

Abstract
Food borne trematodes (FBTs) are an assemblage of platyhelminth parasites transmitted through the food chain, four of which are recognized as neglected tropical diseases (NTDs). Fascioliasis stands out among the other NTDs due to its broad and significant impact on both human and animal health, as Fasciola sp., are also considered major pathogens of domesticated ruminants. Here we present a reference genome sequence of the common liver fluke, Fasciola hepatica isolated from sheep, complementing previously reported isolate from cattle. A total of 14,642 genes were predicted from the 1.14 GB genome of the liver fluke. Comparative genomics indicated that F. hepatica Oregon and related food-borne trematodes are metabolically less constrained than schistosomes and cestodes, taking advantage of the richer millieux offered by the hepatobiliary organs. Protease families differentially expanded between diverse trematodes may facilitate migration and survival within the heterogeneous environments and niches within the mammalian host. Surprisingly, the sequencing of Oregon and Uruguay F. hepatica isolates led to the first discovery of an endobacteria in this species. Two contigs from the F. hepatica Oregon assembly were joined to complete the 859,205 bp genome of a novel Neorickettsia endobacterium (nFh) closely related to the etiological agents of human Sennetsu and Potomac horse fevers. Immunohistochemical studies targeting a Neorickettsia surface protein found nFh in specific organs and tissues of the adult trematode including the female reproductive tract, eggs, the Mehlis’ gland, seminal vesicle, and oral suckers, suggesting putative routes for fluke-to-fluke and fluke-to-host transmission. The genomes of F. hepatica and nFh will serve as a resource for further exploration of the biology of F. hepatica, and specifically its newly discovered trans-kingdom interaction with nFh and the impact of both species on disease in ruminants and humans.
Author Summary
This report presents novel findings revealing (a) the genome sequence of the food-borne trematode Fasciola hepatica (the liver fluke) isolated from sheep, which stands out among neglected tropical diseases due to its zoonotic impact on both human and animal health and (b) the first instance (and the genome) of the rickettsial endobacterium of the genus Neorickettsia in F. hepatica. Using stage-specific gene expression data, we identified liver fluke proteins likely involved in host-parasite interactions, and using immunolocalization, we confirmed Neorickettsia in organs and tissues of the adult trematode. The presence of the bacteria in fluke reproductive tissues and eggs suggests a possible mechanism for vertical transmission, and the presence of bacteria in the oral sucker used to anchor flukes to the lining of the biliary tract suggests a potential mechanism for horizontal transmission to the mammalian host. This is of interest because related Neorickettsia cause severe, even deadly, illness in a variety of species, including humans. This is the first report to localize Neorickettsia endobacteria within the tissues of adult F. hepatica. The discoveries in our manuscript have wide impact for the fields of both the pathophysiology and evolution of Fasciola and related FBTs, and the transmission strategies of Neorickettsia.
Introduction
Food borne trematodes (FBTs) are an assemblage of platyhelminth parasites that are transmitted through the food chain [1]. Among the four major groups of FBT infections recognized as neglected tropical diseases (NTDs) by the World Health Organization [2], fascioliasis stands out due to its zoonotic impact on both human and animal health [3]. Fasciola species are major pathogens of domesticated ruminants, but they infect numerous other species of mammals, including people [4]. Due to the significant burden to livestock globally, with annual losses exceeding US $3.2 billion [5] and public health with ~50 million infected people [4], these parasites are among the most-extensively studied FBTs.
Like other digenetic trematodes, Fasciola hepatica has a complex developmental cycle [1]. The hermaphroditic adult stage resides in the host bile ducts and reproduces sexually, releasing thousands of eggs each day that pass with the bile into the intestines and exit in the fecal stream. Eggs that reach fresh water embryonate over a couple of weeks, hatching a free-swimming miracidium that seeks out and infects a snail of the family Lymnaeidae. Within the snail, the parasite progresses through sporocyst, redia, and daughter redia stages by asexual replication and development, resulting in the release thousands of the cercariae [6]. The free-living, aquatic cercaria encysts as the metacercarial stage on solid substrates, including vegetation at the margins of the watercourse. When infected vegetation (for example, uncooked watercress) are ingested by a suitable host, the metacercaria excysts in the duodenum, transverses the wall of the small intestine, migrates through the peritoneal cavity, and penetrates the Glisson's capsule of the liver [7]. The migration of the juvenile fluke though the liver parenchyma into the biliary ducts damages the liver and provokes reactions associated with the acute phase of the infection. This phase is accompanied by systemic disease including fever, nausea and abdominal pain. Once the adult is established in the bile ducts, anemia, inflammation, fibrosis, cholangitis and biliary stasis may ensue. In this chronic phase adult worms can survive several years in the absence of intervention [8, 9]. Despite its potent and broad action against other human parasitic flatworms the anthelmintic drug praziquantel has no effect on F. hepatica [10]. Triclabendazole (TCBZ) is the drug of choice since its effective against juveniles and adult liver flukes, but resistance to this benzimidazole has emerged in livestock in different countries [11]. There have been recent reports of human fascioliasis refractive to TCBZ treatment in Peru and Chile [12, 13], highlighting a need for alternative drugs and treatments.
In addition to being important pathogens themselves, some digeneans serve as vectors of bacterial pathogens. Neorickettsia (family Anaplasmataceae) belongs to a poorly characterized assemblage of obligate, intracellular α-Proteobacteria associated with serious, even fatal disease in mammals [14]. These bacteria can be horizontally transmitted from the fluke to host tissue invading and multiplying within mammalian cells such as macrophages, monocytes and other cells types, e.g. intestinal epithelium, eventually leading to severe disease. Neorickettsia can be detected by PCR in trematodes spanning the major lineages of the Digenea [15, 16], but it has never been reported from a trematode that is itself a prevalent human and livestock pathogen. Furthermore, the fact that Neorickettsia is not found among all fluke species (or all members of infected species) suggests that these endobacteria are not essential to fluke survival. Indeed, the exact nature of their relationship is remains unclear.
Here we describe the second reported reference genome of the common liver fluke, F. hepatica and the first discovery and genome sequences of the Neorickettsia endobacteria of F. hepatica. In contrast to the previously sequenced isolate from cattle from the UK, the presently described strain, taken from a sheep in Oregon, US, was infected with a Neorickettsia species closely related to the etiological agents of Potomac horse and Sennetsu fevers. Histological, PCR, and gene sequence analyses revealed its presence in tissues of liver fluke isolates from Oregon, and in one of several liver fluke isolates from Uruguay that were screened. Taken together, these genomes represent a benchmark resource for studies of trematode and Neorickettsia biology, pathogenesis and evolution.
Results and Discussion
The general features of the nuclear genome of Fasciola hepatica
The nuclear genome of F. hepatica Oregon was sequenced and assembled with a total length of 1.14 Gb, N50 number of 2,036 and N50 length of 161 kb (S1 Table). Completeness was estimated at 90.6% using the CEGMA method [17]. GC content was similar to other Food Borne Trematodes (FBTs) including Clonorchis sinensis [18] and Opisthorchis viverrini [19], but differed from blood flukes. Intriguingly, the genome of F. hepatica Oregon had a markedly higher repeat content (55.29%, S1 Table) than other FBTs, including the recently published genome of F. hepatica United Kingdom (32.0%) isolated from cattle. We detected > 92 Mb corresponding to LTR elements, 268 Mb corresponding to LINEs, and 235 Mb of unclassified repetitive sequences, values all higher than other trematodes. Functional RNAs including rRNA, tRNA and miRNA (S2 Table) were identified, representing 0.002% of the coding genome, most supported by RNAseq data.
Consistent with other FBTs [19, 20], a very small percentage of the genome assembly was predicted to encode proteins (1.08%, considering only exonic regions). A total of 14,642 protein-coding genes were identified using a combination of de novo and evidence-based methods. Predicted genes had an average of 3.3 exons and 2.3 introns, average footprint of 3,078 bp, and average coding length of 837 bp (S1 Table). Comparisons of protein-coding genes between F. hepatica Oregon and F. hepatica UK (S1 Fig) revealed that most functional elements (e.g. KEGG orthologous groups) were shared, despite the fact that the gene models showed relatively poor overlap. In general, both genome annotations showed a predominance of short genes compared with other trematodes. This could be an indication of incomplete gene models in both assemblies, as the size, complexity, and incompleteness of both hindered gene prediction. Accordingly, long reads from third generation sequencing and additional RNAseq data will be needed to improve gene predictions, as demonstrated for S. mansoni [21] and C. sinensis [20].
Comprehensive functional annotation of the deduced proteins of F. hepatica Oregon is provided in S3 Table, including (a) 3,907 unique InterPro protein domains predicted from 8,609 proteins, associated with 1,147 unique gene ontology (GO) terms, (b) 3,175 proteins associated with 2,685 KEGG orthologous groups, (c) 339 proteins classified as putative proteases, (d) 65 proteins classified as protease inhibitors, and (e) 855 of proteins predicted to be secreted. Majority of the genes (94% of the predicted 13,740/14,642) were supported by RNAseq data from the developmental stages sampled for this study (eggs, metacercariae, and adult flukes; S2 Fig). Of the >6,000 genes expressed in these stages, ~2,500 showed no differential expression, with GO terms related to core cellular functions such as translation, RNA processing, and vesicular transport (S3 Table).
Among the differentially expressed gene sets resulting from the DESeq analysis, stage-specific overexpressed gene sets were identified (e.g., for metacercariae, genes significantly overexpressed in the metacercarial stage relative to the adult and to the egg, but not differentially expressed between adult and egg). Using these criteria, four of the top five most significantly metacercaria-overexpressed genes (2,076 total) were cysteine proteases, including four papain-like family proteases and one C13-family protease (P < 10−15 for all comparisons), while the most significantly adult-overexpressed was also a papain-family cysteine protease (P < 10−38). Among the other 1,169 adult-overexpressed genes, four of the top 11 were tubulin genes (P < 10−20). Fewer genes (259) were egg-overexpressed since adult females contain eggs expressing transcripts, but the most significantly differentially expressed gene in this set (P < 10−13) was a glucose-6-phosphate dehydrogenase.
Comparative analysis reveals phylogenetic conservation and diversification among trematodes
Approximately 88.5% of the 14,642 inferred proteins from the F. hepatica Oregon isolate found at least one BLAST hit (E < 1e-05) to non-Fasciola proteins in the non-redundant database (NR), with most matching sequences from other FBTs (particularly the liver flukes C. sinensis and O. viverrini). 11.5% of genes are Fasciola-specific with respect to NR (S3 Table), 4.9% of which were assigned additional functional annotations (Interpro domains, GO terms or KEGG orthologous groups; compared to 67.7% for genes with non-Fasciola NR matches). The putative Fasciola-specific genes were enriched for GO terms related to cysteine-type endopeptidase inhibitor activity and neurotransmitter secretion (S4 Table).
Protein conservation among flatworm parasites and their hosts was analyzed by clustering predicted proteins from 10 genomes into orthologous protein families (OPFs; Fig 1A), and 7,624 F. hepatica proteins were included in 5,721 unique OPFs with proteins from other flatworms including the free-living planarian Schmidtea mediterranea, trematodes (including schistosomes, the liver flukes C. sinensis and O. viverrini), and mammalian hosts (human, cow and sheep). Some 2,875 F. hepatica proteins (1,451 OPFs) were conserved across the 10 species, and these were more likely than other genes to be differentially expressed across developmental stages of F. hepatica (P = 7 x 10−5, binomial distribution test; S3 Table). In contrast, 393 F. hepatica genes (359 OPFs) were conserved between F. hepatica and at least one other FBT (C. sinensis and/or O. viverrini); these were significantly enriched for GO terms related to microtubule-based processes, cysteine endopeptidase activity and pH regulation (S4 Table). An additional 29 genes (29 OPFs) were conserved with at least one FBT and at least one host species; these were significantly enriched for the GO terms related to phagocytosis and L-ascorbic acid binding.
Fig 1. Protein families in trematodes.

(A) A Venn diagram demonstrating the phylogenetic distribution of orthologous protein families among the trematode species analyzed (B) Differential amplification of cathepsins L and F in trematodes. A maximum likelihood tree of the genes annotated as members of the C1A protease family from trematodes shows that while a single set of cathepsins F are detected in Schistosomes and F. hepatica strains, an expansion of cathepsins F is observed in O. viverrini and C. sinensis (green arch). Similarly, the cathepsins L-like of the trematodes (yellow arch) show a basal node more related to the mammalian enzymes and several independent amplifications in schistosomes, opistorchiids and Fasciola. Most known cathepsins variants are supported by expression data in different stages (red, green and blue bars), and proteomic data (yellow, green and blue dots) from a recent report [34]. Several putative novel variants are indicated, most of them not expressed at the stages analyzed. (C) Cathepsin B subfamily of the C1A protease family. A basal cathepsin B node and differential expansions in schistosomatids (red arch), fasciolids (blue arch) and opisthorchiids (purple arch) is observed. As in cathepsins L novel isoforms are identified, some of them supported by expression data. (D) Legumain is differentially amplified in food borne trematodes. Maximum likelihood tree of the genes annotated as members of the C13 protease family. While a single gene corresponding to the glycosyl-phosphatidyl-inositol anchor transamidase exists in all trematodes, amplification of the legumains was evident in the food borne trematodes F. hepatica, C. sinensis and O. viverrini, but not in the blood flukes S. mansoni and S. japonicum. These amplification events are independent in the different lineages. Species included in the tree are color coded (human, emerald dots, planaria (grey) cestodes (yellow) S. japonicum (dark red) S. mansoni (orange) C. sinensis (purple) O. viverrini (pink), F. hepatica Oregon strain (sky blue) F. hepatica Liverpool strain (navy).
OPF analysis also enabled the identification of gene sets specific to schistosomes; 1,365 OPFs conserved among at least two of the three species of Schistosoma were analyzed. Like the FBTs, these were significantly enriched for GO terms related to microtubule activity and cysteine endopeptidases (see below). This suggests that certain functions are conserved across the platyhelminth clades despite clear divergence at the sequences level, a possible indication of rapid evolution [22].
Expanded families of secreted/excreted proteases suggest a role in host-parasite interaction
Excreted and secreted proteins (ESPs) play a crucial role in parasitism. We identified 855 (5.8%) proteins with computationally predicted signal peptides but no transmembrane domains (S3 Table), indicating they may be secreted/ excreted. These proteins were significantly enriched for GO terms related to proteolysis (S4 Table). Again, the most significantly enriched molecular process GO term was “cysteine-type endopeptidase activity” (GO:0004197). Secreted cysteine proteases have a well-defined role in the biology of F. hepatica and liver fluke disease [23]. Cathepsin L’s are predominant in adult ESPs, where they participate in feeding, immune evasion and immune modulation. Distinct suites of cathepsin L’s and cathepsin B’s are abundant in the juvenile fluke, participating in excystment, migration through gut wall and liver capsule, and immune evasion [24, 25]. Although it was known that liver fluke cathepsins constitute a multigene family [26], the complexity and diversity within the family is now apparent. In addition to the six known cathepsin L’s, other isoforms were detected consistently in both the Oregon and the UK isolates, raising the total count to 14; most of these overexpressed in the adult stage (Fig 1B). Independent amplifications of cathepsin L’s occurred in schistosomes and the opisthorchiids, but the resulting gene copy number is less than in F. hepatica. In contrast, Cathepsin F’s showed a divergent pattern in these lineages, with single enzymes in Fasciola and schistosomes, and an amplified family in the carcinogenic fish-borne liver flukes (Fig 1B). A similar pattern of independent amplifications among trematodes was observed for cathepsin B’s (Fig 1C), again with a distinct expansion in F. hepatica. In contrast to cathepsin Ls, cathepsin Bs were overexpressed in metacercariae (MC), confirming biochemical, genetic and proteomic evidence of differential expression along the life cycle [27]. Interestingly, within both the cathepsins L and B, a clade comprising a single enzyme from each trematode species and vertebrates was identified, which might be basal to all the lineage-specific expansions. F. hepatica enzymes of this clade have not been described yet, and, notably, they are expressed in eggs (Fig 1B and 1C).
The remarkable amplification and diversity of secreted cysteine proteases in trematode lineages suggests key roles during parasite adaptation. Diverse trematodes express different (and amplified) subfamilies of cathepsins, reflecting their host parasite relationships, including host niche, organ sites, and transmission strategies. For example, cathepsins B and an L3 (CL3) participate in transit of the juvenile liver fluke through the gut wall with collagenolysis [28, 29], whereas juvenile of the fish-borne liver flukes ascend into the biliary tree through the ampulla of Vater [30]. In turn, cathepsins F and the aspartic protease cathepsin D are characteristically overrepresented in these carcinogenic liver flukes [19, 31]. The blood flukes, on the other hand, invade the skin of their hosts, with conserved serine proteases essential to this process, with the cysteine protease cathepsin B providing critical activities in some species [32].
Asparaginyl endopeptidases, Class C13 (also known as legumain) [33] were expanded in F. hepatica with ≥10 members, and were also differentially expanded among the trematodes; 3 copies in S. mansoni, 5 in S. japonicum, 4 in C. sinensis and ~ 100 in O. viverrini. (Fig 1D). These proteases might participate in the activation of cathepsins and the digestion of infected host tissues, liberating essential amino acids.
A recent proteomic study of ESPs from juvenile and adult F. hepatica provide further support for the differential expression of these gene families [34], confirming our transcriptomic data (S3 Table). For example, within the cathepsin B family, members of 10 out of 13 clusters are detected by LC-MS/MS in ESPs; while 3 isoforms are exclusively expressed by adults, 2 are characteristic of juveniles (also detected by RNAseq in metacercariae), and 5 are expressed in both stages but clearly predominant in juveniles (Fig 1C). Similarly, a predominance of expression of cathepsin Ls variants in adults is observed at proteomic level consistent with our transcriptomic data (Fig 1B). Within these some of the novel clades here described were detected as being expressed.
FBTs are metabolically less constrained than blood flukes and cestodes
Metabolic pathways predicted in the F. hepatica Oregon strain were compared to those of other sequenced flatworms. All parasitic flatworms showed a significant reduction in metabolic capabilities compared to free living platyhelminth species, including planaria (Fig 2). As shown previously [35], parasitic flatworms depend on the hosts for provision of fatty acids. Unlike the blood flukes, however, F. hepatica and the other liver flukes possess enzymatic pathways for fatty acid elongation by reversal of beta-oxidation (S3A Fig) and fatty acid catabolism (S3B Fig), allowing them to take advantage of the fatty acid rich environment of bile.
Fig 2. Metabolic pathway reduction in parasitic flatworms.
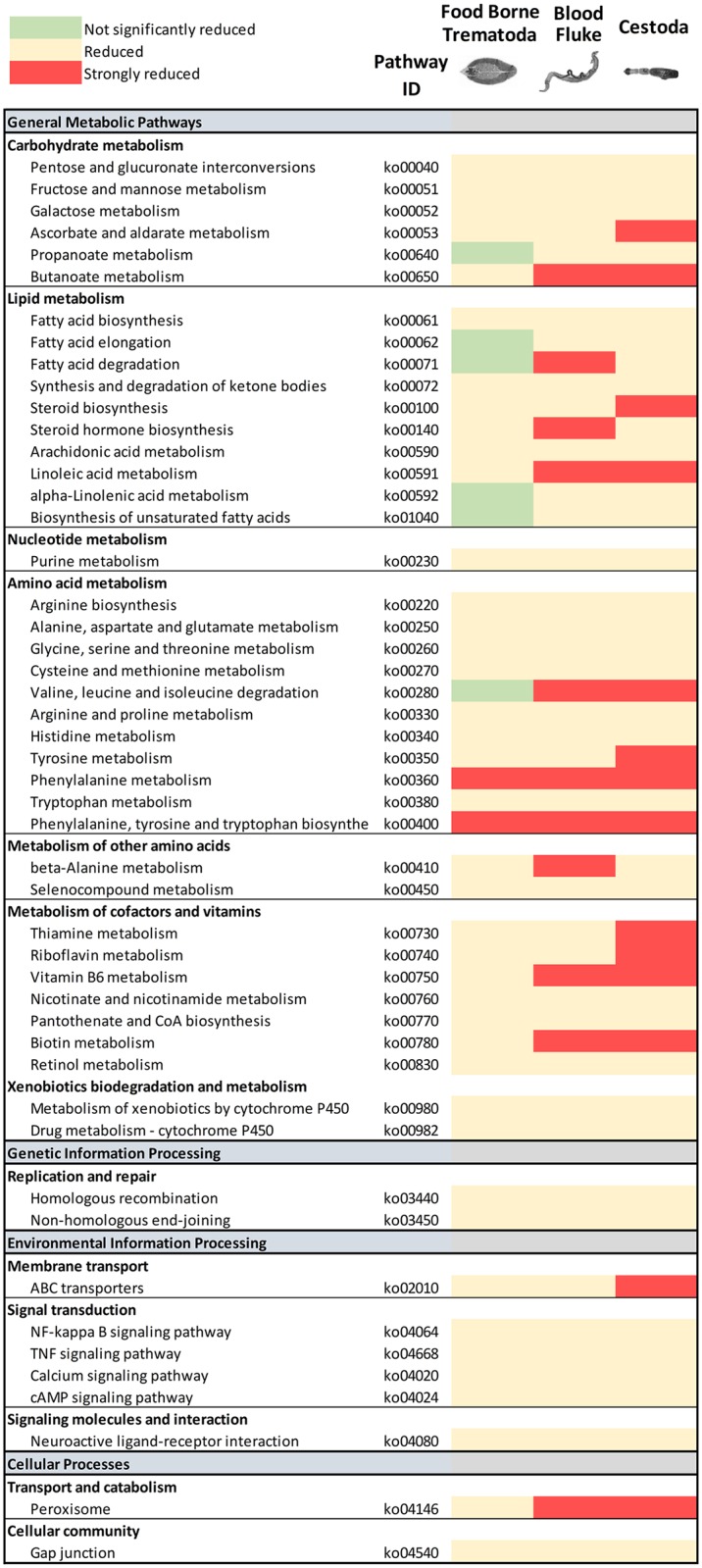
The global number of proteins assigned to different metabolic pathway was compared between different parasitic flatworms, the free-living planaria and cattle predicted proteomes. Those pathways showing a significant reduction in the parasitic species in relation to those present in planaria are indicated (strongly reduced: less than 30% conservation; reduced: conservation between 30 and 80%; not significantly reduced: more than 80% conservation). While in general several pathways are reduced in all parasitic species, some pathways are differential between food borne trematodes (F. hepatica, O. viverrini, C. sinensis), blood flukes (S. mansoni and S. japonicum) and cestodes (E. multilocularis, H. microstoma and T. solium). Most notably some lipid metabolism and amino acid pathways (i.e. aliphatic amino acid degradation) are not reduced in FBT (green) while they are reduced in the other groups. In general, FBT seem to be less constrained than blood flukes, with cestodes being the most restricted metabolically.
Additional differences between blood and liver flukes were evident in amino acid metabolism. Inabilities to synthesize several amino acids were generally observed in neodermatan flatworms, including Fasciola. However, the liver flukes operate a complete catabolic pathway of aliphatic amino acids, and enzymes of these pathways (e.g. branched chain amino acid aminotransferase [BCAT, EC. 2.6.1.42]) are missing in schistosomes (S3C Fig). Aliphatic amino acids are more abundant in the bile than in blood, which may have been exploited since it facilitates access to protein synthesis precursors and alternative energy sources. While expanded families of secreted proteases are involved in protein digestion, conserved oligopeptide transporters that mediate the uptake of di- and tri-peptides in metazoans were not identified [36], suggesting the lack of these specific transporters in trematodes at large. Consequently, protein digestion up to individual amino acids may occur extracellularly, explaining the rare presence of usually cytoplasmic enzymes as leucine aminopeptidase in the secreted products and vesicles released by the parasite [37].
Further metabolic differences may have evolved in liver flukes compared to blood flukes in relation to an environment characterized by low oxygen tension. Flukes switch from aerobic to anaerobic metabolism in the low oxygen environment of the bile duct, but instead of fermenting carbohydrates to lactate, the parasite exploits the more energy-efficient malate dismutation pathway [38], where phosphoenolpyruvate from glycolysis is converted to oxaloacetate via the phosphoenolpyruvate kinase (PEPCK), and further reduced to malate. After entering the mitochondria, some malate is oxidized to acetate, and some is reduced to succinate and transformed to propionate, in a series of reactions that reverts the Krebs cycle (Fig 3A), providing a source of electrons for the respiratory chain finally yielding five ATP molecules per glucose molecule. While the whole pathway was precisely described biochemically, we now for the first time identify the cognate enzymes (Fig 3A).
Fig 3. Metabolic pathways in F. hepatica.

(A) Energy metabolism in anaerobic mitochondria of F. hepatica by malate dismutase. While the classical anaerobic fermentation to lactate is present (enzymes indicated in gray) when oxygen tension is low, the malate dismutation pathway is preferred (blue arrows). Phosphenol pyruvate reduction to malate occurs in the cytoplasm (orange). Within the mitochondria, part of the malate is oxidized to acetate (yellow) while other fraction reduced to succinate and further transformed to propionate (blue). Genes predicted for key enzymes involved in anaerobic respiration are indicated. Abbreviations: PEP, phosphenolpyruvate; OXAC, oxaloacetate; MAL, malate; FUM, fumarate; SUCC, succinate; PYR, pyruvate; AcCoA, acetyl-CoA; CITR, citrate. Enzymes indicated are: PK, pyruvate kinase; LDH, lactate dehydrogenase, PEPCK, phosphenolpyruvate carboxykinase (ATP dependent); MDH, malate dehydrogenase; ME, malic enzyme; PDH, pyruvate dehydrogenase; ASCT, acetate:succinate CoA-transferase; SCS, succinyl-CoA synthetase; FH, fumarate hydratase; FRD, fumarate reductase; SDH, succinate dehydrogenase, MMM methylmalonyl-CoA mutase; PCC, propionyl-CoA carboxylase. (B) Parasite specific enzyme usage in TCA cycle. The KEGG module for TCA cycle (M00009) is shown with groups of orthologous enzymes indicated using KEGG orthology (KO) IDs. An interesting example of alternate enzyme usage is shown for fumarate hydratase (reaction R01082), catalyzed by a class II enzyme (EC 4.2.1.2B; K01679) in both the host and parasite. However, F. hepatica also has a Platyhelminthes-specific class I fumarate hydratase (EC 4.2.1.2A; K01676), not annotated in any of the host proteomes. Such knowledge can be leveraged to design worm specific therapies with potentially low (or no) impact on the host health.
To provide further insight into the metabolism of F. hepatica, we compared the metabolic pathway modules present and complete in F. hepatica, the carcinogenic liver flukes C. sinensis and O. viverrini, the blood fluke S. mansoni and mammalian hosts of F. hepatica (sheep, cow and human) were incorporated in the analysis (S4A Fig). Twenty-five KEGG pathway modules were complete in our assembly (i.e., contain the complement of KO’s necessary to convert the initial substrate to the final product based on strict completion [39]). These values are similar in other trematodes, but far lower than in mammals. Use of a lenient completion criterion of ≤2 missing steps in a module extended these values, with similar trends (S4B Fig). The analysis also identified modules that differ between the liver flukes and S. mansoni. Module M00020 (serine biosynthesis) also showed differences consistent with those observed in amino acid metabolism. Further differences in inositol phosphate metabolism were detected with two steps missing in S. mansoni (R03427 and R04372, INPP1 and INPP4 phosphatases). Module M00087 (beta-oxidation) occurs in liver flukes but is absent from schistosomes. Notably, this module revealed differences with the host, since in step 2 (R04738), F. hepatica shares two KOs with mammals, but there is an additional, putative platyhelminth-specific ortholog (K01692, EC 4.2.1.17) corresponding to enoyl coA-hydratase, which warrants investigation in flatworms. Differences between liver fluke and mammal were evident in module M00009m, corresponding to the tricarboxylic acid cycle (Fig 3B). The fumarate forming reaction (R01082) is dependent on fumarate hydratase class II enzyme (EC 4.2.1.2B, K01679) by the hosts, while a second fumarate hydratase, class I enzyme for this step (EC 4.2.1.2A, K01676), was detected in trematodes, an enzyme that might participate in the reverse step of the malate dismutation pathway (above).
A complete Neorickettsia genome identified in F. hepatica Oregon and Uruguay
The most striking feature of the F. hepatica Oregon isolate was an apparent infection with Neorickettsia endobacteria (nFh). Alpha-protobacterial sequences were first identified among “contaminating” sequences in the F. hepatica genome and the presence of Neorickettsia was confirmed and validated by both PCR and 16S rRNA sequencing (S5 Fig). The genome of nFh was assembled from 241,957 2x100bp read pairs that were identified during the sequencing of F. hepatica Oregon. A single 859,205 bp scaffold with average 56.3x sequence coverage was constructed from two contigs joined by 189 bp of inferred gaps (Fig 4). This novel Neorickettsia genome was similar in size and GC content to those of previously sequenced Neorickettsia species [40, 41]. Full genome alignments indicated that, with the exception of a small inversion, it shared nearly complete synteny with the genomes of Neorickettsia risticii and N. sennetsu (Fig 5A). Synteny among Neorickettsia species may reflect the lethality of large genome rearrangements due to a reduced set of DNA repair genes, but this may have increased the genetic variation in a stable intra-trematode environment by accumulation of mutations in non-essential genes [41].
Fig 4. The genome of the Neorickettsia endobacterium of Fasciola hepatica.

The first track (from outside to inside) represents the 859,205bp genome of the Neorickettsia endobacterium of Fasciola hepatica (100 kb major ticks, 10 kb minor ticks). The genome shows nearly complete synteny with Neorickettsia risticii and Neorickettsia sennetsu with the exception of an inversion (shaded in grey). The second and third tracks represent the 744 inferred protein coding genes on the plus and minus strands, respectively. Genes are coded based on their NCBI Clusters of Orthologous Groups of proteins database classification. The fourth track represents RNA coding genes, including 3 ribosomal (red), 33 transfer (black), and 1 short, noncoding (blue). The fifth track depicts the G-C skew [(G-C)/(G+C)] calculated over 500bp windows.
Fig 5. Phylogenetic affinities of the Neorickettsia symbiont of Fasciola hepatica.

(A) The genomes of the four fully sequenced Neorickettsia species were aligned. Syntenic blocks are colored. The genome of the Neorickettsia of Fasciola hepatica (PRJNA295290) shares almost complete synteny with the genomes of Neorickettsia risticii (PRJNA19099) and Neorickettsia sennetsu (PRJNA357), with the exception of a small inversion that is also present in Neorickettsia helminthoeca (PRJNA187358). (B) Bayesian inference phylogenetic analysis based on available 16S ribosomal RNA sequences of Neorickettsia species, retrieved from [15]. While the resolution of the sub-clade consisting of Neorickettsia risticii and Neorickettsia sennetsu is sub-optimal, the tree indicates that the Neorickettsia of Fasciola hepatica (nFh) may be most closely related to a strain that occurs in Metagonimoides species and an agent of Sennetsu fever. (C) A Bayesian inference phylogenetic tree based on the protein sequences of 473 single-copy, orthologous protein families conserved in the represented species clearly indicates that nFh is more closely related to N. risticii and N. sennetsu than N. helminthoeca or other species of the family Anaplasmataceae. NCBI GenBank accession numbers are indicated. Trematode hosts or other defining features are indicated for uncharacterized species.
Table 1 outlines the inferred features of nFh. Similar to related species, nFh encodes 33 tRNA genes and one copy each of 5S, 16S, and 23S rRNA genes. A total of 744 protein-coding genes were predicted, slightly fewer than N. risticii and N. sennetsu (Table 1). Gene conservation analysis among representative bacterial species of the Anaplasmataceae identified three orthologous protein families (OPFs) that were conserved in all analyzed species except nFh (S5 Table). Closer inspection revealed that these genes may be present and intact, although absent from the gene calls. Two additional OPFs were conserved in all sequenced Neorickettsia except nFh (S5 Table), but in both of these cases the corresponding sequences were identified but stop codons appeared to disrupt them.
Table 1. The sequenced genomes of Neorickettsia species.
| Neorickettsia of F. hepatica | Neorickettsia sennetsu | Neorickettsia risticii | Neorickettsia helminthoeca | |
|---|---|---|---|---|
| Trematode host | F. hepatica | Unknown | Acanthatrium orgonense | Nanophyetus salmincola |
| Vertebrate host | Unknown | Human | Horse, bat | Canids |
| Disease | Unknown | Sennetsu fever | Potomac horse fever | Salmon poisoning of dogs |
| RefSeq Accession | NZ_AGCN 00000000.1 |
NC_007798.1 | NC_013009.1 | NZ_CP007481.1 |
| Assembly size | 859,205 bp | 859,006 bp | 879,977 bp | 884,232 bp |
| Length of inferred gaps | 189 bp | --- | --- | --- |
| GC content | 41.4% | 41.1% | 41.3% | 41.7% |
| rRNA | 3 | 3 | 3 | 3 |
| tRNA | 33 | 33 | 33 | 33 |
| ncRNA | 1 | 3 | 1 | 1 |
| Pseudo genes | 8 | 2 | 11 | 17 |
| Protein coding genes | 744 | 753 | 759 | 772 |
| Average CDS length | 968 bp | 966 bp | 962 | 970 |
| Minimum CDS length | 4,761 bp | 5,766 bp | 4,761 bp | 4,776 bp |
| Maximum CDS length | 156 bp | 156 bp | 147 bp | 135 bp |
| % coding | 83.8% | 84.7% | 82.9% | 84.6% |
S5 Table presents a functional annotation of the 744 predicted proteins of nFh, including (a) 1,453 unique InterPro protein domains predicted from 620 proteins and associated with 596 unique gene ontology (GO) terms, (b) 720 proteins associated with 509 KEGG orthologous groups, further binned into 120 enzymatic pathways and 101 pathway modules, (c) 25 proteins classified as putative proteases, (d) two protease inhibitors, and (e) 25 proteins with secretion signals (which were enriched for biological process GO terms related to proteolysis and protein transport; S4 Table). Protein transporters such as porins, identified in previous proteomic studies might transport nutrients from the host cytoplasm [42]. Whether Neorickettsia enzymes interact with those of the fluke is a fascinating but unresolved question. Revealingly, however, N. risticii synthesizes nucleotides, vitamins, and cofactors that the fluke cannot, raising the possibility that they may be harvested by the trematode for their mutual advantage [41].
While sequencing reads from the previously reported UK strains (SRA Project ID: ERP006249) did not map to our nFh genome (Fig 6), suggesting that no Neorickettsia DNA was present in the samples, one of our Uruguay isolate (out of five that were screened) tested positive for the presence of Neorickettsia by 16S rDNA PCR (S5 Fig). Whole genome sequencing of this sample recovered the genome of nFh (99.9% breadth of genome coverage; S6 Table), allowing a comparative analysis of sequence variation in both the Neorickettsia and the fluke genomes. In total, 15 single nucleotide variants were identified between the two nFh genomes, 11 of which occurred within the coding regions (7 non-synonymous and 4 synonymous variants; S7 Table). Notably, 2-C-methyl-D-erythritol 4-phosphate cytidylyltransferase (EC 2.7.7.60; AS219_00645), a key enzyme in the MEP pathway of isoprenoid biosynthesis [43], was found among the genes that harbored non-synonymous SNPs. It has been hypothesized that the Wolbachia endosymbionts of Brugia malayi and Dirofilaria immitis rely on their helminth host for the completion of the MEP pathway [44, 45]. Additionally, in many pathogenic and opportunistic bacteria, the MEP pathway intermediate (HMB-PP) has the capacity to modulate vertebrate host immune response [46], suggesting an interesting possibility of the involvement of the isoprenoid biosynthesis pathway in the host-parasite-endosymbiont interactions. Overall, the genetic distance (1-ibs, identity by state) between the Oregon (US) and Uruguay (UY) nFh genomes (1.75 × 10−5) was three orders of magnitude lower than that estimated between the respective nuclear genomes of F. hepatica (1.08 × 10−2; S8 Table). The observed level of genetic divergence between the US and UY isolates indicated that these flukes are not substantially more closely related to each other than either is to the five published UK isolates (S8 Table) although their Neorickettsia endosymbionts are genetically close to each other.
Fig 6. Immunofluorescence detection of Neorickettsia in adult Fasciola hepatica using polyclonal anti-serum raised against a recombinant surface protein of Neorickettsia of P. elegans (PeNsp-3, green labeling).

DAPI (blue) and wheat hemagglutinin (red) were used to detect double stranded DNA and plasma membranes, respectively. (A) No green labeling was seen in the tegument of F. hepatica from Uruguay that were known to be devoid of Neorickettsia. (B) Clusters of Neorickettsia (arrows) in the tegument close to tegumental nuclei in F. hepatica from Oregon. (C) Numerous ‘donut’-shaped endobacteria (arrows) in the parenchyma in F. hepatica from Oregon. (D) Labeling of large numbers of Neorickettsia in the Mehlis’ gland and labeling of single endobacterium in the ootype or intrauterine eggs of F. hepatica from Oregon. (E) Magnification of a region proximal to (D) showing granular staining of single endobacterium in intrauterine eggs. (F) Clusters of Neorickettsia rods (arrows) in the cytoplasm of Mehlis’ cells of F. hepatica Oregon. (G) Individual Neorickettsia endobacteria (arrows) in a vitelline follicle with different stages of vitelline cells of F. hepatica Oregon. (H) No green staining indicative of Neorickettsia were found in the testis of F. hepatica from Uruguay tested negative for Neorickettsia by PCR. (I) Neorickettsia endobacteria (arrows) in the testis of F. hepatica Oregon with spermatogonia in the periphery and developing spermatozoa in the center. N, nucleus; Ts, tegument spine; P, parenchyma; Mg, Mehlis’ gland; Ot, ootype; Ut, uterus; Mc, Mehlis’ cell; Sg1/2, primary and secondary spermatogonia; Bar corresponds in A-B, D-I to 100 μm and in C to 1 μm.
Phylogenetic affinities of Neorickettsia endobacteria of F. hepatica
A phylogenetic analysis was undertaken using the 16S rRNA sequences from nFh and 16 other species and isolates; the findings were similar to those previous reports [15]. Based on the 16S rRNA locus, nFh is closely related to the agent of Sennetsu fever (a strain of N. sennetsu) and a Neorickettsia isolate isolated from species of Metagonimoides (Heterophyidae) (Fig 5B). A complementary phylogenetic analysis was undertaken with conserved, single-copy homologues from sequenced species of Neorickettsia and representatives of the Anaplasmataceae (Fig 5C); in regard to this collection of 473 gene families, nFh appeared to be closer to N. risticii and N. sennetsu than to N. helminthoeca, the agent of salmon poisoning in dogs, consistent with synteny-based observations.
Approximately 97% of the predicted proteins of nFh showed BLASTP matches in NR (e-value ≤ 1e-05, S5 Table). The top hits were to N. risticii or other Neorickettsia species. Indeed, most nFh genes (721 of 744) belonged to 719 OPFs shared with other Neorickettsia species (S5 Table). Of the 22 genes that were excluded from OPFs, 17 failed to find a match in NR and most lacked other functional annotations; the other five matched to hypothetical proteins from N. risticii and N. sennetsu. A single OPF was identified with members from nFh, Wolbachia species, Anaplasma phagocytophilum and Ehrlichia chaffeensis. The nFh gene assigned to this OPF was annotated as a replicative DNA helicase. Further assessment will be needed to validate these genes and explore their roles.
A set of 625 OPFs contained members common to all four sequenced Neorickettsia genomes; 83 of these OPFs were specific to Neorickettsia. The nFh proteins included in the Neorickettsia-specific OPFs were enriched for cellular component GO terms related to the outer membrane and biological process GO terms related to transport (S4 Table). The association with the cell surface suggested a role in endobacterial-digenean host interactions.
Neorickettsia-like endobacteria localized within tissues of F. hepatica Oregon
Although reports of Neorickettsia-infected trematodes have emerged, they relied on detection by PCR; localization within the trematode was poorly established. Gibson et al. [47] employed Ig from the serum of a horse infected with N. risticii to detect Neorickettsia in eggs from the bat-infecting trematode Acanthatrium oregonense to support the hypothesis of vertical transmission. The same serum was used to localize Neorickettsia in discrete developmental stages of Plagiorchis elegans, a trematode of rodents and birds [14]. We attempted to use the horse serum to localize nFh in adult F. hepatica Oregon, but background staining interfered with interpretation of the signals. However, polyclonal antibodies raised against recombinant surface protein-3 of P. elegans Neorickettsia (PeNsp-3) provided useful to support localization studies (Fig 6). The PeNsp-3 protein (Genbank KX082665) and of nFh (AS219_03540; S5 Table) share 98% identity. Minimal background signal occurred with the PeNsp-3 antisera, and nFh were sensitively detected as a ‘donut’-shaped structure surrounding the blue DAPI-stained nucleus, consistent with the staining pattern expected for surface proteins (Fig 6C) [48]. Whereas staining of Neorickettsia surface protein was not observed in Neorickettsia-negative (as confirmed by PCR; Fig 6A and 6H) F. hepatica from Uruguay endobacteria were detected in six of six individual adult F. hepatica worms from Oregon.
Because of the size of F. hepatica (~2 cm; F) and because Neorickettsia may be transmitted vertically, analysis focused on intra-uterine eggs and reproductive tissues. Endobacteria were frequently detected in varying numbers in the ovary, ootype, Mehlis’ gland, vitelline glands and in intrauterine eggs (Fig 6D–6G) as well as mature eggs isolated from liver tissue (Fig 7). The presence of nFh in female reproductive tissue is highly suggestive of vertical transmission. Furthermore, we analyzed by PCR adult flukes obtained after an experimental infection with Oregon metacercariae, detecting a few individual worms positive for the presence of nFh (S7 Fig). More interestingly, eggs collected from this assay were both PCR positive and presented the characteristic images of Neorickettsia supports the notion of vertical transmission.
Fig 7. Immunofluorescence detection of Neorickettsia in eggs of F. hepatica from Oregon using polyclonal anti-serum raised against a recombinant surface protein of Neorickettsia of P. elegans (PeNsp-3, green labeling, D-F).

(A) Unstained eggs recovered from the liver by regular light microcopy. (B) and (C) Unstained eggs recovered from the liver by immunofluorescence microscopy using different filters demonstrating auto-fluorescence. (D-F) Cross-sections of eggs showing various amounts of Neorickettsia (arrows). Bar corresponds to 50 μm.
Surprisingly, we also observed nFh in the testis and other parts of the male reproductive organs (Fig 6I and S8 Fig). Although F. hepatica is a hermaphrodite, cross-fertilization is assumed to be the usual reproductive strategy [49]. The presence of Neorickettsia in spermatozoa and seminal fluid could provide an alternative route for fluke-to-fluke transmission, as it was described for tick-borne pathogens [50], though further studies will be needed to explore this possibility.
The somatic tissues of F. hepatica Oregon were mostly Neorickettsia-free. Clusters of nFh were occasionally seen in the tegument adjacent to some syncytial nuclei (Fig 6B) and in intestinal tissue, particularly near the oral suckers. Liver flukes use the oral suckers to penetrate the host tissues and anchor themselves to the bile ducts, thus providing a potential mechanism for fluke-to-host transmission of nFh. Several infectious diseases described in the medical and veterinary literature are attributable to Neorickettsia carried by digenean parasites; among the more relevant are the ‘Salmon Poisoning Disease’ (SPD) of dogs and the Elokomin fluke fever (EFF) of fish-eating mammals in the west coast of North America, Sennetsu fever described in humans mainly in Japan and southeast Asia, and Potomac Horse Fever (PHF) in the east coast of North America [15, 16]. Notably, PHF, also known as ‘churrido equino’, has been described in horses in the Lake Merin region of Uruguay and Brazil (reviewed in [51]). Several species of Neorickettsia based on pathology, serology, antigen profile and/or genomic sequence, are considered the causative agents for these diseases; in particular, Neorickettsia (Ehrlichia) sennetsu causes acute, debilitating, mononucleosis-like disease [52], and has been implicated as a significant cause of human fevers of unknown etiology in southeastern Asia [53, 54].
Whereas the disease potential of Neorickettsia found in F. hepatica remains to be established, the F. hepatica-nFh association should be explored as a cryptic rickettsial pathogen of humans and ruminants in regions endemic for fasciolosis [55]. Additionally, more thorough studies of both the vertical transmission of nFh among the developmental stages of the liver fluke, and the potential horizontal transmission to the mammalian host might shine a light on the mechanisms behind the pathology induced by Neorickettsia endosymbionts of digenean parasites.
Methods
Liver flukes
Two isolates of Fasciola hepatica were analyzed: adult worms collected from livers of naturally infected sheep from a commercial slaughterhouse in Oregon (provided by Baldwin Aquatics Inc., Monmouth, Oregon), i.e. Oregon isolate; and worms isolated from livers of naturally infected sheep obtained from a commercial slaughterhouse in Montevideo, Uruguay, i.e. Uruguay isolate. For transcriptomic analysis, total RNAs were obtained from the egg, metacercarial and adult developmental stages (in duplicate). Eggs were collected from gall bladder of naturally infected sheep. Metacercariae were purchased from Baldwin Aquatics Inc. (Monmouth, Oregon). Tissue sections for the histological analysis were prepared from adult worms of the Oregon and Uruguay isolates.
DNA & RNA isolation, sequencing, assembly, annotation of the genome of F. hepatica Oregon isolate and transcriptome analysis
Fresh or ethanol-preserved adult worms were fragmented using a scalpel blade, and genomic DNA (gDNA) was extracted and purified using the kit E.Z.N.A. SQ Tissue DNA Kit (Omega Bio-tek), and the yield and purified assessed by Bio-Analyzer. Whole genome shotgun fragment and paired-end sequencing libraries (3 kb and 8 kb) were constructed from the gDNAs, as described [39, 56], and sequenced on the Illumina HiSeq2000 platform.
Linker and adapter sequences were trimmed, and cleaned reads were assembled using ALLPATHS-LG [57]. Pygap, an in-house assembly improvement tool, was used to join and extend contigs using unassembled reads when possible. Annotation of different features present in the assembly was done as previously described [58] and outlined in S1 Text.
Total RNA was extracted from eggs and adults from the gall bladder of naturally infected sheep and metacercariae (Baldwin Aquatics Inc., Monmouth, Oregon) using TRIzol reagent (Invitrogen/Life Technologies, Carlsbad, CA) according to the manufacturer’s instructions, and treated with Ambion Turbo DNase (Ambion/Applied Biosystems, Austin, TX). As previously described [59], RNA quality and yield were assessed, the purified RNA was poly(A) selected, reverse transcribed, paired-end cDNA libraries were generated, sequenced on the Illumina HiSeq 2000 platform and reads were analytically processed. Remaining, high-quality RNAseq reads (from one egg, two metacercariae and two adult biological replicates) were aligned to the genome assembly and constitutively expressed and differentially expressed genes were identified using standard protocols outlined in S1 Text.
Identification, assembly and annotation of the genome of Neorickettsia from F. hepatica Oregon, isolate nFh
A total of 126 contigs were identified as being from bacterial origin in the F. hepatica genome assembly, and BLAST analyses indicated significant homology to Neorickettsia species. The total complement of raw reads were re-mapped to the 126 Neorickettsia contigs using BWA-MEM version 7.10 with default parameters [60], and matching reads were assembled and assembly improved using standard protocols (see S1 Text). The genome assembly was annotated via the NCBI prokaryotic genome annotation pipeline [61].
Functional annotation of deduced proteins of F. hepatica and nFh, and MultiParanoid analyses
Deduced protein sequences were subjected to BLASTP against informative databases, including NCBI NR, InterPro, gene ontology (GO), KEGG, MEROPS using default cutoffs and release versions as specified in the S1 Text. Module completion was assessed as described [39], and transmembrane domains and classical secretion peptides were predicted using standard protocols (see S1 Text).
Inferred protein sequences of F. hepatica were compared to proteins from other trematodes & cognate mammalian hosts and from Neorickettsia were compared to proteins from representative species from the Anaphasmataceae, included all four fully sequenced Neorickettsia (accession numbers are provided in S1 Text). Orthologous protein families (OPFs) were constructed from pairwise InParanoid comparisons using MultiParanoid [62]. More detailed phylogenetic analyses at a level of rRNA sequences or single copy genes were performed as outlined in the S1 Text. F. hepatica Oregon and F. hepatica UK gene sets were compared using orthologs identified by Orthofinder v. 0.7.1 [63].
Genetic variation in Neorickettsia and F. hepatica nuclear genomes
We sequenced the genomic DNA of an F. hepatica isolate from Uruguay that was PCR-positive for Neorickettsia using the Illumina platform (2 × 100bp paired-end sequencing), as previously described [56]. We included the published genomes of the United Kingdom isolates (SRA Project ID: ERP006249) in the variant analysis to help contextualize our data. Genomic reads were mapped against the combined Oregon reference assembly of Neorickettsia and F. hepatica using bwa v0.7.15 [60], followed by removal of PCR and optical duplicates using picard tools v2.6.0 [64]. Single-nucleotide variants were called via local de-novo assembly of haplotypes using the GATK pipeline v3.6 [65]. The following set of quality filters were applied to obtain high-confidence SNP calls: DP (maximum depth) > median depth+(median absolute deviation×1.4826)×2; QD (variant confidence divided by the unfiltered depth of non-reference samples) < 2.0; FS (Phred-scaled p-value using Fisher’s Exact Test to detect strand bias in the reads) > 60.0; MQ (Root Mean Square of the mapping quality of the reads across all samples) < 40.0; MQRankSum (Mann-Whitney Rank Sum Test for mapping qualities) < -12.5; ReadPosRankSum (Mann-Whitney Rank Sum Test for the distance from the end of the read for reads with the alternate allele) < -8.0. Using SnpEff [66], variants were annotated based on their genomic locations and predicted coding effects. The genetic distance between isolates (1-ibs, identity by state) were computed using PLINK v1.90 after excluding loci with missing genotypes in any of the isolates.
Detection of Neorickettsia by PCR
To investigate vertical transmission of nFh, genomic DNA was extracted from individual worms obtained after two experimental infections in bovines (Bos taurus) performed at the Experimental Farm of the Institute of Hygiene, Montevideo, Uruguay, following international standards for care of research animals, and approved by the National Committee of Experimental Animal Health (CHEA). Polled Hereford calves negative to F. hepatica by fecal egg count and ELISA (Piacenza et al., 1999) and treated orally with ivemectin 1% (Mexiver, Laboratorios Santa Elena), were used in immunization studies that included challenge infection by mouth with 400 metacercariae (MCs). The MCs were obtained from Baldwin Aquatics, Oregon (assay 1) or DILAVE, Uruguay (assay 2). The cattle were euthanized at a commercial abattoir on week 20, and adult flukes were recovered from the liver of each of the calves, and flukes from each calf stored separately in >70% ethanol. Liver fluke eggs from the gall bladder of the calves also were recovered, and stored in the immunization group. DNA was extracted from individual worms of each assay, and from the pooled eggs, as described above. The presence of Neorickettsia within the F. hepatica adult flukes was investigated by nested PCR directed to the 16s rRNA gene, as described [67].
Histological examination of F. hepatica and its Neorickettsia endobacterium
Oregon strain flukes from sheep (and Neorickettsia-negative flukes from Uruguay) were fixed first in 70% ethanol and then in 10% buffered formalin overnight, tissue processed (Shandon 1000 Tissue Processor, Thermo Scientific, Waltham, MA, USA), embedded in paraffin, sectioned at 5 μm. Serial sections were used for immunohistochemical studies and hematoxylin & eosin staining (S6 Fig). The sections stained with hematoxylin & eosin (according to standard technique) were used to assess morphology and determine the anatomical structures to be expected on adjacent slides used for immunohistochemical localization of nFh. Unstained tissue sections were rehydrated and blocked with 5% bovine serum albumin (Sigma, St. Louis MO, USA) for 30 min to prevent non-specific antibody binding. Polyclonal mouse antisera raised against a recombinant Neorickettsia surface protein from Plagiorchis elegans (Genbank Accession KX082665, PeNsp-3) diluted 1:250 in phosphate buffered saline containing 0.1% Triton-X and 1% bovine serum albumin was used as the primary antibody. Anti-mouse IgG Alexa Fluor 488 (Invitrogen) was used as a secondary antibody for fluorescence microscopy. Wheat germ agglutinin 633 (200 μg/ml, Invitrogen, Carlsbad, CA, USA) and DAPI (Prolong Antifade with DAPI, Molecular Probes by Life Technologies, Carlsbad, CA, USA) were used to label membranes and double-stranded DNA, respectively. Sections were examined using a wide field fluorescence microscope (WFFM, Zeiss Axios Imager Upright Fluorescence Microscope) with plan-apochromat 100X oil, 63X or 40X objectives. Fluorescence microscopy was performed at the Washington University Molecular Microbiology Imaging Facility (http://micro.imaging.wustl.edu/).
Supporting Information
(A) Histogram indicates the size distribution of the predicted protein coding sequences of both Fasciola genomes. F. hepatica UK contains an abundance of very short genes (as small as 3bp) but more large (>600bp) genes. While the proteins predicted from the two genomes do not correspond well with one another (B), functional elements appear to be shared (C). KEGG Orthologous groups (KO) shared among the F. hepatica genomes.
(PDF)
Gene expression was profiled in eggs, metacercariae (MC), and sexually mature adults (hermaphrodites). (A) Clustering of samples based on gene expression (fragments per kilobase per million reads mapped, FPKM) indicated that eggs were more closely related to adults (which, themselves, contain eggs) than to metacercariae. (B) Differential expression of F. hepatica genes between the diverse egg, metacercariae and adult life cycle stages. Differentially expressed genes were significantly more likely than other genes to be phylogenetically conserved across all species test (P = 0.006) and more likely to contain transmembrane domains (P = 0.015). Genes with higher expression in metacercariae were enriched for several GO terms related to signal transduction and organismal development (S4 Table), and were less likely to be conserved with other FBTs (P = 1 x 10−7), which is not surprising given that F. hepatica metacercariae encyst on plants rather than within fish or crustaceans. In contrast, the 3,811 genes upregulated in adult flukes were enriched for microtubule based movement, redox regulation, and metabolic processes (S4 Table), as previously found in expression studies of F. hepatica from the UK [68]. The genes overexpressed in adults compared to metacercariae were more likely to be FBT conserved and specific (P = 2 x 10−7) and to have orthologs in mammals but not the free-living platyhelminth S. mediterranea (P = 6 x 10−10), suggesting potential roles in host interaction. (C) Summary of Illumina RNAseq reads, and SRA accessions.
(TIF)
(A) Fatty acid elongation by reversal of beta-oxidation (B) Fatty acid degradation (C) Aliphatic amino acid catabolism. Enzymes present in F. hepatica are identified in yellow, and those present in S. mansoni in blue. Image generated with the KEGGscan_pathway at trematode.net (http://trematode.net).
(TIF)
Clustering based on complete (A) and lenient (B) completion of KEGG metabolic pathways modules in each species (light green is “incomplete with < 3 reaction steps. Modules with 2 steps have been manually filled in after combining “strict” and “lenient” results). Oa = Ovis aries, Bt = Bos taurus, Hs = Homo sapiens, FhOREGON = F. hepatica, Oregon strain, FhUK = F. hepatica, UK strain, Sm = Schistosoma mansoni, Cs = Clonorchis sinensis, Ov = Opisthorchis viverrini.
(TIF)
Nested PCR for the bacterial 16s RNA gene in five different F. hepatica flukes from Uruguay (lanes 3–7) and the reference Oregon strain (8). nFh-positive signals were observed in one sample from Uruguay and the Oregon isolate.
(TIF)
(A) Cross-section of the proximal part of F. hepatica. (B) Cross-section of the distal part of F. hepatica. (C) Longitudinal section of F. hepatica. Bar corresponds to 1 mm.
(TIF)
Primary (top panel) and secondary (nested) PCR for the bacterial 16s RNA gene following protocol previously described [65]. DNA from nFh-negative sample from Uruguay (lane 11) and nFh-positive sample from Oregon (lane 13). To further test if the bacteria might have been transmitted through the parasite, we tested by PCR individual flukes isolated after two different experimental infections performed with metacercariae from Uruguay (lanes 3–10) and Oregon (lanes 16–23) respectively, and eggs collected from these experimental infections (lanes 1–2, and 14–15). Since nested PCR was performed using dilution of primary amplicons without band purification, carry over of first round primers occurred, visible as a doublet band below the primary amplification, corresponding to the expected nested product (lower band) and byproducts between the external and internal primers. The identity of the nFh 16s rRNA gene was confirmed by nucleotide sequencing.
(TIF)
(A) Cross-section of the vas deferens with overlay of the individual stains for plasma membranes (wheat germ agglutinin, WGA), double stranded DNA (DAPI) and Neorickettsia (Nsp). (B) Individual stain for plasma membranes (red). (C) Individual stain for DNA. Note the strong blue stain of spermatozoa and the lighter bluish stain of low DNA content Neorickettsia (arrows). D. Individual green stain for Neorickettsia (arrows). Bar corresponds to 100 μm.
(TIF)
(DOCX)
(DOCX)
(XLSX)
(XLSX)
(XLSX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
Acknowledgments
We thank Mr. Norman Baldwin, Baldwin Aquatics Inc. for developmental stages F. hepatica, Dr. Vasyl Tkach (University of North Dakota) for providing Neorickettsia-infected P. elegans material, and Dr. Yasuko Rikihisa (Ohio State University) for serum from a horse infected with Neorickettsia.
Data Availability
All sequence data from this project is available at NCBI. Raw reads from F. hepatica have been deposited in the GenBank sequence read archive (SRA), as BioProject PRJNA179522. Raw RNAseq reads from F. hepatica were submitted to the GenBank sequence read archive (SRA) under the same BioProject id, with the following accession numbers: SRX1037419, SRX1037422, SRX1037423, SRX1037421, SRX1037418. The F. hepatica Neorickettsia genome and predicted features are available under accession RefSeq NZ_AGCN00000000.1. All other relevant data are within the paper and its Supporting Information files.
Funding Statement
Sequencing of the genomes of Fasciola/Neorickettsia was supported by the ‘Sequencing the etiological agents of the Food-Borne Trematodiases’ project (NIH-NHGRI award number U54HG003079). The Barnes Jewish Hospital Foundation supported the immunolocalization studies. The Comisión Sectorial de Investigación Científica (CSIC-UDELAR) financially supported the Uruguayan co-authors. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Keiser J, Utzinger J. Food-borne trematodiases. Clin Microbiol Rev. 2009;22(3):466–83. 10.1128/CMR.00012-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hotez PJ, Alvarado M, Basanez MG, Bolliger I, Bourne R, Boussinesq M, et al. The global burden of disease study 2010: interpretation and implications for the neglected tropical diseases. PLoS Negl Trop Dis. 2014;8(7):e2865 10.1371/journal.pntd.0002865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Torgerson PR, Devleesschauwer B, Praet N, Speybroeck N, Willingham AL, Kasuga F, et al. World Health Organization Estimates of the Global and Regional Disease Burden of 11 Foodborne Parasitic Diseases, 2010: A Data Synthesis. PLoS Med. 2015;12(12):e1001920 10.1371/journal.pmed.1001920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nyindo M, Lukambagire AH. Fascioliasis: An Ongoing Zoonotic Trematode Infection. Biomed Res Int. 2015;2015:786195 10.1155/2015/786195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Charlier J, Vercruysse J, Morgan E, van Dijk J, Williams DJ. Recent advances in the diagnosis, impact on production and prediction of Fasciola hepatica in cattle. Parasitology. 2014;141(3):326–35. Epub 2013/11/16. 10.1017/S0031182013001662 [DOI] [PubMed] [Google Scholar]
- 6.Rondelaud D, Belfaiza M, Vignoles P, Moncef M, Dreyfuss G. Redial generations of Fasciola hepatica: a review. J Helminthol. 2009;83(3):245–54. 10.1017/S0022149X09222528 [DOI] [PubMed] [Google Scholar]
- 7.Burden DJ, Bland AP, Hammet NC, Hughes DL. Fasciola hepatica: migration of newly excysted juveniles in resistant rats. Experimental parasitology. 1983;56(2):277–88. Epub 1983/10/01. [DOI] [PubMed] [Google Scholar]
- 8.Lopez M, White AC Jr., Cabada MM. Burden of Fasciola hepatica Infection among children from Paucartambo in Cusco, Peru. The American journal of tropical medicine and hygiene. 2012;86(3):481–5. Epub 2012/03/10. 10.4269/ajtmh.2012.11-0448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marcos L, Terashima A, Leguia G, Canales M, Espinoza J, Gotuzzo E. La infección por Fasciola Hepática en el Perú: una enfermedad emergente. Revista de Gastroenterología del Perú. 2007;27:389–96. [PubMed] [Google Scholar]
- 10.Chai JY. Praziquantel treatment in trematode and cestode infections: an update. Infection & chemotherapy. 2013;45(1):32–43. Epub 2013/11/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kelley JM, Elliott TP, Beddoe T, Anderson G, Skuce P, Spithill TW. Current Threat of Triclabendazole Resistance in Fasciola hepatica. Trends Parasitol. 2016. [DOI] [PubMed] [Google Scholar]
- 12.Cabada MM, Lopez M, Cruz M, Delgado JR, Hill V, White AC Jr. Treatment Failure after Multiple Courses of Triclabendazole among Patients with Fascioliasis in Cusco, Peru: A Case Series. PLoS Negl Trop Dis. 2016;10(1):e0004361 10.1371/journal.pntd.0004361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gil LC, Diaz A, Rueda C, Martinez C, Castillo D, Apt W. Resistant human fasciolasis: report of four patients. Rev Med Chil. 2014;142(10):1330–3. 10.4067/S0034-98872014001000014 [DOI] [PubMed] [Google Scholar]
- 14.Greiman SE, Rikihisa Y, Cain J, Vaughan JA, Tkach VV. Germs within worms: localization of Neorickettsia sp. within life cycle stages of digenean plagiorchis elegans. Applied and environmental microbiology. 2016. Epub 2016/02/14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Greiman SE, Tkach VV, Pulis E, Fayton TJ, Curran SS. Large scale screening of digeneans for Neorickettsia endosymbionts using real-time PCR reveals new Neorickettsia genotypes, host associations and geographic records. PLoS One. 2014;9(6):e98453 10.1371/journal.pone.0098453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vaughan JA, Tkach VV, Greiman SE. Neorickettsial endosymbionts of the digenea: diversity, transmission and distribution. Adv Parasitol. 2012;79:253–97. 10.1016/B978-0-12-398457-9.00003-2 [DOI] [PubMed] [Google Scholar]
- 17.Parra G, Bradnam K, Korf I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics. 2007;23(9):1061–7. 10.1093/bioinformatics/btm071 [DOI] [PubMed] [Google Scholar]
- 18.Wang X, Chen W, Huang Y, Sun J, Men J, Liu H, et al. The draft genome of the carcinogenic human liver fluke Clonorchis sinensis. Genome Biol. 2011;12(10):R107 Epub 2011/10/26. 10.1186/gb-2011-12-10-r107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Young ND, Nagarajan N, Lin SJ, Korhonen PK, Jex AR, Hall RS, et al. The Opisthorchis viverrini genome provides insights into life in the bile duct. Nature communications. 2014;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang Y, Chen W, Wang X, Liu H, Chen Y, Guo L, et al. The carcinogenic liver fluke, Clonorchis sinensis: new assembly, reannotation and analysis of the genome and characterization of tissue transcriptomes. PLoS One. 2013;8(1):e54732 Epub 2013/02/06. 10.1371/journal.pone.0054732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Protasio AV, Tsai IJ, Babbage A, Nichol S, Hunt M, Aslett MA, et al. A Systematically Improved High Quality Genome and Transcriptome of the Human Blood Fluke Schistosoma mansoni. PLoS Negl Trop Dis. 2012;6(1):e1455 10.1371/journal.pntd.0001455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Irving JA, Spithill TW, Pike RN, Whisstock JC, Smooker PM. The evolution of enzyme specificity in Fasciola spp. J Mol Evol. 2003;57(1):1–15. 10.1007/s00239-002-2434-x [DOI] [PubMed] [Google Scholar]
- 23.Stack C, Dalton JP, Robinson MW. The phylogeny, structure and function of trematode cysteine proteases, with particular emphasis on the Fasciola hepatica cathepsin L family. Adv Exp Med Biol. 2011;712:116–35. 10.1007/978-1-4419-8414-2_8 [DOI] [PubMed] [Google Scholar]
- 24.Cancela M, Ruetalo N, Dell'Oca N, da Silva E, Smircich P, Rinaldi G, et al. Survey of transcripts expressed by the invasive juvenile stage of the liver fluke Fasciola hepatica. BMC Genomics. 2010;11:227 10.1186/1471-2164-11-227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cancela M, Acosta D, Rinaldi G, Silva E, Duran R, Roche L, et al. A distinctive repertoire of cathepsins is expressed by juvenile invasive Fasciola hepatica. Biochimie. 2008;90(10):1461–75. Epub 2008/06/25. 10.1016/j.biochi.2008.04.020 [DOI] [PubMed] [Google Scholar]
- 26.Robinson MW, Tort JF, Lowther J, Donnelly SM, Wong E, Xu W, et al. Proteomics and phylogenetic analysis of the cathepsin L protease family of the helminth pathogen Fasciola hepatica: expansion of a repertoire of virulence-associated factors. Mol Cell Proteomics. 2008;7. [DOI] [PubMed] [Google Scholar]
- 27.Lowther J, Robinson MW, Donnelly SM, Xu W, Stack CM, Matthews JM, et al. The Importance of pH in Regulating the Function of the Fasciola hepatica Cathepsin L1 Cysteine Protease. PLoS Negl Trop Dis. 2009;3(1):e369 10.1371/journal.pntd.0000369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Robinson MW, Corvo I, Jones PM, George AM, Padula MP, To J, et al. Collagenolytic activities of the major secreted cathepsin L peptidases involved in the virulence of the helminth pathogen, Fasciola hepatica. PLoS Negl Trop Dis. 2011;5(4):e1012 10.1371/journal.pntd.0001012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Corvo I, Cancela M, Cappetta M, Pi-Denis N, Tort JF, Roche L. The major cathepsin L secreted by the invasive juvenile Fasciola hepatica prefers proline in the S2 subsite and can cleave collagen. Mol Biochem Parasitol. 2009;167. [DOI] [PubMed] [Google Scholar]
- 30.Kim TI, Yoo WG, Kwak BK, Seok JW, Hong SJ. Tracing of the Bile-chemotactic migration of juvenile Clonorchis sinensis in rabbits by PET-CT. PLoS Negl Trop Dis. 2011;5(12):e1414 10.1371/journal.pntd.0001414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kang JM, Bahk YY, Cho PY, Hong SJ, Kim TS, Sohn WM, et al. A family of cathepsin F cysteine proteases of Clonorchis sinensis is the major secreted proteins that are expressed in the intestine of the parasite. Mol Biochem Parasitol. 2010;170(1):7–16. 10.1016/j.molbiopara.2009.11.006 [DOI] [PubMed] [Google Scholar]
- 32.Dvořák J, Mashiyama ST, Braschi S, Sajid M, Knudsen GM, Hansell E, et al. Differential use of protease families for invasion by schistosome cercariae. Biochimie. 2008;90(2):345–58. 10.1016/j.biochi.2007.08.013. [DOI] [PubMed] [Google Scholar]
- 33.Rawlings ND, Barrett AJ, Finn R. Twenty years of the MEROPS database of proteolytic enzymes, their substrates and inhibitors. Nucleic acids research. 2016;44(D1):D343–50. Epub 2015/11/04. 10.1093/nar/gkv1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Di Maggio LS, Tirloni L, Pinto AF, Diedrich JK, Yates Iii JR, Benavides U, et al. Across intra-mammalian stages of the liver fluke Fasciola hepatica: a proteomic study. Scientific reports. 2016;6:32796 10.1038/srep32796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsai IJ, Zarowiecki M, Holroyd N, Garciarrubio A, Sanchez-Flores A, Brooks KL, et al. The genomes of four tapeworm species reveal adaptations to parasitism. Nature. 2013;496(7443):57–63. 10.1038/nature12031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Daniel H, Spanier B, Kottra G, Weitz D. From bacteria to man: archaic proton-dependent peptide transporters at work. Physiology (Bethesda). 2006;21:93–102. [DOI] [PubMed] [Google Scholar]
- 37.Marcilla A, De la Rubia JE, Sotillo J, Bernal D, Carmona C, Villavicencio Z, et al. Leucine Aminopeptidase Is an Immunodominant Antigen of Fasciola hepatica Excretory and Secretory Products in Human Infections. Clinical and Vaccine Immunology: CVI. 2008;15(1):95–100. 10.1128/CVI.00338-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tielens AG, van Grinsven KW, Henze K, van Hellemond JJ, Martin W. Acetate formation in the energy metabolism of parasitic helminths and protists. Int J Parasitol. 2010;40(4):387–97. 10.1016/j.ijpara.2009.12.006 [DOI] [PubMed] [Google Scholar]
- 39.Tyagi R, Joachim A, Ruttkowski B, Rosa BA, Martin JC, Hallsworth-Pepin K, et al. Cracking the nodule worm code advances knowledge of parasite biology and biotechnology to tackle major diseases of livestock. Biotechnology advances. 2015;33(6 Pt 1):980–91. Epub 2015/06/01. 10.1016/j.biotechadv.2015.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dunning Hotopp JC, Lin M, Madupu R, Crabtree J, Angiuoli SV, Eisen JA, et al. Comparative genomics of emerging human ehrlichiosis agents. PLoS Genet. 2006;2(2):e21 10.1371/journal.pgen.0020021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lin M, Zhang C, Gibson K, Rikihisa Y. Analysis of complete genome sequence of Neorickettsia risticii: causative agent of Potomac horse fever. Nucleic acids research. 2009;37(18):6076–91. 10.1093/nar/gkp642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gibson K, Kumagai Y, Rikihisa Y. Proteomic analysis of Neorickettsia sennetsu surface-exposed proteins and porin activity of the major surface protein P51. J Bacteriol. 2010;192(22):5898–905. 10.1128/JB.00632-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tsang A, Seidle H, Jawaid S, Zhou W, Smith C, Couch RD. Francisella tularensis 2-C-Methyl-D-Erythritol 4-Phosphate Cytidylyltransferase: Kinetic Characterization and Phosphoregulation. PLoS One. 2011;6(6):e20884 10.1371/journal.pone.0020884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Luck AN, Evans CC, Riggs MD, Foster JM, Moorhead AR, Slatko BE, et al. Concurrent transcriptional profiling of Dirofilaria immitis and its Wolbachia endosymbiont throughout the nematode life cycle reveals coordinated gene expression. BMC Genomics. 2014;15(1):1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Foster J, Ganatra M, Kamal I, Ware J, Makarova K, Ivanova N, et al. The Wolbachia genome of Brugia malayi: endosymbiont evolution within a human pathogenic nematode. PLoS Biol. 2005;3(4):e121 10.1371/journal.pbio.0030121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Eberl M, Hintz M, Reichenberg A, Kollas AK, Wiesner J, Jomaa H. Microbial isoprenoid biosynthesis and human gammadelta T cell activation. FEBS letters. 2003;544(1–3):4–10. Epub 2003/06/05. [DOI] [PubMed] [Google Scholar]
- 47.Gibson KE, Rikihisa Y, Zhang C, Martin C. Neorickettsia risticii is vertically transmitted in the trematode Acanthatrium oregonense and horizontally transmitted to bats. Environmental microbiology. 2005;7(2):203–12. Epub 2005/01/22. 10.1111/j.1462-2920.2004.00683.x [DOI] [PubMed] [Google Scholar]
- 48.Fischer K, Beatty WL, Jiang D, Weil GJ, Fischer PU. Tissue and stage-specific distribution of Wolbachia in Brugia malayi. PLoS Negl Trop Dis. 2011;5(5):e1174 10.1371/journal.pntd.0001174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Peoples RC, Fried B. Form and function in the digenea. Adv Exp Med Biol. 2014;766:3–20. Epub 2014/06/07. 10.1007/978-1-4939-0915-5_1 [DOI] [PubMed] [Google Scholar]
- 50.Gaber MS, Khalil GM, Hoogstraal H. Borrelia crocidurae: venereal transfer in Egyptian Ornithodoros erraticus ticks. Exp Parasitol. 1982;54(2):182–4. Epub 1982/10/01. [DOI] [PubMed] [Google Scholar]
- 51.Dutra F, Schuch LF, Delucchi E, Curcio BR, Coimbra H, Raffi MB, et al. Equine monocytic Ehrlichiosis (Potomac horse fever) in horses in Uruguay and southern Brazil. J Vet Diagn Invest. 2001;13(5):433–7. Epub 2001/10/03. [DOI] [PubMed] [Google Scholar]
- 52.Rikihisa Y. The tribe Ehrlichieae and ehrlichial diseases. Clin Microbiol Rev. 1991;4(3):286–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Newton PN, Rolain JM, Rasachak B, Mayxay M, Vathanatham K, Seng P, et al. Sennetsu neorickettsiosis: a probable fish-borne cause of fever rediscovered in Laos. Am J Trop Med Hyg. 2009;81(2):190–4. [PMC free article] [PubMed] [Google Scholar]
- 54.Walker DH. Sennetsu neorickettsiosis: a potentially prevalent, treatable, acutely incapacitating tropical infectious disease. Am J Trop Med Hyg. 2009;81(2):187–8. [PubMed] [Google Scholar]
- 55.Walker DH, Ismail N. Emerging and re-emerging rickettsioses: endothelial cell infection and early disease events. Nat Rev Microbiol. 2008;6(5):375–86. 10.1038/nrmicro1866 [DOI] [PubMed] [Google Scholar]
- 56.McNulty SN, Strube C, Rosa BA, Martin JC, Tyagi R, Choi YJ, et al. Dictyocaulus viviparus genome, variome and transcriptome elucidate lungworm biology and support future intervention. Scientific reports. 2016;6:20316 Epub 2016/02/10. 10.1038/srep20316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gnerre S, Maccallum I, Przybylski D, Ribeiro FJ, Burton JN, Walker BJ, et al. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc Natl Acad Sci U S A. 2011;108(4):1513–8. 10.1073/pnas.1017351108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tang YT, Gao X, Rosa BA, Abubucker S, Hallsworth-Pepin K, Martin J, et al. Genome of the human hookworm Necator americanus. Nat Genet. 2014. Epub 2014/01/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McNulty SN, Fischer PU, Townsend RR, Curtis KC, Weil GJ, Mitreva M. Systems biology studies of adult paragonimus lung flukes facilitate the identification of immunodominant parasite antigens. PLoS Negl Trop Dis. 2014;8(10):e3242 Epub 2014/10/21. 10.1371/journal.pntd.0003242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–60. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Angiuoli SV, Gussman A, Klimke W, Cochrane G, Field D, Garrity G, et al. Toward an online repository of Standard Operating Procedures (SOPs) for (meta)genomic annotation. OMICS. 2008;12(2):137–41. 10.1089/omi.2008.0017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Alexeyenko A, Tamas I, Liu G, Sonnhammer EL. Automatic clustering of orthologs and inparalogs shared by multiple proteomes. Bioinformatics. 2006;22(14):e9–15. 10.1093/bioinformatics/btl213 [DOI] [PubMed] [Google Scholar]
- 63.Emms DM, Kelly S. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015;16:157 Epub 2015/08/06. 10.1186/s13059-015-0721-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43(5):491–8. 10.1038/ng.806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–303. Epub 2010/07/21. 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin). 2012;6(2):80–92. Epub 2012/06/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Greiman SE, Tkach VV, Vaughan JA. Transmission rates of the bacterial endosymbiont, Neorickettsia risticii, during the asexual reproduction phase of its digenean host, Plagiorchis elegans, within naturally infected lymnaeid snails. Parasit Vectors. 2013;6:303 10.1186/1756-3305-6-303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cwiklinski K, Dalton JP, Dufresne PJ, La Course J, Williams DJ, Hodgkinson J, et al. The Fasciola hepatica genome: gene duplication and polymorphism reveals adaptation to the host environment and the capacity for rapid evolution. Genome Biol. 2015;16:71 10.1186/s13059-015-0632-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Histogram indicates the size distribution of the predicted protein coding sequences of both Fasciola genomes. F. hepatica UK contains an abundance of very short genes (as small as 3bp) but more large (>600bp) genes. While the proteins predicted from the two genomes do not correspond well with one another (B), functional elements appear to be shared (C). KEGG Orthologous groups (KO) shared among the F. hepatica genomes.
(PDF)
Gene expression was profiled in eggs, metacercariae (MC), and sexually mature adults (hermaphrodites). (A) Clustering of samples based on gene expression (fragments per kilobase per million reads mapped, FPKM) indicated that eggs were more closely related to adults (which, themselves, contain eggs) than to metacercariae. (B) Differential expression of F. hepatica genes between the diverse egg, metacercariae and adult life cycle stages. Differentially expressed genes were significantly more likely than other genes to be phylogenetically conserved across all species test (P = 0.006) and more likely to contain transmembrane domains (P = 0.015). Genes with higher expression in metacercariae were enriched for several GO terms related to signal transduction and organismal development (S4 Table), and were less likely to be conserved with other FBTs (P = 1 x 10−7), which is not surprising given that F. hepatica metacercariae encyst on plants rather than within fish or crustaceans. In contrast, the 3,811 genes upregulated in adult flukes were enriched for microtubule based movement, redox regulation, and metabolic processes (S4 Table), as previously found in expression studies of F. hepatica from the UK [68]. The genes overexpressed in adults compared to metacercariae were more likely to be FBT conserved and specific (P = 2 x 10−7) and to have orthologs in mammals but not the free-living platyhelminth S. mediterranea (P = 6 x 10−10), suggesting potential roles in host interaction. (C) Summary of Illumina RNAseq reads, and SRA accessions.
(TIF)
(A) Fatty acid elongation by reversal of beta-oxidation (B) Fatty acid degradation (C) Aliphatic amino acid catabolism. Enzymes present in F. hepatica are identified in yellow, and those present in S. mansoni in blue. Image generated with the KEGGscan_pathway at trematode.net (http://trematode.net).
(TIF)
Clustering based on complete (A) and lenient (B) completion of KEGG metabolic pathways modules in each species (light green is “incomplete with < 3 reaction steps. Modules with 2 steps have been manually filled in after combining “strict” and “lenient” results). Oa = Ovis aries, Bt = Bos taurus, Hs = Homo sapiens, FhOREGON = F. hepatica, Oregon strain, FhUK = F. hepatica, UK strain, Sm = Schistosoma mansoni, Cs = Clonorchis sinensis, Ov = Opisthorchis viverrini.
(TIF)
Nested PCR for the bacterial 16s RNA gene in five different F. hepatica flukes from Uruguay (lanes 3–7) and the reference Oregon strain (8). nFh-positive signals were observed in one sample from Uruguay and the Oregon isolate.
(TIF)
(A) Cross-section of the proximal part of F. hepatica. (B) Cross-section of the distal part of F. hepatica. (C) Longitudinal section of F. hepatica. Bar corresponds to 1 mm.
(TIF)
Primary (top panel) and secondary (nested) PCR for the bacterial 16s RNA gene following protocol previously described [65]. DNA from nFh-negative sample from Uruguay (lane 11) and nFh-positive sample from Oregon (lane 13). To further test if the bacteria might have been transmitted through the parasite, we tested by PCR individual flukes isolated after two different experimental infections performed with metacercariae from Uruguay (lanes 3–10) and Oregon (lanes 16–23) respectively, and eggs collected from these experimental infections (lanes 1–2, and 14–15). Since nested PCR was performed using dilution of primary amplicons without band purification, carry over of first round primers occurred, visible as a doublet band below the primary amplification, corresponding to the expected nested product (lower band) and byproducts between the external and internal primers. The identity of the nFh 16s rRNA gene was confirmed by nucleotide sequencing.
(TIF)
(A) Cross-section of the vas deferens with overlay of the individual stains for plasma membranes (wheat germ agglutinin, WGA), double stranded DNA (DAPI) and Neorickettsia (Nsp). (B) Individual stain for plasma membranes (red). (C) Individual stain for DNA. Note the strong blue stain of spermatozoa and the lighter bluish stain of low DNA content Neorickettsia (arrows). D. Individual green stain for Neorickettsia (arrows). Bar corresponds to 100 μm.
(TIF)
(DOCX)
(DOCX)
(XLSX)
(XLSX)
(XLSX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
Data Availability Statement
All sequence data from this project is available at NCBI. Raw reads from F. hepatica have been deposited in the GenBank sequence read archive (SRA), as BioProject PRJNA179522. Raw RNAseq reads from F. hepatica were submitted to the GenBank sequence read archive (SRA) under the same BioProject id, with the following accession numbers: SRX1037419, SRX1037422, SRX1037423, SRX1037421, SRX1037418. The F. hepatica Neorickettsia genome and predicted features are available under accession RefSeq NZ_AGCN00000000.1. All other relevant data are within the paper and its Supporting Information files.