Gastroenterol 18: 5862–5869, 2012. doi: 10.3748/wjg.v18.i41.5862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Duggan P, O’Brien M, Kiely M, McCarthy J, Shanahan F, Cashman KD. Vitamin K status in patients with Crohn’s disease and relationship to bone turnover. Am J Gastroenterol 99: 2178–2185, 2004. doi: 10.1111/j.1572-0241.2004.40071.x. [DOI] [PubMed] [Google Scholar]
- 21.Fiocchi C. Inflammatory bowel disease: etiology and pathogenesis. Gastroenterology 115: 182–205, 1998. doi: 10.1016/S0016-5085(98)70381-6. [DOI] [PubMed] [Google Scholar]
- 22.Franchimont N, Putzeys V, Collette J, Vermeire S, Rutgeerts P, De Vos M, Van Gossum A, Franchimont D, Fiasse R, Pelckmans P, Malaise M, Belaiche J, Louis E. Rapid improvement of bone metabolism after infliximab treatment in Crohn’s disease. Aliment Pharmacol Ther 20: 607–614, 2004. doi: 10.1111/j.1365-2036.2004.02152.x. [DOI] [PubMed] [Google Scholar]
- 23.Frazier TH, DiBaise JK, McClain CJ. Gut microbiota, intestinal permeability, obesity-induced inflammation, and liver injury. JPEN J Parenter Enteral Nutr 35, Suppl: 14S–20S, 2011. doi: 10.1177/0148607111413772. [DOI] [PubMed] [Google Scholar]
- 24.Gat-Yablonski G, Phillip M. Leptin and regulation of linear growth. Curr Opin Clin Nutr Metab Care 11: 303–308, 2008. doi: 10.1097/MCO.0b013e3282f795cf. [DOI] [PubMed] [Google Scholar]
- 25.Gokhale R, Favus MJ, Karrison T, Sutton MM, Rich B, Kirschner BS. Bone mineral density assessment in children with inflammatory bowel disease. Gastroenterology 114: 902–911, 1998. doi: 10.1016/S0016-5085(98)70309-9. [DOI] [PubMed] [Google Scholar]
- 26.Hamdani G, Gabet Y, Rachmilewitz D, Karmeli F, Bab I, Dresner-Pollak R. Dextran sodium sulfate-induced colitis causes rapid bone loss in mice. Bone 43: 945–950, 2008. doi: 10.1016/j.bone.2008.06.018. [DOI] [PubMed] [Google Scholar]
- 27.Hamdy O, Porramatikul S, Al-Ozairi E. Metabolic obesity: the paradox between visceral and subcutaneous fat. Curr Diabetes Rev 2: 367–373, 2006. doi: 10.2174/1573399810602040367. [DOI] [PubMed] [Google Scholar]
- 28.Harris L, Senagore P, Young VB, McCabe LR. Inflammatory bowel disease causes reversible suppression of osteoblast and chondrocyte function in mice. Am J Physiol Gastrointest Liver Physiol 296: G1020–G1029, 2009. doi: 10.1152/ajpgi.90696.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heuschkel R, Salvestrini C, Beattie RM, Hildebrand H, Walters T, Griffiths A. Guidelines for the management of growth failure in childhood inflammatory bowel disease. Inflamm Bowel Dis 14: 839–849, 2008. doi: 10.1002/ibd.20378. [DOI] [PubMed] [Google Scholar]
- 30.Hyun JG, Mayer L. Mechanisms underlying inflammatory bowel disease. Drug Discov Today Dis Mech 3: 457–462, 2006. doi: 10.1016/j.ddmec.2006.11.010. [DOI] [Google Scholar]
- 31.Ito R, Shin-Ya M, Kishida T, Urano A, Takada R, Sakagami J, Imanishi J, Kita M, Ueda Y, Iwakura Y, Kataoka K, Okanoue T, Mazda O. Interferon-gamma is causatively involved in experimental inflammatory bowel disease in mice. Clin Exp Immunol 146: 330–338, 2006. doi: 10.1111/j.1365-2249.2006.03214.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jurjus AR, Khoury NN, Reimund JM. Animal models of inflammatory bowel disease. J Pharmacol Toxicol Methods 50: 81–92, 2004. doi: 10.1016/j.vascn.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 33.Kappelman MD, Rifas-Shiman SL, Porter CQ, Ollendorf DA, Sandler RS, Galanko JA, Finkelstein JA. Direct health care costs of Crohn’s disease and ulcerative colitis in U.S. children and adults. Gastroenterology 135: 1907–1913, 2008. doi: 10.1053/j.gastro.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kugathasan S, Judd RH, Hoffmann RG, Heikenen J, Telega G, Khan F, Weisdorf-Schindele S, San Pablo W Jr, Perrault J, Park R, Yaffe M, Brown C, Rivera-Bennett MT, Halabi I, Martinez A, Blank E, Werlin SL, Rudolph CD, Binion DG; Wisconsin Pediatric Inflammatory Bowel Disease Alliance . Epidemiologic and clinical characteristics of children with newly diagnosed inflammatory bowel disease in Wisconsin: a statewide population-based study. J Pediatr 143: 525–531, 2003. doi: 10.1067/S0022-3476(03)00444-X. [DOI] [PubMed] [Google Scholar]
- 35.Laroui H, Ingersoll SA, Liu HC, Baker MT, Ayyadurai S, Charania MA, Laroui F, Yan Y, Sitaraman SV, Merlin D. Dextran sodium sulfate (DSS) induces colitis in mice by forming nano-lipocomplexes with medium-chain-length fatty acids in the colon. PLoS One 7: e32084, 2012. doi: 10.1371/journal.pone.0032084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Larsen S, Bendtzen K, Nielsen OH. Extraintestinal manifestations of inflammatory bowel disease: epidemiology, diagnosis, and management. Ann Med 42: 97–114, 2010. doi: 10.3109/07853890903559724. [DOI] [PubMed] [Google Scholar]
- 37.Lieberman BY, Fiocchi C, Youngman KR, Sapatnekar WK, Proffitt MR. Interferon gamma production by human intestinal mucosal mononuclear cells. Decreased levels in inflammatory bowel disease. Dig Dis Sci 33: 1297–1304, 1988. doi: 10.1007/BF01536683. [DOI] [PubMed] [Google Scholar]
- 38.McCabe LR. Understanding the pathology and mechanisms of type I diabetic bone loss. J Cell Biochem 102: 1343–1357, 2007. doi: 10.1002/jcb.21573. [DOI] [PubMed] [Google Scholar]
- 39.Melgar S, Bjursell M, Gerdin AK, Svensson L, Michaëlsson E, Bohlooly-Y M. Mice with experimental colitis show an altered metabolism with decreased metabolic rate. Am J Physiol Gastrointest Liver Physiol 292: G165–G172, 2007. doi: 10.1152/ajpgi.00152.2006. [DOI] [PubMed] [Google Scholar]
- 40.Melgar S, Karlsson A, Michaëlsson E. Acute colitis induced by dextran sulfate sodium progresses to chronicity in C57BL/6 but not in BALB/c mice: correlation between symptoms and inflammation. Am J Physiol Gastrointest Liver Physiol 288: G1328–G1338, 2005. doi: 10.1152/ajpgi.00467.2004. [DOI] [PubMed] [Google Scholar]
- 41.Melgar S, Karlsson L, Rehnström E, Karlsson A, Utkovic H, Jansson L, Michaëlsson E. Validation of murine dextran sulfate sodium-induced colitis using four therapeutic agents for human inflammatory bowel disease. Int Immunopharmacol 8: 836–844, 2008. doi: 10.1016/j.intimp.2008.01.036. [DOI] [PubMed] [Google Scholar]
- 42.Melgar S, Yeung MM, Bas A, Forsberg G, Suhr O, Oberg A, Hammarstrom S, Danielsson A, Hammarstrom ML. Over-expression of interleukin 10 in mucosal T cells of patients with active ulcerative colitis. Clin Exp Immunol 134: 127–137, 2003. doi: 10.1046/j.1365-2249.2003.02268.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miheller P, Muzes G, Rácz K, Blázovits A, Lakatos P, Herszényi L, Tulassay Z. Changes of OPG and RANKL concentrations in Crohn’s disease after infliximab therapy. Inflamm Bowel Dis 13: 1379–1384, 2007. doi: 10.1002/ibd.20234. [DOI] [PubMed] [Google Scholar]
- 44.Mizoguchi A. Healing of intestinal inflammation by IL-22. Inflamm Bowel Dis 18: 1777–1784, 2012. doi: 10.1002/ibd.22929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mohanty SK, Shivakumar P, Sabla G, Bezerra JA. Loss of interleukin-12 modifies the pro-inflammatory response but does not prevent duct obstruction in experimental biliary atresia. BMC Gastroenterol 6: 14, 2006. doi: 10.1186/1471-230X-6-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moss AC. The meaning of low-grade inflammation in clinically quiescent inflammatory bowel disease. Curr Opin Gastroenterol 30: 365–369, 2014. doi: 10.1097/MOG.0000000000000082. [DOI] [PubMed] [Google Scholar]
- 47.Motyl KJ, Botolin S, Irwin R, Appledorn DM, Kadakia T, Amalfitano A, Schwartz RC, McCabe LR. Bone inflammation and altered gene expression with type I diabetes early onset. J Cell Physiol 218: 575–583, 2009. doi: 10.1002/jcp.21626. [DOI] [PubMed] [Google Scholar]
- 48.Neurath M, Fuss I, Strober W. TNBS-colitis. Int Rev Immunol 19: 51–62, 2000. doi: 10.3109/08830180009048389. [DOI] [PubMed] [Google Scholar]
- 49.Okayasu I, Hatakeyama S, Yamada M, Ohkusa T, Inagaki Y, Nakaya R. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology 98: 694–702, 1990. doi: 10.1016/0016-5085(90)90290-H. [DOI] [PubMed] [Google Scholar]
- 50.Osta B, Benedetti G, Miossec P. Classical and paradoxical effects of TNF-α on bone homeostasis. Front Immunol 5: 48, 2014. doi: 10.3389/fimmu.2014.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Paganelli M, Albanese C, Borrelli O, Civitelli F, Canitano N, Viola F, Passariello R, Cucchiara S. Inflammation is the main determinant of low bone mineral density in pediatric inflammatory bowel disease. Inflamm Bowel Dis 13: 416–423, 2007. doi: 10.1002/ibd.20039. [DOI] [PubMed] [Google Scholar]
- 52.Papadakis KA, Targan SR. Role of cytokines in the pathogenesis of inflammatory bowel disease. Annu Rev Med 51: 289–298, 2000. doi: 10.1146/annurev.med.51.1.289. [DOI] [PubMed] [Google Scholar]
- 53.Pappa H, Thayu M, Sylvester F, Leonard M, Zemel B, Gordon C. Skeletal health of children and adolescents with inflammatory bowel disease. J Pediatr Gastroenterol Nutr 53: 11–25, 2011. doi: 10.1097/MPG.0b013e31821988a3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pappa HM, Gordon CM, Saslowsky TM, Zholudev A, Horr B, Shih MC, Grand RJ. Vitamin D status in children and young adults with inflammatory bowel disease. Pediatrics 118: 1950–1961, 2006. doi: 10.1542/peds.2006-0841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Perše M, Cerar A. Dextran sodium sulphate colitis mouse model: traps and tricks. J Biomed Biotechnol 2012: 1–13, 2012. doi: 10.1155/2012/718617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dresner Pollak R, Karmeli F, Eliakim R, Ackerman Z, Tabb K, Rachmilewitz D. Femoral neck osteopenia in patients with inflammatory bowel disease. Am J Gastroenterol 93: 1483–1490, 1998. doi: 10.1111/j.1572-0241.1998.468_q.x. [DOI] [PubMed] [Google Scholar]
- 57.Puntis J, McNeish AS, Allan RN. Long-term prognosis of Crohn’s disease with onset in childhood and adolescence. Gut 25: 329–336, 1984. doi: 10.1136/gut.25.4.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rosati R, Horan GS, Pinero GJ, Garofalo S, Keene DR, Horton WA, Vuorio E, de Crombrugghe B, Behringer RR. Normal long bone growth and development in type X collagen-null mice. Nat Genet 8: 129–135, 1994. doi: 10.1038/ng1094-129. [DOI] [PubMed] [Google Scholar]
- 59.Rosen MJ, Dhawan A, Saeed SA. Inflammatory bowel disease in children and adolescents. JAMA Pediatr 169: 1053–1060, 2015. doi: 10.1001/jamapediatrics.2015.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rosenberg L, Lawlor GO, Zenlea T, Goldsmith JD, Gifford A, Falchuk KR, Wolf JL, Cheifetz AS, Robson SC, Moss AC. Predictors of endoscopic inflammation in patients with ulcerative colitis in clinical remission. Inflamm Bowel Dis 19: 779–784, 2013. doi: 10.1097/MIB.0b013e3182802b0e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Semeao EJ, Jawad AF, Zemel BS, Neiswender KM, Piccoli DA, Stallings VA. Bone mineral density in children and young adults with Crohn’s disease. Inflamm Bowel Dis 5: 161–166, 1999. doi: 10.1097/00054725-199908000-00003. [DOI] [PubMed] [Google Scholar]
- 62.Semeao EJ, Stallings VA, Peck SN, Piccoli DA. Vertebral compression fractures in pediatric patients with Crohn’s disease. Gastroenterology 112: 1710–1713, 1997. doi: 10.1016/S0016-5085(97)70055-6. [DOI] [PubMed] [Google Scholar]
- 63.Sentongo TA, Semaeo EJ, Stettler N, Piccoli DA, Stallings VA, Zemel BS. Vitamin D status in children, adolescents, and young adults with Crohn's disease. Am J Clin Nutr 76: 1077–1081, 2002. [DOI] [PubMed] [Google Scholar]
- 64.Shamir R, Phillip M, Levine A. Growth retardation in pediatric Crohn’s disease: pathogenesis and interventions. Inflamm Bowel Dis 13: 620–628, 2007. doi: 10.1002/ibd.20115. [DOI] [PubMed] [Google Scholar]
- 65.Shi J, Huang Z, Wang Y, Huang Y. The protective effects of exclusive enteral nutrition formulas on growth factor expression and the proximal tibial epiphyseal growth plate in a TNBS-induced IBD rat model. Dig Dis Sci 60: 1931–1940, 2015. doi: 10.1007/s10620-015-3582-3. [DOI] [PubMed] [Google Scholar]
- 66.Shikhare G, Kugathasan S. Inflammatory bowel disease in children: current trends. J Gastroenterol 45: 673–682, 2010. doi: 10.1007/s00535-010-0241-5. [DOI] [PubMed] [Google Scholar]
- 67.Siffledeen JS, Fedorak RN, Siminoski K, Jen H, Vaudan E, Abraham N, Steinhart H, Greenberg G. Randomized trial of etidronate plus calcium and vitamin D for treatment of low bone mineral density in Crohn’s disease. Clin Gastroenterol Hepatol 3: 122–132, 2005. doi: 10.1016/S1542-3565(04)00663-9. [DOI] [PubMed] [Google Scholar]
- 68.Siffledeen JS, Siminoski K, Steinhart H, Greenberg G, Fedorak RN. The frequency of vitamin D deficiency in adults with Crohn’s disease. Can J Gastroenterol 17: 473–478, 2003. doi: 10.1155/2003/391308. [DOI] [PubMed] [Google Scholar]
- 69.Sugimoto K, Ogawa A, Mizoguchi E, Shimomura Y, Andoh A, Bhan AK, Blumberg RS, Xavier RJ, Mizoguchi A. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J Clin Invest 118: 534–544, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sweeney E, Roberts D, Jacenko O. Altered matrix at the chondro-osseous junction leads to defects in lymphopoiesis. Ann NY Acad Sci 1237: 79–87, 2011. doi: 10.1111/j.1749-6632.2011.06227.x. [DOI] [PubMed] [Google Scholar]
- 71.Sylvester FA. IBD and skeletal health: children are not small adults! Inflamm Bowel Dis 11: 1020–1023, 2005. doi: 10.1097/01.MIB.0000188341.96726.15. [DOI] [PubMed] [Google Scholar]
- 72.Sylvester FA, Davis PM, Wyzga N, Hyams JS, Lerer T. Are activated T cells regulators of bone metabolism in children with Crohn's disease? J Pediatr 148: 461–466, 2006. doi: 10.1016/j.jpeds.2005.12.027. [DOI] [PubMed] [Google Scholar]
- 73.Sylvester FA, Gordon CM, Thayu M, Burnham JM, Denson LA, Essers J, Ferrari S, Gupta N, Hewison M, Koletzko S, McCabe L, Pappa H, Sanderson I, Ward L, Zanotti S. Report of the CCFA pediatric bone, growth and muscle health workshop, New York City, November 11-12, 2011, with updates. Inflamm Bowel Dis 19: 2919–2926, 2013. doi: 10.1097/MIB.0b013e3182a5a004. [DOI] [PubMed] [Google Scholar]
- 74.Sylvester FA, Wyzga N, Hyams JS, Davis PM, Lerer T, Vance K, Hawker G, Griffiths AM. Natural history of bone metabolism and bone mineral density in children with inflammatory bowel disease. Inflamm Bowel Dis 13: 42–50, 2007. doi: 10.1002/ibd.20006. [DOI] [PubMed] [Google Scholar]
- 75.Teitelbaum SL. Osteoclasts: what do they do and how do they do it? Am J Pathol 170: 427–435, 2007. doi: 10.2353/ajpath.2007.060834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Vaisman N, Dotan I, Halack A, Niv E. Malabsorption is a major contributor to underweight in Crohn’s disease patients in remission. Nutrition 22: 855–859, 2006. doi: 10.1016/j.nut.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 77.van Hogezand RA, Hamdy NA. Skeletal morbidity in inflammatory bowel disease. Scand J Gastroenterol Suppl 41, Suppl. 243: 59–64, 2006. doi: 10.1080/00365520600664276. [DOI] [PubMed] [Google Scholar]
- 78.van Staa TP, Cooper C, Brusse LS, Leufkens H, Javaid MK, Arden NK. Inflammatory bowel disease and the risk of fracture. Gastroenterology 125: 1591–1597, 2003. doi: 10.1053/j.gastro.2003.09.027. [DOI] [PubMed] [Google Scholar]
- 79.Vengellur A, LaPres JJ. The role of hypoxia inducible factor 1alpha in cobalt chloride induced cell death in mouse embryonic fibroblasts. Toxicol Sci 82: 638–646, 2004. doi: 10.1093/toxsci/kfh278. [DOI] [PubMed] [Google Scholar]
- 80.Waldner MJ, Neurath MF. Master regulator of intestinal disease: IL-6 in chronic inflammation and cancer development. Semin Immunol 26: 75–79, 2014. doi: 10.1016/j.smim.2013.12.003. [DOI] [PubMed] [Google Scholar]
- 81.Weghofer M, Karlic H, Haslberger A. Quantitative analysis of immune-mediated stimulation of tumor necrosis factor-alpha in macrophages measured at the level of mRNA and protein synthesis. Ann Hematol 80: 733–736, 2001. doi: 10.1007/s00277-001-0383-x. [DOI] [PubMed] [Google Scholar]
- 82.White A, Wallis G. Endochondral ossification: a delicate balance between growth and mineralisation. Curr Biol 11: R589–R591, 2001. doi: 10.1016/S0960-9822(01)00359-1. [DOI] [PubMed] [Google Scholar]
- 83.Wirtz S, Neufert C, Weigmann B, Neurath MF. Chemically induced mouse models of intestinal inflammation. Nat Protoc 2: 541–546, 2007. doi: 10.1038/nprot.2007.41. [DOI] [PubMed] [Google Scholar]
- 84.Wirtz S, Neurath MF. Mouse models of inflammatory bowel disease. Adv Drug Deliv Rev 59: 1073–1083, 2007. doi: 10.1016/j.addr.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 85.Yen D, Cheung J, Scheerens H, Poulet F, McClanahan T, McKenzie B, Kleinschek MA, Owyang A, Mattson J, Blumenschein W, Murphy E, Sathe M, Cua DJ, Kastelein RA, Rennick D. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Invest 116: 1310–1316, 2006. doi: 10.1172/JCI21404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Karow M, Flavell RA. Interleukin-22 but not interleukin-17 provides protection to hepatocytes during acute liver inflammation. Immunity 27: 647–659, 2007. doi: 10.1016/j.immuni.2007.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Stevens S, Flavell RA. Innate and adaptive interleukin-22 protects mice from inflammatory bowel disease. Immunity 29: 947–957, 2008. doi: 10.1016/j.immuni.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhang P, Peterson M, Su GL, Wang SC. Visceral adiposity is negatively associated with bone density and muscle attenuation. Am J Clin Nutr 101: 337–343, 2015. doi: 10.3945/ajcn.113.081778. [DOI] [PMC free article] [PubMed] [Google Scholar]